- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Cell Type-specific mRNA Purification in Caenorhabditis elegans via Translating Ribosome Affinity Purification
Published: Vol 9, Iss 15, Aug 5, 2019 DOI: 10.21769/BioProtoc.3328 Views: 6090
Reviewed by: Anand Ramesh PatwardhanSaumik BasuAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Cell type-specific molecular profiling is widely used to gain new insights into the diverse cell types that make up complex biological tissues. Translating ribosome affinity purification (TRAP) is a method in which the cell type-specific expression of epitope-tagged ribosomal subunits allows one to purify actively translating mRNAs without the need for cell sorting or fixation. We adapted this method for use in C. elegans to identify novel transcripts in single cell types or to identify the effects of environmental changes on the transcriptomes of larger cohorts of cells. In this protocol, we describe methods to generate transgenic animals bearing tagged ribosomes in cells of interest, prepare these animals for immunoprecipitation, purify ribosome-mRNA complexes, and obtain purified mRNA for next-generation sequencing.
Keywords: RNA purificationBackground
With the advent of next-generation sequencing technologies, transcriptome profiling of defined cell populations and single cells has reached new heights of popularity. Many new techniques to isolate mRNA from select cells have arisen as a consequence. Translating Ribosome Affinity Purification (TRAP), or ribotagging, is a method to purify polysomal RNA from genetically-defined cell types (Heiman et al., 2008; Sanz et al., 2009; Heiman et al., 2014). In this method, epitope-tagged ribosomes are expressed in cells of interest, then tissue is extracted in the presence of cycloheximide, which stalls the ribosomes on the mRNAs that they are translating and thereby preserves a snapshot of translation. Tagged ribosomes are then affinity purified and the isolated RNA can be analyzed via quantitative PCR, microarrays, or RNA sequencing. This technique is advantageous because it does not require fixation or sorting of cells, has a high degree of sensitivity, and detects only the translated pool of mRNAs.
We recently adapted the TRAP methodology for use in the nematode Caenorhabditis elegans. We have used this method to identify novel transcripts that are enriched in the serotonergic neurosecretory motor (NSM) neuron (Rhoades et al., 2019) and to identify transcripts that vary with environmental changes in the entire nervous system (unpublished results). TRAP is particularly valuable in C. elegans because it is relatively easy to generate new transgenic lines for any cell type of interest, and because it can provide a snapshot of gene expression following an experimental manipulation. TRAP complements previous methods for neuron-specific mRNA isolation in larval and adult C. elegans, such as fluorescence-activated cell sorting (FACS) of dissociated cells (Spencer et al., 2014, Kaletsky et al., 2018) and affinity purification with epitope-tagged poly(A) binding protein (Roy et al., 2002, Takayama et al., 2010).
A TRAP protocol has previously been described in C. elegans (Gracida and Calarco, 2017). Our protocols share many similarities as both are derived from original protocols developed for mouse tissue and cell culture. Some key differences are the use of a distinct ribosomal subunit (tagged rpl-1 versus rpl-22 here) and epitope tag (GFP versus HA here). Both of these approaches have been successfully used in mammalian systems and appear to be sensitive and robust in C. elegans. The method that we describe here has successfully isolated mRNA from single C. elegans neuronal cell types (Rhoades et al., 2019).
Materials and Reagents
- 15 cm Petri dishes (Corning, catalog number: 351058)
- RNase-free pipette tips
- Glass slides (ThermoFisher, catalog number: 3011-002)
- 1.5 ml low-adhesion RNAse-free microcentrifuge tubes (USA Scientific, catalog number: 1415-2600)
- 50 ml conical centrifuge tubes (Corning, catalog number: 352098)
- 15 ml conical centrifuge tubes (Corning, catalog number: 352097)
- RNase-free 20 µl filter tips (Rainin, catalog number: RT-20F)
- RNase-free 200 µl filter tips (Rainin, catalog number: RT-200F)
- RNase-free 1,000 µl filter tips (Rainin, catalog number: RT-1000F)
- RNase-free disposable serological pipettes (Corning, catalog number: EF20361E)
- RNAse-free disposable pellet pestles with tubes (Thermo Fisher Scientific, catalog number: 12-141-368)
- Caenorhabditis elegans (100 young adult animals)
- rpl-22-3xHA plasmid (Addgene, catalog number: 122203)
- FseI and AscI restriction enzymes (New England Biolabs, catalog numbers: R0588, R0558)
- Liquid nitrogen
- 1,2-diheptanoyl-sn-glycero-3-phosphocholine (DHPC; Avanti, catalog number: 850306P)
- Dynabeads Protein G (Thermo Fisher Scientific, catalog number: 10004D)
- Monoclonal anti-HA antibody, purified, clone HA-7 (Sigma-Aldrich, catalog number: H3663-200UL)
- 1 M MgCl2 (Ambion, catalog number: 9503G)
- 2 M KCl (Ambion, catalog number: 9604G)
- Nuclease-free H2O (1 L: Ambion, catalog number: 4387936; 100 ml: Ambion, catalog number: 9939)
- RNAse Zap (Ambion, catalog number: AM9782)
- RNAsin RNAse inhibitor (Promega, catalog number: N2515)
- Complete Mini, EDTA-free Protease Inhibitor Tablets (Roche, catalog number:4693124)
- DTT (Sigma-Aldrich, catalog number:D9779-5G)
- Cycloheximide (Sigma-Aldrich, catalog number: C7698-5G)
- Igepal Ca-630 (NP-40) (Sigma-Aldrich, catalog number: I8896-50ML)
- Tetramethyl Sulfolane (for use with Absolutely RNA Nanoprep Kit) (Sigma-Aldrich, catalog number: T2209-100G)
- Absolutely RNA Nanoprep Kit (Agilent, catalog number: 400753). Kit includes the beta-mercaptoethanol (BME).
- Ribonucleoside Vanadyl Complex (VRC) (New England Biolabs, catalog number: S1402S)
- 1 M HEPES, pH 7.3 (Thermo Fisher Scientific, catalog number: J16924AE)
- Methanol (Sigma-Aldrich, catalog number: 494437)
- Ethanol (for use with Absolutely RNA Nanoprep Kit) (Sigma-Aldrich, catalog number: E7023)
- Standard laboratory reagents for C. elegans maintenance, molecular cloning, and transgenesis
- Sodium Hypochlorite Solution (Fisher, catalog number: SS290-1)
- Enriched-peptone nematode growth medium (NGM) plates (see Recipes)
- Liquid NGM (standard NGM without agar) (see Recipes)
- 100 mg/ml cycloheximide (see Recipes)
- 300 mM DHPC stock (see Recipes)
- Homogenization buffer (stock) (see Recipes)
- Homogenization buffer with fresh ingredients (see Recipes)
- 150 mM KCl wash buffer (stock) (see Recipes)
- 150 mM KCl wash buffer with fresh ingredients (see Recipes)
- 350 mM KCl wash buffer (stock) (see Recipes)
- 350 mM KCl wash buffer with fresh ingredients (see Recipes)
Equipment
- Pipettes
- -80 °C freezer
- Benchtop microcentrifuge (Thermo Fisher Scientific, catalog number: 75002411 or equivalent)
- Benchtop centrifuge (Thermo Fisher Scientific, catalog number: 75004504 or equivalent)
- Dynamag-2 magnetic rack for 1.5 ml microcentrifuge tubes (Thermo Fisher Scientific, catalog number: 12321D)
- Dynamag-15 magnetic rack for 15 ml conical tubes (optional; Thermo Fisher Scientific, catalog number: 12301D)
- Nanodrop One Spectrophotometer (Thermo Fisher Scientific, model: ND-ONE-W)
- Cold room or refrigerator with access to electrical outlet (4 °C)
- Forceps (Fine Science Tools)
- Autoclave
- Standard laboratory equipment for C. elegans maintenance, molecular cloning, and transgenesis
Procedure
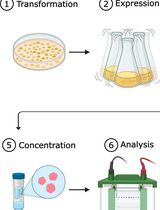
Note: This protocol assumes basic familiarity with C. elegans maintenance and transgenesis.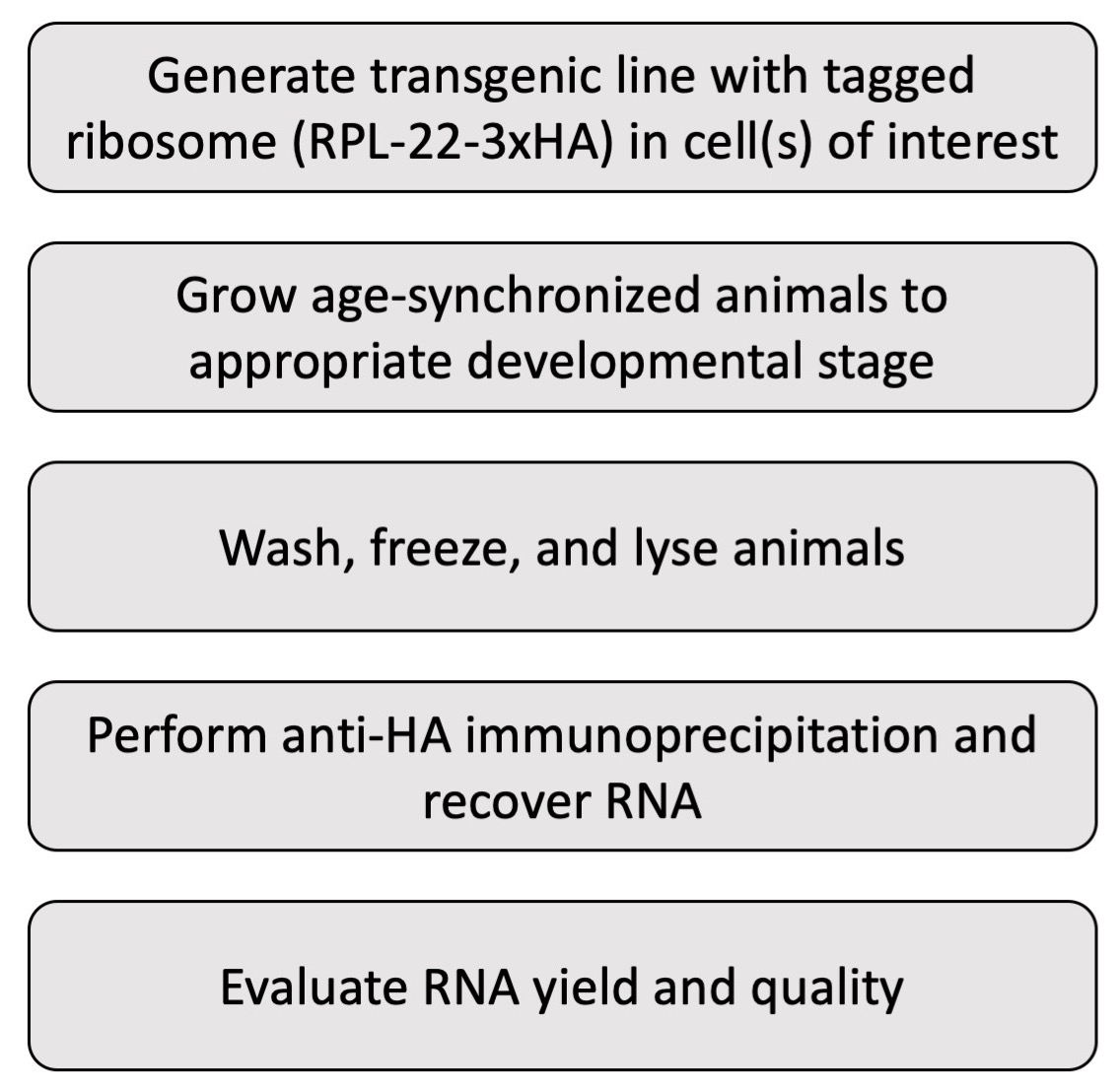
Figure 1. Workflow for the described protocol. This protocol consists of several steps, each described in detail in the main text.
- Prepare a transgenic line expressing the tagged ribosome in cell(s) of interest
- C. elegans TRAP requires expressing a tagged ribosomal subunit (rpl-22-3xHA cDNA) in cells of interest (Figure 2). Using standard molecular cloning techniques, generate a promoter::rpl-22-3xHA plasmid with any cell-specific promoter of interest.
A tph-1p::rpl-22-3xHA expression plasmid that drives expression of RPL-22-3xHA in serotonergic neurons is available via Addgene. In this plasmid, the tph-1 promoter is flanked by a 5′ FseI site and 3′ AscI site, so another promoter of choice can easily be swapped in with a FseI/AscI restriction digest.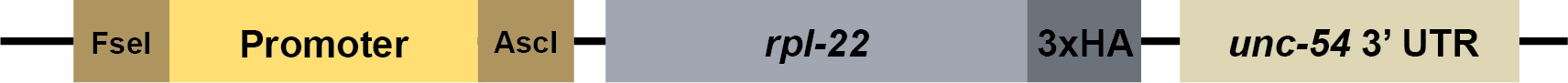
Figure 2. Schematic of the rpl-22-3xHA expression vector. The promoter can be replaced by amplifying a cell-specific promoter of interest by PCR with a 5′ FseI restriction site and 3′ AscI restriction site. - Inject the promoter::rpl-22-3xHA plasmid into adult worms with promoter::GFP as a co-injection marker to generate transgenic lines carrying an extrachromosomal array.
- Inject the plasmid at half the normal concentration typically used for the promoter (if known) to reduce the likelihood of off-target expression in other tissues.
- Using a plasmid that drives GFP expression under the same promoter as the rpl-22 construct as the sole co-injection marker eliminates the possibility of off-target expression of rpl-22 in other tissues as a consequence of plasmid recombination.
- Integrate one line by UV and backcross 5x to remove background mutations. We have typically selected transgenic lines based on co-injection marker expression, but anti-HA immunostaining or immunoblot analysis could also be conducted if necessary. We have not attempted TRAP on transgenic animals with extra-chromosomal arrays or MosSCI insertions, but it is possible that these may work.
- C. elegans TRAP requires expressing a tagged ribosomal subunit (rpl-22-3xHA cDNA) in cells of interest (Figure 2). Using standard molecular cloning techniques, generate a promoter::rpl-22-3xHA plasmid with any cell-specific promoter of interest.
- Grow age-synchronized animals to the appropriate developmental stage
- Pour 15 cm enriched-peptone NGM plates and allow them to dry at room temperature overnight. These plates allow for robust bacterial growth that can support a very dense nematode culture.
- Seed enriched-peptone NGM plates (one per condition) with 1 ml OP50 and allow to dry. Drip the OP50 over as much of the plate surface as possible, but do not spread it.
- Pick 100 young adult animals onto each plate and allow them to grow for 3 days, until there are many adult progeny and eggs on the plate. We typically prepare four parallel plates to recover enough animals for single-cell TRAP (150,000 animals; see below).
- Wash each plate with 6 ml M9 and transfer animals into a 15 ml conical tube with a serological pipette. To bleach-synchronize animals, add 3 ml bleach and 1 ml 5 N NaOH, then invert occasionally for 5 min until adult animals are broken up.
- Centrifuge at room temperature at 1,300 x g for 1 min to pellet eggs, aspirate the solution without disturbing the pellet, and wash with 10 ml M9. Repeat wash 2 x and resuspend in a small volume of M9.
- Eggs obtained from the above procedure can be plated directly on a freshly-seeded enriched-peptone plate. We plate all of the eggs recovered from one bleached 15 cm plate on to a single freshly-seeded enriched-peptone 15 cm NGM plate.
Note: We typically avoid plating arrested L1s due to concerns that developmental arrest could affect gene expression. However, animals are not perfectly synchronized if L1 starvation is omitted. If perfect synchronization is required, we suggest including this L1 starvation step. - Allow worms to grow at 20 °C tothe desired developmental stage. Animals will reach adulthood roughly 60 h after plating. Ensure that animals are not starved on the day of the experiment, unless this is desired as part of the experimental design.
- Wash, freeze, and lyse animals in the presence of cycloheximid
- At least one day in advance, prepare liquid NGM, homogenization buffer stock, and wash buffer stocks (see Recipes). Liquid NGM can be stored at room temperature, and homogenization and wash buffers are stable at 4 °C for one month.
- On the day of harvesting, add fresh reagents (DTT, protease inhibitor, RNasin, and VRC) to an aliquot of homogenization buffer (1 ml per plate).
- Prepare cycloheximide stock and add the stock to the following solutions at a final concentration of 0.8 mg/ml (125x dilution): liquid NGM (20ml per plate), homogenization buffer (10 ml per plate), and homogenization buffer with fresh reagents (1 ml per plate). Please note that cycloheximide is hazardous and should be handled with proper precautions.
- Clean your workstation, equipment, and gloves thoroughly with RNase ZAP.
- Place the cardboard insert from a microcentrifuge tube cryo box at the bottom of the ice bucket, and place RNase-free tubes (six tubes per plate) from the mortar/pestle set in the insert (Figure 3).

Figure 3. Photograph of set-up for grinding frozen pellets. Fill an ice bucket and conical tube with liquid nitrogen. Cool pestle in a microcentrifuge tube and use it to grind the frozen sample. To cool the sample, use forceps to grip a microcentrifuge tube, and use it to transfer liquid nitrogen from the conical tube to the sample. - Add liquid nitrogen to the ice bucket, filling the RNase-free tubes and the bottom of the bucket. Do not fill the bucket beyond the height of the tubes; only add enough extra liquid nitrogen to cool the outside of the tubes and prevent the evaporation of the liquid nitrogen in the tubes.
- Wash worms off each plate in 10 ml liquid NGM + cycloheximide and transfer them into a 15 ml conical tube with a serological pipette. Take care not to disrupt the bacterial lawn during the wash to avoid transferring excess bacteria to the tube.
- Spin at room temperature at 1,300 x g for 30 s, aspirate solution, then wash worms with 10 ml liquid NGM + cycloheximide.
- Spin at 1,300 x g for 30 s, aspirate solution, then wash worms with 10 ml homogenization buffer + cycloheximide and transfer the solution to a fresh 15 ml conical tube.
- Spin at 1,300 x g for 30 s, aspirate solution, and add 1 ml homogenization buffer with cycloheximide and fresh reagents.
- Using an RNase free pipette, drip the worm suspension into the RNase free tubes filled with liquid nitrogen, splitting the worm suspension evenly across five tubes. Place a small drop in a sixth tube that will be later used to estimate worm concentration. The worm suspension will bead up and freeze immediately upon contact with the liquid nitrogen. You do not need to record the volumes of these drops, as this will be estimated in a subsequent step.
Note: Do not close the tubes until all of the liquid nitrogen has evaporated or been poured off. - Store tubes at -80 °C.
- Estimation of worm concentration
- To estimate worm concentration, thaw the extra tube of frozen worms, then dilute it 10x and pipet 10 µl onto a glass slide.
- Count the number of worms present in the droplet.
- Repeat this 2 x and calculate the average to estimate the number of worms per µl in each frozen sample.
- Grind the frozen worm pellets using the RNase-free pestles
- Add liquid nitrogen to an RNase-free ice bucket with a cardboard insert, as before. Fill a 50 ml RNase-free conical tube with liquid nitrogen to act as a reservoir (Figure 3).
- Cool a pestle by dipping it into an RNase-free microcentrifuge tube filled with liquid nitrogen.
- Remove one tube from the -80 °C freezer and place it in the bucket. Grind the sample thoroughly with a pestle, ensuring that it remains frozen and will be ground up into a fine powder. Whenever the sample begins to thaw slightly, cool it again by pouring liquid nitrogen over it.
Notes:- It is easiest to cool the sample by using forceps to dip an RNase-free microcentrifuge tube into the reservoir of liquid nitrogen, then pouring it over the sample. Continually cool the pestle as well.
- This process can take quite some time; be patient and continue to cool the sample, ensuring that it does not thaw. Avoid overly aggressive grinding, as this could break the tube or cause the sample to fly out of the tube.
- Repeat for each tube until all samples have been ground into powder.
- Place the tubes back at -80 °C for storage. Samples can be stored at -80 °C for weeks before proceeding to immunoprecipitation step.
- It is easiest to cool the sample by using forceps to dip an RNase-free microcentrifuge tube into the reservoir of liquid nitrogen, then pouring it over the sample. Continually cool the pestle as well.
- Stock buffer preparation
Prepare the liquid NGM, homogenization buffer stock, 150 mM KCl wash buffer stock, and 350 mM KCl wash buffer stock at least one day before performing the immunoprecipitation. The homogenization buffer and wash buffers can be stored for up to one month at 4 °C - Perform immunoprecipitation
- Clean your workstation and all equipment thoroughly with RNAse Zap. All of the following steps should be performed on ice, keeping the samples cold throughout.
- Thaw the worm powder by diluting it into cold homogenization buffer (with all fresh ingredients added) to a concentration of 8 worms/µl and transfer it to a 15 ml conical tube. Based on the total volume of extract used, we estimate that our typical starting material for single cell type pull downs is ∼150,000 animals (typically pooled from several plates). Ensure that samples remain cold to prevent RNA degradation.
Notes:- Use a small known volume of homogenization buffer to gently resuspend the worm powder in each microcentrifuge tube. Once all powder is transferred to the 15 ml tube, measure the displacement with a serological pipette to determine the amount of powder added to the tube, then add extra homogenization buffer to bring the solution to the correct concentration.
- This dilution is very important, high concentrations of animals can lead to substantial RNA degradation, presumably due to endogenous RNases.
- Add 1/10 volume of 10% NP-40 and 1/9 volume of 300 mM DHPC to the 15 ml tube. Incubate on ice for 10 min, inverting occasionally.
- Transfer the solution to RNase-free microcentrifuge tubes and spin for 12 min at 15,000 x g at 4 °C. Transfer the supernatant into fresh tubes, then centrifuge again, and pool the supernatant into a fresh 15 ml RNase-free conical tube.
- While centrifuging the samples, wash the Protein G beads in 150 mM KCl Wash Buffer. Add 1 ml of 150 mM KCl Wash Buffer to 150 µl beads (per IP; use 300 µl beads per IP if pre-clearing), collect the beads with a magnetic rack, aspirate wash buffer, and repeat 2 x.
- (OPTIONAL) To pre-clear the lysate, add 150 µl washed Protein G beads to the lysate and rotate at 4 °C for 30 min, then collect the beads with the magnetic rack and transfer the cleared supernatant to a fresh 15 ml conical tube. We have typically applied this step when performing TRAP on single cells, but have skipped it when performing TRAP on large numbers of cells (for example, during pan-neuronal TRAP).
- Collect a 100 µl “input” sample at this point and mix with 100 µl of lysis buffer from the Agilent Absolutely RNA Nanoprep Kit. Ensure that beta-mercaptoethanol (BME) has been added to the lysis buffer, as per kit protocol. Vortex, let sit at room temperature for 10 min, vortex again, then store at -80 °C.
- Add 3.5 µl monoclonal anti-HA antibody to pre-cleared lysate and rotate at 4 °C for 30 min. For large numbers of cells (pan-neuronal TRAP), we have used 10 µl of antibody.
- Add 150 µl Protein G beads to the lysate and rotate at 4 °C for 30 min.
- Place the 15 ml conical tube on the magnetic rack and collect a 100 µl “flowthrough” sample and process in the same manner as the “input” material. This sample is valuable to check if there was any RNA degradation during the incubation with antibody and beads.
- Wash twice with 350 mM KCl Wash Buffer, each time gently inverting the tube then placing it on the magnetic rack to aspirate supernatant. Resuspend the beads in 350 mM KCl Wash Buffer and transfer to a clean RNase-free microcentrifuge tube. Wash once more with 350 mM KCl Wash Buffer, place on the magnetic rack, and aspirate supernatant (in total, the beads should be washed 4 x with 350 mM KCl Wash Buffer).
- Elute RNA by adding 100µl lysis buffer with BME (from the Agilent Absolutely RNA Nanoprep Kit), vortex, and let the sample sit for 10 min at room temperature with occasional agitation. Place the tube on the magnetic rack, collect liquid in a fresh, chilled RNase-free microcentrifuge tube, and store at -80 °C. Beads can be discarded after elution.
- RNA purification
Purify RNA with the Agilent Absolutely RNA Nanoprep Kit as per the kit protocol, including the on-column DNase treatment. Typical RNA concentrations (measured by nanodrop) are 30-200 ng/µl for input samples and 4-10 ng/µl for IP samples in a volume of 10 µl.
Data analysis
- Check for RNA degradation by running the input, flowthrough, and IP samples on an Agilent Bioanalyzer chip (nano or pico, depending on concentration). RNA integrity number (RIN) values should be greater than 7.5 to proceed, and values greater than 8.5 are typical in experienced hands. We find that the IP samples can show relatively lower RNA degradation regardless of the quality of the input, so pay particular attention to input and flowthrough quality to ensure that the procedure went well and that the IP sample is representative.
- Quantify positive control genes via RT-qPCR. Reverse transcribe RNA with a Superscript II kit with random hexamers to create a cDNA sample for qPCR. For low RNA amounts, we have also used Nugen Ovation RNA amplification system v2 to perform RT reactions. Use a 2x dilution of this cDNA for qPCR. We have used cdc-42 and unc-37 as ubiquitously-expressed control genes, and rab-3 and tag-168 as pan-neuronal genes. Ensure that you see enrichment for known genes in your IP sample relative to the input before proceeding to RNA sequencing.
Notes
- The details of preparing and analyzing RNA-seq data sets are beyond the scope of this protocol, but we typically prepare libraries with a Takara SMART-Seq Ultra Low Input RNA Kit for sequencing on an Illumina platform.
- We and other laboratories have found that standard oligo-dT priming is not sufficient to deplete ribosomal RNA from C. elegans libraries. Nevertheless, we typically recover enough non-rRNA reads to have sufficient sequencing depth for most purposes (> 20 million non-rRNA reads per library).
Recipes
- Enriched-peptone nematode growth medium (NGM)
- Per 1 L of NGM, add to a 2 L or larger Erlenmeyer flask:
22 g agar
20 g peptone
3 g NaCl
975 ml H2O - Autoclave, allow to cool to 55 °C, and add:
1 ml 1M CaCl2
1 ml 1M MgSO4
25 ml 1M KPO4
1 ml 5 mg/ml cholesterol in ethanol - Pour 50 ml per 15 cm Petri dish and allow to set completely. Store inverted at 4 °C
- Per 1 L of NGM, add to a 2 L or larger Erlenmeyer flask:
- M9 Solution
3 g KH2PO4
6 g Na2HPO4
5 g NaCl
Suspend in 1 L of H2O and autoclave
After cooling, add 1 ml of sterile 1 M MgSO4 - Liquid NGM (standard NGM without agar)
- Per 1 L of liquid NGM, add to a capped bottle:
2.5 g peptone
3 g NaCl
975 ml H2O - Autoclave, allow to cool to 55 °C, and add:
1 ml 1 M CaCl2
1 ml 1 M MgSO4
25 ml 1 M KPO4
1 ml 5 mg/ml cholesterol in ethanol - Store at room temperature
- Per 1 L of liquid NGM, add to a capped bottle:
- 100mg/ml cycloheximide
- Add 500 µl methanol to 50 mg cycloheximideb.
- Make a fresh stock each day, do not store
- 300 mM DHPC stock
- Add 1.38 ml nuclease-free H2O to 200 mg DHPC, and allow > 60 min to fully resuspend
- Store at 4 °C up to one month
- Homogenization buffer (stock)
- Remove 9 ml from a 100 ml bottle of nuclease-free H2O
- Add:
1 ml 1 M HEPES, pH 7.3 (10 mM final conc.)
7.5 ml 2 M KCl (150 mM final conc.)
0.5 ml 1 M MgCl2 (5 mM final conc.) - Store at 4 °C up to one month
- Homogenization buffer with fresh ingredients
Add to 10 ml homogenization buffer stock:
5 µl 1 M DTT (0.5 mM final conc.)
100 µl RNAsin RNAse inhibitor (note that this is a higher concentration than wash buffers)
0.5 ml Ribonucleoside Vanadyl Complex (VRC)
10 µl 100 mg/ml cycloheximide (100 µg/ml final conc.)
Note: We use a higher concentration of cycloheximide (0.8 mg/ml final concentration) in the liquid NGM and homogenization buffer applied to live animals on the day of freezing. Then, we use a lower concentration of cycloheximide (100 µg/ml final concentration) in subsequent lysis and immunoprecipitation steps.
1 protease inhibitor tablet - 150 mM KCl wash buffer (stock)
- Remove 19 ml from a 100 ml bottle of nuclease-free H2O
- Add:
1 ml 1 M HEPES, pH 7.3 (10 mM final conc.)
7.5 ml 2M KCl (150 mM final conc.)
0.5 ml 1M MgCl2 (5 mM final conc.)
10 ml 10% NP-40 (1% final conc.) - Store at 4 °C up to one month
- 150 mM KCl wash buffer with fresh ingredients
Add to 10 ml 150 mM KCl wash buffer stock:
5 µl 1 M DTT (0.5 mM final conc.)
10 µl RNAsin RNase inhibitor
10 µl 100 mg/ml cycloheximide (100 μg/ml final conc.)
Note: Prepare just before use. - 350 mM KCl wash buffer (stock)
- Remove 29 ml from a 100 ml bottle of nuclease-free H2O
- Add:
1 ml 1 M HEPES, pH 7.3 (10 mM final conc.)
17.5 ml 2M KCl (350 mM final conc.)
0.5 ml 1M MgCl2 (5 mM final conc.)
10 ml 10% NP-40 (1% final conc.) - Store at 4 °C up to one month
- 350 mM KCl wash buffer with fresh ingredients
Add to 10 ml 350 mM KCl wash buffer stock:
5 µl 1 M DTT (0.5 mM final conc.)
10 µl RNAsin RNase inhibitor
10 µl 100 mg/ml cycloheximide (100 µg/ml final conc.)
Note: Prepare just before use.
Acknowledgments
We thank Cori Bargmann, Eric Schmidt, Ivo Spiegel, and Myriam Heiman for help during early phases of developing this protocol. We also thank Max Heiman for discussions about effective cycloheximide concentrations in C. elegans. Howard Hughes Medical Institute supported early phases of this project. I.G.M. was supported by a JPB Postdoctoral Fellowship. S.W.F. acknowledges support from the JPB Foundation, PIIF, and PNDRF.
Competing interests
There are no conflicts of interest or competing interests.
References
- Gracida, X. and Calarco, J. A. (2017). Cell type-specific transcriptome profiling in C. elegans using the Translating Ribosome Affinity Purification technique. Methods 126: 130-137.
- Heiman, M., Schaefer, A., Gong, S., Peterson, J. D., Day, M., Ramsey, K. E., Suarez-Farinas, M., Schwarz, C., Stephan, D. A., Surmeier, D. J., Greengard, P. and Heintz, N. (2008). A translational profiling approach for the molecular characterization of CNS cell types. Cell 135(4): 738-748.
- Heiman, M., Kulicke, R., Fenster, R. J., Greengard, P. and Heintz, N. (2014). Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP). Nat Protoc 9(6): 1282-1291.
- Kaletsky, R., Yao, V., Williams, A., Runnels, A. M., Tadych, A., Zhou, S., Troyanskaya, O. G. and Murphy, C. T. (2018). Transcriptome analysis of adult Caenorhabditis elegans cells reveals tissue-specific gene and isoform expression. PLoS Genet 14(8): e1007559.
- Rhoades, J. L., Nelson, J. C., Nwabudike, I., Yu, S. K., McLachlan, I. G., Madan, G. K., Abebe, E., Powers, J. R., Colón-Ramos, D. A. and Flavell, S. W. (2019). ASICs mediate food responses in an enteric serotonergic neuron that controls foraging behaviors. Cell 176:85-97.e14.
- Roy, P. J., Stuart, J. M., Lund, J. and Kim, S. K. (2002). Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature 418:975-979.
- Sanz, E., Yang, L., Su, T., Morris, D. R., McKnight, G. S. and Amieux, P. S. (2009). Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A 106:13939-13944.
- Spencer, W. C., McWhirter, R., Miller, T., Strasbourger, P., Thompson, O., Hillier, L. W., Waterston, R. H., and Miller, D. M. (2014). Isolation of specific neurons from C. elegans larvae for gene expression profiling. PLoS ONE 9: e112102.
- Takayama, J., Faumont, S., Kunitomo, H., Lockery, S.R. and Iino, Y. (2010). Single-cell transcriptional analysis of taste sensory neuron pair in Caenorhabditis elegans. Nucleic Acids Res 38: 131-142.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
McLachlan, I. G. and Flavell, S. W. (2019). Cell Type-specific mRNA Purification in Caenorhabditis elegans via Translating Ribosome Affinity Purification. Bio-protocol 9(15): e3328. DOI: 10.21769/BioProtoc.3328.
Category
Molecular Biology > RNA > RNA purification
Biochemistry > Protein > Isolation and purification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.