- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

QUEEN-based Spatiotemporal ATP Imaging in Budding and Fission Yeast
Published: Vol 9, Iss 15, Aug 5, 2019 DOI: 10.21769/BioProtoc.3320 Views: 7081
Reviewed by: Samantha E. R. DundonUdita UpadhyayGunjan Mehta
Original research article
The authors used this protocol in:

Apr 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Yeasts have provided an exceptional model for studying metabolism and bioenergetics in eukaryotic cells. Among numerous metabolites, adenosine triphosphate (ATP) is a major metabolite that is essential for all living organisms. Therefore, a clearer understanding of ATP dynamics in living yeast cells is important for deciphering cellular energy metabolism. However, none of the methods currently available to measure ATP, including biochemical analyses and ATP indicators, have been suitable for close examinations of ATP concentrations in yeast cells at the single cell level. Using the recently developed ATP biosensor QUEEN, which is suitable for yeasts and bacteria, a protocol was described herein to visualize ATP concentrations in living budding and fission yeast cells. This simple method enables the easy and reliable examination of ATP dynamics in various yeast mutants, thereby providing novel molecular insights into cellular energy metabolism.
Keywords: ATPBackground
Adenosine triphosphate (ATP) is one of the major and indispensable metabolites for all living organisms. In addition to being useful “energy currency” for various cellular processes, ATP serves as an intracellular and extracellular signaling molecule as well as a phosphate donor for protein phosphorylation (Lehninger et al., 2010). Recent studies suggested that ATP itself solubilizes protein as a “biological hydrotrope” (Patel et al., 2017) and influences the aggregation state of intracellular proteins (Pu et al., 2019; Sridharan et al., 2019). Due to the obvious importance of ATP and its wide involvement in many cellular events, the spatial distributions and temporal dynamics of ATP in living organisms need to be clarified in order to elucidate energy metabolism and signal transductions at the molecular level. To date, a number of methods have been developed to measure ATP concentrations in vitro. However, these biochemical methods have only a limited ability to examine ATP dynamics in vivo because of their insufficient time resolution relative to the rapid turnover of ATP (less than one minute) (Mortensen et al., 2011; Takaine et al., 2019). Moreover, although these biochemical analyses detect the average ATP level of a cell population, they are incapable of showing the intracellular or intratissue distribution of ATP.
To overcome these difficulties, several types of ATP indicators have been developed for the direct visualization of ATP in living organisms (Dong and Zhao, 2016). Among them, genetically encodable ATP indicators are advantageous because their introduction into a cell population is non-invasive and highly specific. A series of fluorescence resonance energy transfer (FRET)-based genetically encoded ATP indicators, named “the ATeam”, was developed for the first time a decade ago by the pioneering work of Imamura’s group (Imamura et al., 2009). The ATeam comprises the ATP-binding domain of the ε subunit of bacterial FoF1 ATP synthase and flanking cyan and yellow fluorescent proteins (FPs), and is now widely and successfully used to monitor intracellular ATP concentrations in various types of cells ranging from plants to mammals. However, the ATeam composed of two FPs may be susceptible to proteolysis and, thus, yield immature sensor molecules because of differences in the maturation times of the two FPs. These features manifest when the ATeam is expressed in fast-growing microorganisms, in which protein turnover is very rapid, such as yeasts or bacteria, and become problematic because the presence of a dysfunctional sensor inevitably affects the FRET signal. To circumvent the drawbacks associated with ATeam, the second-generation ATP indicator “QUEEN” (quantitative evaluator of cellular energy) was recently developed by Imamura’s group (Yaginuma et al., 2014). QUEEN is a dual-excitation ratiometric ATP biosensor composed of a single FP and, thus, is more resistant to degradation and matures more rapidly than ATeam, yielding the reliable quantification of ATP, even in actively proliferating bacterial cells.
Yeasts have provided an exceptional model for studying metabolism and bioenergetics in eukaryotic cells. Yeast carbon metabolism has long been studied because of its industrial importance for producing many valuable chemicals (yeast metabolites) (Borodina and Nielsen, 2014). Energy metabolism in yeasts also provides a tractable model for studying energy metabolism in tumor cells because they are similar in that they synthesize the majority of ATP through glycolysis and not oxidative phosphorylation, even in the presence of oxygen, and this is known as “aerobic glycolysis” or “the Warburg effect” (Diaz-Ruiz et al., 2011). Therefore, a close examination of ATP dynamics in living yeast cells has been of great significance, but waits to be embarked due to the absence of an appropriate ATP biosensor.
In a recent study, QUEEN was applied to both the budding yeast Saccharomyces cerevisiae and fission yeast Schizosaccharomyces pombe for the first time and intracellular ATP levels and its dynamicity were visualized in wild-type cells (Takaine et al., 2019). The use of QUEEN enables the easy and reliable examination of ATP dynamics in living yeast cells, providing novel insights into cellular energy metabolism. The protocols used to observe and analyze the QUEEN fluorescence signal in yeast cells have been described herein. The constitutive and strong expression of QUEEN did not induce any noticeable defects in cell growth, meiosis, or sporulation. Thus, ATP concentrations in various yeast mutants under a number of conditions may be observed, as well as ATP in wild-type cells, using the same method. Moreover, this method employs a conventional wide-field fluorescence microscope and free image processing software for image calculations, and, thus, is accessible to a broad range of researchers.
Materials and Reagents
- 35-mm glass-bottomed dish (IWAKI, catalog number: 3971-035, No. 1.5 thickness)
- Cell culture dish, sterilized (SANSEI MEDICAL CO., LTD., catalog number: 01-013)
- Pipette tips (Watson® Bio Lab, catalog numbers: 123R-755YS [200 µl], 123R-757CS [1,000 µl])
- Sterile culture tube (16 ml, polypropylene) (EVERGREEN, catalog number: 222-2393-080)
- Toothpicks (6.5-18 cm) (any brand)
Note: Sterilize before use. Reusable until they break. - Budding yeast cells expressing QUEEN-2m:
e.g., MTY3261 (MATa his3∆1::3×pRS303-PTEF1-QUEEN-2m-TCYC1 leu2∆0 ura3∆0) from Takaine et al., 2019. This strain was derived from a slightly modified BY4741 parental strain (MATa his3∆1 leu2∆0 ura3∆0). - Fission yeast cells expressing QUEEN-2m:
e.g., KSP3769 (h- leu1-32::Ptif51-QUEEN-2m::leu1+) from Ito et al., 2019.
Note: Both yeast strains expressing QUEEN-2m and plasmids for expressing QUEEN-2m in budding yeast are available from the Yeast Genetic Resource Centre Japan (YGRC, http://yeast.nig.ac.jp/yeast/). - Concanavalin A (Sigma-Aldrich, catalog number: C-7275)
- Soybean lectin (Sigma-Aldrich, catalog number: L-1395)
- Yeast nitrogen base w/o amino acids (YNB) (Invitrogen, catalog number: Q300-09)
- Yeast extract (BD, catalog number: 212750)
- Bacto-peptone (BD, catalog number: 211677)
- D(+)-Glucose (FUJIFILM Wako, catalog number: 049-31165)
- 2-Deoxy-D-glucose (2DG) (FUJIFILM Wako, catalog number: 046-06483)
Note: 2DG is a potent inhibitor of glycolysis and has been used to deplete ATP in yeast cells (Serrano, 1977; Xu and Bretscher, 2014; Takaine et al., 2019). - Agar (FUJIFILM Wako, catalog number: 010-08725)
- Potassium hydrogen phthalate (FUJIFILM Wako, catalog number: 163-03822)
- Na2HPO4·12H2O (Kishida Chemical Co., Ltd., catalog number: 000-72535)
- Ethanol (Kishida Chemical Co., Ltd., catalog number: 140-28553)
- NH4Cl (FUJIFILM Wako, catalog number: 017-02995)
- Uracil (FUJIFILM Wako, catalog number: 212-00062)
- Myo-inositol (FUJIFILM Wako, catalog number: 096-00285)
- L-Glutamic acid (FUJIFILM Wako, catalog number: 072-00501)
- Adenine sulfate (FUJIFILM Wako, catalog number: 018-19613)
- L-Leucine (PEPTIDE INSTITUTE INC., catalog number: 2713)
- L-Histidine (FUJIFILM Wako, catalog number: 082-00683)
- L-Lysine·HCl (PEPTIDE INSTITUTE INC., catalog number: 2714)
- L-Methionine (PEPTIDE INSTITUTE INC., catalog number: 2715)
- L-Phenylalanine (PEPTIDE INSTITUTE INC., catalog number: 2717)
- L-Serine (PEPTIDE INSTITUTE INC., catalog number: 2719)
- L-Threonine (PEPTIDE INSTITUTE INC., catalog number: 2720)
- MgCl2·6H2O (FUJIFILM Wako, catalog number: 132-00175)
- CaCl2·2H2O (FUJIFILM Wako, catalog number: 033-25035)
- KCl (FUJIFILM Wako, catalog number: 163-03545)
- Na2SO4 (FUJIFILM Wako, catalog number: 195-03341)
- H3BO3 (FUJIFILM Wako, catalog number: 021-02195)
- MnSO4·5H2O (FUJIFILM Wako, catalog number: 139-00825)
- ZnSO4·7H2O (FUJIFILM Wako, catalog number: 268-00405)
- FeCl3·6H2O (FUJIFILM Wako, catalog number: 095-00875)
- Na2MoO4·2H2O (FUJIFILM Wako, catalog number: 198-02471)
- KI (FUJIFILM Wako, catalog number: 166-03971)
- CuSO4·5H2O (FUJIFILM Wako, catalog number: 031-04411)
- Citric acid (FUJIFILM Wako, catalog number: 038-05521)
- Nicotinic acid (FUJIFILM Wako, catalog number: 142-01232)
- Biotin (TOKYO CHEMICAL INDUSTRY CO., LTD., catalog number: B0463)
- Sodium pantothenate (FUJIFILM Wako, catalog number: 198-05651)
- 100x Amino acids mix (see Recipes)
- 100x L-Leucine stock (see Recipes)
- 50x Uracil stock (see Recipes)
- SC-His liquid medium (see Recipes)
- YPD agar plate (see Recipes)
- YELA agar plate (see Recipes)
- 100x 5 Low-supplements (see Recipes)
- 50x Salt stock (see Recipes)
- 1,000x Vitamins (see Recipes)
- 10,000x Minerals (see Recipes)
- EMM liquid medium (see Recipes)
- 2-Deoxy-D-glucose (2DG) medium (see Recipes)
- Glass-bottomed dish coated with concanavalin A (see Recipes)
- Glass-bottomed dish coated with soybean lectin (see Recipes)
Equipment
- Inverted fluorescent microscope (Nikon, Eclipse Ti-E) equipped with:
- A 100x objective lens (Nikon, Apo TIRF 100x Oil DIC N2/NA 1.49)
- A high-pressure mercury lamp (Nikon, Intensilight C-HGFIE 130W)
- An electron multiplying charge-coupled device camera (Andor-Oxford Instruments, iXon3 DU897E-CS0-#BV80)
- A FITC filter set (Nikon, Ex465-495/DM505/BA515-555)
- A custom-made Ex409 filter set (Semrock, Ex393-425/DM506/BA516-556)
- Neutral density (ND) filters (Nikon, ND4 [MBN21804] and ND8 [MBN21808])
- Bench-top aspirator (INTEGRA, VACUSIP)
- Inverted routine microscope (Nikon, Eclipse Ts2) equipped with a 40× objective lens (Nikon, CFI Achromat LWD ADL 40XF/NA 0.55, catalog number: MRP46402)
- Autoclave (TOMY SEIKO CO., LTD, LBS-325)
- Pure water system (Merck Millipore, Milli-Q® Integral 15)
- Erlenmeyer flasks (50 ml) (HARIO, catalog number: 82-0144)
- Silicone stopper (for a 50-ml flask) (Shin-Etsu Polymer, SILICOSEN T22)
- Incubator (TAITEC, INCUBATE BOX M-200F)
- Culture rotator (TAITEC, RT-50)
- Rotary shaker (NISSIN, NX-20)
- Pipettes (BM EQUIPMENT Co., LTD, PipetPAL®, model: PAL-200 [20-200 µl], PAL-1000 [100-1,000 µl])
Software
- NIS-Element AR ver. 4.30.01 (Nikon, https://www.microscope.healthcare.nikon.com/ja_JP/)
- Fiji ver. 2.0.0-rc-69/1.52n (Fiji contributors [Schindelin et al., 2012], http://fiji.sc/#)
- Microsoft® Excel for Mac 2011 ver. 14.7.3 (Microsoft, https://products.office.com/ja-jp/excel)
- KaleidaGraph ver. 4.5.1 (HULINKS, https://www.hulinks.co.jp/software/stat_graph/kaleida)
Procedure
- Cell preparation
- Streak yeast cells from a frozen stock using a pipette tip on a YPD (for S. cerevisiae) or YELA (for S. pombe) agar plate, and incubate at 30 °C for 1-2 days.
- Preparation of the preculture:
- For budding yeast, inoculate 2 ml of SC-H with a loopful of cells in a 16-ml culture tube using a sterilized toothpick, and culture at 30 °C overnight with rotation at approximately 40 rpm using a rotator.
- For fission yeast, inoculate 2 ml of EMM with a loopful of cells in a 16-ml culture tube using a sterilized toothpick, and culture at 30 °C overnight with shaking at 150 rpm using a rotary shaker.
- Preparation of the main culture:
- For budding yeast, dilute 0.15 ml of the overnight culture with 2.85 ml SC-H (1:20) in a 16-ml culture tube, and culture at 30 °C with rotation at approximately 40 rpm.
- For fission yeast, dilute 0.5 ml of the overnight culture with 9.5 ml EMM in a 50-ml flask (1:20), and culture at 30 °C with shaking at 150 rpm.
- Grow yeast cells to the mid-log phase. Approximately 3 h is needed for budding yeast and approximately 5 h for fission yeast under the experimental conditions described herein. The dilution ratio and incubation time may be optimized according to individual culture systems.
- Dilute the cell culture with the same medium by 10-fold and place 100 µl of cells onto a 35-mm glass-bottomed dish coated by concanavalin A for S. cerevisiae or soybean lectin for S. pombe (see Recipes).
- Wait for 5 min, remove most of the medium with gentle aspiration, and wash cells immobilized on the glass surface with 300 µl of fresh medium three times.
- Observe the yeast cells immobilized on the glass-bottomed dish with a 40x objective lens using an inverted routine microscope to check cell density.
- Repeat Steps A2-A3 if cells are too sparse.
- Fill the dish with 4-5 ml of medium and incubate the dish in a room for fluorescent microscopy (maintained at 25 °C) for at least 30 min before microscopic observations.
- Optional: To deplete intracellular ATP, replace the medium with 300 µl of 2DG medium three times. Acquire images of QUEEN fluorescence within 10 min.
- Image acquisition
- Power on the fluorescent microscope system at least 1 h before imaging to reach a stable and constant temperature of approximately 25 °C.
- Observe the yeast cells immobilized on the glass-bottomed dish with a 100x objective lens using an inverted fluorescence microscope by bright-field imaging and focus the cells.
- Take a bright-field image and two fluorescence images of QUEEN using FITC (ex480 image) and Ex409 (ex409 image) filter sets from a single z-plane.
- Regarding time-lapse imaging, reduce the intensity of the excitation light and exposure times (see Table 1 for details) to prevent the photobleaching of QUEEN fluorescence and minimize damage to cells.
- Save the combined images (bright-field and QUEEN) as an nd2 file.
Table 1. Parameters for the fluorescence microscopy of QUEEN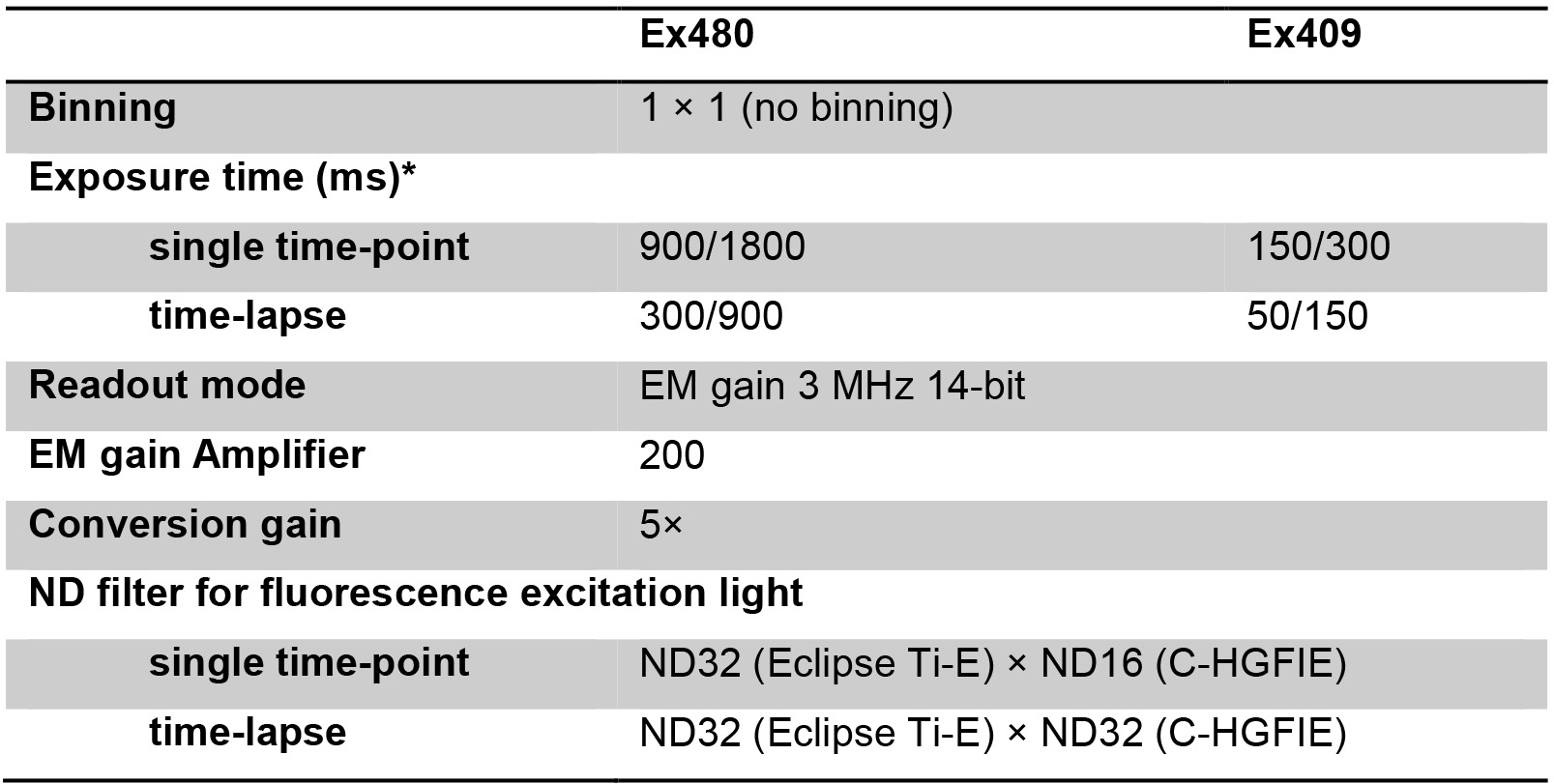
*Exposure time for the acquisition of images of QUEEN expressed in budding/fission yeast.
Data analysis
In the following sections, menu commands and options in Fiji software are expressed in bold font.
- Calculation of the QUEEN ratio (single time-point imaging)
- Open an nd2 file using Fiji software as a hyperstack file (multi-channels and one time frame) and split the hyperstack into each channel image (one bright-field image, two QUEEN images): Image -> Color -> Split Channels (Figure 1A, raw image).
- Subtract the background (pixel values in an area outside the cells) of QUEEN images using the rolling-ball algorithm (Figure 1A, background-subtracted): Process -> Subtract Background. Set the rolling ball radius 20 (for S. cerevisiae) or 50 (for S. pombe) pixels and keep all the check boxes unchecked.
- Convert QUEEN images to a signed 32-bit floating-point grayscale: Image -> Type -> 32-bit
- Assess the maximum and minimum thresholds of the images using the modified IsoData algorithm (default algorithm): Image -> Adjust -> Threshold. Check the Dark background check box and click Auto and Apply. Check Set background pixels to NaN to set background (non-thresholded) pixels to the NaN (Not a Number) value (Figure 1A, thresholded).
- Divide the pixel values of the ex409 image by those of the ex480 image to calculate the QUEEN ratio at each pixel: Process -> Image calculator, Image1: ex409 image, Operation: Divide, Image2: ex480 image.
- Apply the rainbow smooth look-up table (or any other table if preferable) to the ratio image to express the QUEEN ratio in pseudocolor: Image -> Lookup Tables -> Rainbow Smooth.
- Adjust the display range to include the minimum and the maximum values of the QUEEN ratios in the image: Image -> Adjust -> Brightness/Contrast -> Set. Set Minimum displayed value 0.5 and Maximum displayed value 3, respectively, for example (Figure 1B, ratio image).
- Save the ratio image and the bright-field image as a tiff file: File -> Save as...
- Optional: steps 1-7 may be automated by running the following macro: Plugins -> Macros -> Install…, select and open stack2ratio.jim macro. Open an nd2 file and apply Run on the macro window.
// Macro starts here
title = getTitle();
run("Split Channels");
c1 = "C1-" + title; // channel #1: bright-field image
c2 = "C2-" + title; // channel #2: ex480 image
c3 = "C3-" + title; // channel #3: ex409 image
selectWindow(c2);
run("Subtract Background...", "rolling=20 stack");
run("32-bit");
setAutoThreshold("Default dark");
run("NaN Background", "stack");
selectWindow(c3);
run("Subtract Background...", "rolling=20 stack");
run("32-bit");
setAutoThreshold("Default dark");
run("NaN Background", "stack");
imageCalculator("Divide create stack",c3,c2);
run("Rainbow Smooth");
setMinAndMax(0.5, 3);//
rename("Ratio-" + title);
// Macro ends here
Note: The optimal rolling ball radius depends on the spatial resolution of the imaging system (0.16 µm/pixel in our system) and the average size of yeast cells examined. Therefore, readers need to adjust the rolling ball radius according to their own imaging system by trial and error.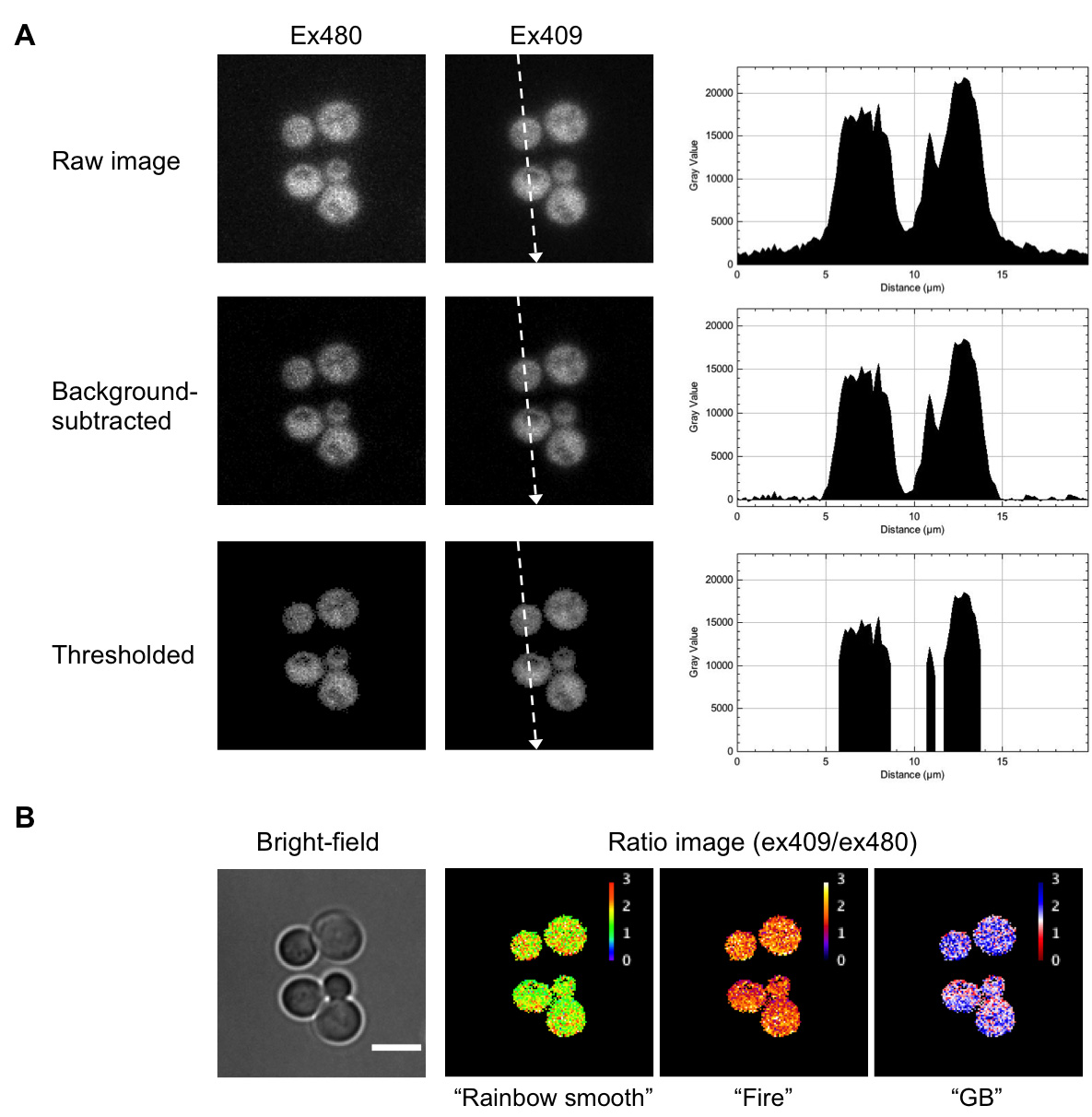
Figure 1. Generation of a QUEEN ratio image. A. Image processing of images of budding yeast cells expressing QUEEN. Right panels show the line profile of pixel values along dotted lines indicated in the ex409 images. B The bright-field image and QUEEN ratio images calculated from the ex480 and ex409 images shown in (A) expressed in three different lookup tables. White scale bar = 5 µm. - Calculation of the QUEEN ratio (time-lapse imaging)
- Open the nd2 file using Fiji software as a hyperstack file (multi-channels and multiple time frames).
- By choosing a Rectangle selection tool (see Figure 2A), draw the region of interest (ROI) to include a cell (and the progeny cells if necessary) throughout the time course.
- Create a new hyperstack file composed of the ROI: Image -> Duplicate. Check the Duplicate hyperstack check box and preserve all Channels (c) and Frames (t).
- Split the hyperstack into each channel stack (containing time frames): Image -> Color -> Split Channels.
- Subtract the background using the rolling-ball algorithm as described in step A2. Process all the images in the stack.
- Convert the QUEEN stacks to a signed 32-bit floating-point grayscale: Image -> Type -> 32-bit
- Display the last time frame: Image -> Stacks -> Set Slice. Enter the last slice number.
Note: Against a stack of 32-bit images, the maximum and minimum thresholds are calculated using the currently displayed image (slice), and not calculated for each image. Assessments of thresholds using the last time frame were appropriate under the experimental conditions described herein. - Create thresholded stacks as described in step A4.
- Calculate and save the QUEEN ratio stack as described in steps A5-A8.
- Measurement of the mean QUEEN ratio in individual cells (single time-point imaging)
- Open the QUEEN ratio image using Fiji software (Figure 2B).
- By choosing the Rectangle or Polygon selection tool (Figure 2A), draw the ROI to include a living cell and register the ROI in the ROI manager: Analyze -> Tools -> ROI manager. Click Add or press t to register the ROI (Figure 2C).
- Repeat step 2.
- Set the measurement: Analyze -> Set Measurements..., check the Mean Gray Value check box.
- Select all ROI stored in the ROI manager and measure the mean of pixel values (QUEEN ratios) in ROIs: ROI manager -> Deselect -> Measure
- Save the data table as a text file in the comma-separated value (csv) format: Activate the Results window (Figure 2D), then File -> Save as...
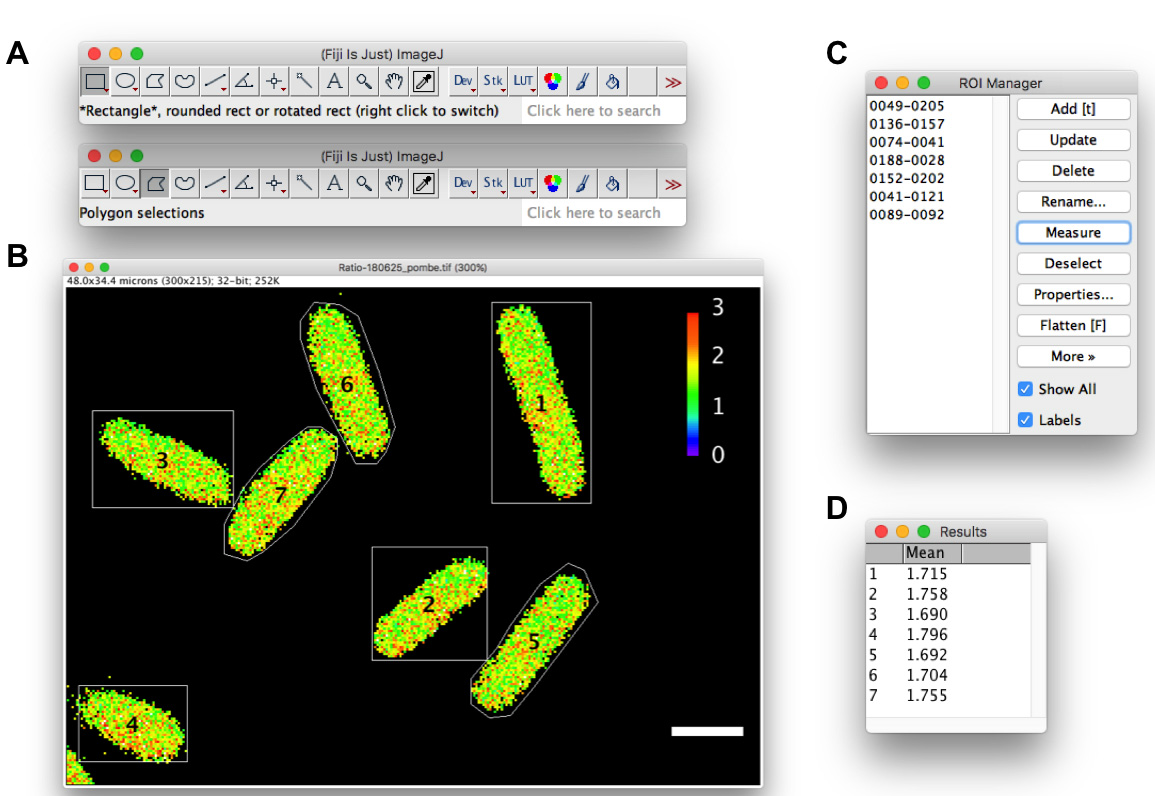
Figure 2. Measurement of the mean QUEEN ratio in yeast cells. Measurements in fission yeast cells are shown as an example. A. Rectangle or Polygon selection tool. B. A QUEEN ratio image of fission yeast cells. Outlines of cells are drawn using the rectangle (cell #1-4) or polygon (cell #5-7) selection tool. C. ROI manager. D. Results window. White scale bar = 5 µm.
- Measurement of the mean QUEEN ratio in a cell (time-lapse imaging)
- Open the QUEEN ratio stack using Fiji software.
- By choosing the Rectangle or Polygon selection tool, draw the ROI to include a cell and register the ROI in the ROI manager.
- Repeat step 2 if necessary.
- Select the ROI stored in the ROI manager and measure the mean of pixel values in the ROI throughout time series: ROI manager -> More -> Multi Measure. Check the Measure all the slices and One row per slice check boxes.
- Save the result as described in step C6.
- Data analysis
- Import data into Microsoft® Excel, and calculate averages and standard deviations.
- Calculate the P-value using an unpaired one-tailed Welch’s t-test in Excel software, and evaluate the significance of differences between two sets of data.
- Plot data as a dot plot using KaleidaGraph software. An example of a dot plot is shown in Figure 3.
Note: The presentation of data as a dot plot appears to be the most effective way to convey distribution characteristics in extensive detail. Data generally follow a normal distribution even though there are some outliers.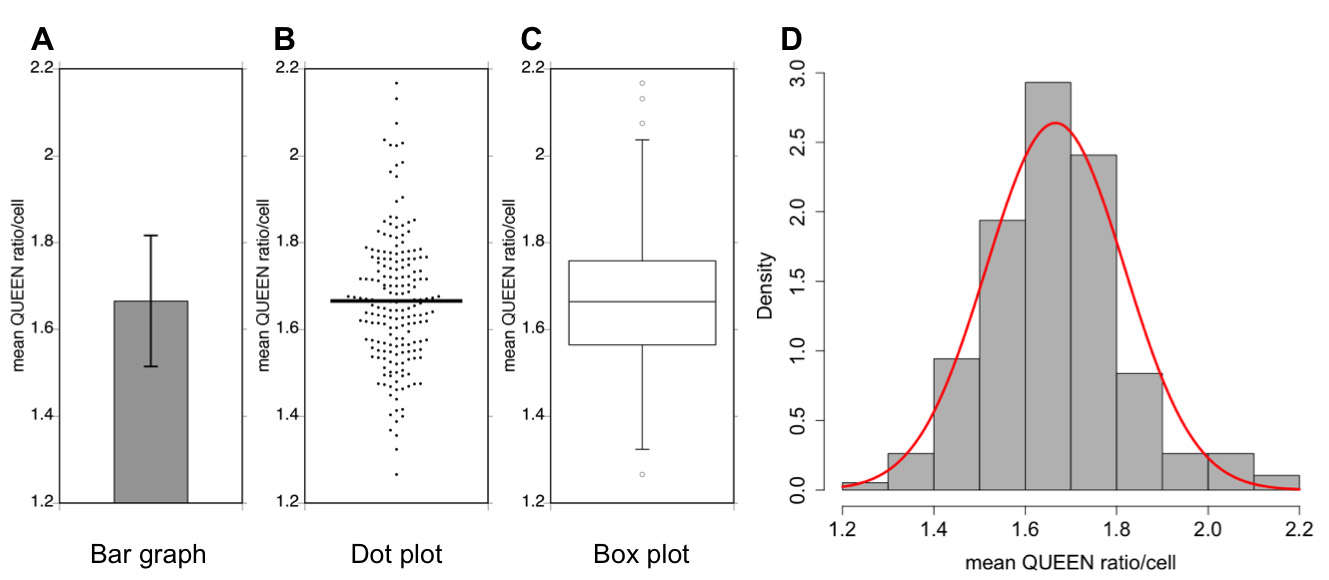
Figure 3. Examples of data analyses. A data set of measurements of the mean QUEEN ratio in budding yeast cells is represented in four different types of graphs. Sample size = 191 cells, the mean value = 1.67, variance = 0.0228, and the standard deviation (SD) = 0.151. A. Bar graph. Data are shown as the mean of the population ± 1× SD (error bar). B. Dot plot. Each dot corresponds to the mean QUEEN ratio inside a cell. The horizontal bar indicates the mean of the population. C. Box plot. The plot shows the 75th and 25th percentiles of the data (interquartile range) as the upper and lower edges of the box. A horizontal bar in the box indicates the median of the population. Whiskers and circles indicate the 1.5× interquartile range and outliers, respectively. D. Histogram. Data are shown as probability density. A normal distribution with the same mean and variance to the data is shown as a red curve.
Notes
- The mean QUEEN ratio in a cell is calculated based on (1) the intracellular ATP concentration, (2) the affinity of QUEEN for ATP, and (3) the settings and conditions of the imaging system. Thus, many factors affect the QUEEN ratio including the conditions of the cell culture, the batch of the culture medium, temperature (as described in Procedure A), the power output of the fluorescence excitation lamp, and the conditions of optical filters (excitation, emission, and neutral density) for fluorescence microscopy. These factors vary slightly day to day and are difficult to strictly control. Therefore, comparisons of data obtained from experiments performed on the same day and not pooling data from experiments performed on different days are recommended.
- The readers need to optimize the imaging parameters of fluorescence microscopy for their own imaging systems. Since an excitation light of approximately 409 nm is cytotoxic and may affect the fluorescence emission spectrum of QUEEN, its intensity and exposure time need to be minimized. Adjustments to imaging parameters in order for the mean QUEEN ratio in cells depleted of ATP by 2DG, corresponding to the minimum mean QUEEN ratio, to be calculated as 0.7-0.8 are recommended. Under preferable conditions, the dynamic range of QUEEN-2m (the ratio between the maximum and minimum mean QUEEN ratios) is expected to reach 3.0-fold (Yaginuma et al., 2014; Takaine et al., 2019).
- In addition to the calculation of the QUEEN ratio, measurements of the mean QUEEN ratio (the steps C1-C5) may also be automated by running a macro. Moreover, steps A1-A8 and C1-C5 in the Data analysis may be batch-processed: multiple nd2 files are automatically and sequentially processed by running a batch macro. However, the automatic drawing of cell outlines and batch processing are both beyond the scope of this study and, thus, were omitted. These macros are available from the author upon request.
Recipes
- 100x Amino acids mix
- Mix L-glutamic acid 10 g, L-lysine 12 g, L-methionine 4 g, L-phenylalanine 5 g, L-serine 38 g, L-threonine 20 g, and Myo-inositol 0.6 g in 1,000 ml of deionized water
- Autoclave at 120 °C for 20 min and store at room temperature
- 100x Leucine stock
- Dissolve L-leucine 11 g in 1,000 ml of deionized water
- Autoclave at 120 °C for 20 min and store at room temperature
- 50x Uracil stock
- Dissolve uracil 2 g in 1,000 ml of deionized water
- Autoclave at 120 °C for 20 min and store at room temperature
- SC-His (according to Hanscho et al., 2012)
- Mix YNB 3.4 g, glucose 20 g, 100x amino acid mix 10 ml, 100x leucine stock 10 ml, and 50x uracil stock 20 ml in 1,000 ml of deionized water
- Autoclave at 120 °C for 20 min and store at room temperature
- YPD agar plate
- Mix yeast extract 10 g, glucose 20 g, Bacto-peptone 20 g, and agar 20 g in 1,000 ml of deionized water
- Autoclave at 120 °C for 20 min
- Pour the medium into a cell culture dish (approximately 20 ml/dish)
- Solidify the plates for 2-3 days before use, wrap in plastic wrap, and store at room temperature
- YELA agar plate
- Mix yeast extract 10 g, glucose 20 g, adenine sulfate 0.04 g, and agar 20 g in 1,000 ml of deionized water
- Autoclave at 120 °C for 20 min
- Pour the medium into a cell culture dish (approximately 20 ml/dish)
- Solidify the plates for 2-3 days before use, wrap in plastic wrap, and store at room temperature
- 100x 5 Low-supplements
- Mix adenine sulfate 10 g, L-lysine 3 g, L-leucine 6 g, L-histidine 2 g, and uracil 2 g in 1,000 ml of deionized water
- Autoclave at 120 °C for 20 min and store at room temperature
- 50x Salt stock
- Mix MgCl2·6H2O 53.3 g, CaCl2·2H2O 0.735 g, KCl 50 g, and Na2SO4 2 g in 1,000 ml of deionized water
- Autoclave at 120 °C for 20 min and store at room temperature
- 1,000x Vitamins
Mix nicotinic acid 0.1 g, myo-inositol 0.1 g, biotin 0.1 mg, and sodium pantothenate 10 mg in 100 ml of absolute ethanol and store at -20 °C
Note: These components do not completely dissolve. - 10,000x minerals
- Dissolve H3BO3 0.05 g, MnSO4·5H2O 0.057 g, ZnSO4·7H2O 0.04 g, FeCl3·6H2O 0.02 g, Na2MoO4·2H2O 0.016 g, KI 0.01 g, CuSO4·5H2O 0.004 g, and citric acid 0.1 g in 100 ml of deionized water
- Sterilize by filtration and store at room temperature
- EMM
- Mix potassium hydrogen phthalate 3 g, Na2HPO4·12H2O 5.54 g, glucose 20 g, 50x salt stock 20 ml, and NH4Cl 5 g, and 10,000x minerals 0.1 ml in 1,000 ml of deionized water
- Autoclave at 120 °C for 20 min and cool down
- Add 1,000x vitamins 1 ml and store at 4 °C
- Aliquot and prewarm before use
- 2DG medium
The same as for SC-His or EMM except for the addition of 3.28 g of 2DG (final 20 mM) instead of glucose - Glass-bottomed dish coated with concanavalin A
- Dissolve concanavalin A with sterilized deionized water at 2 mg/ml and aliquot. The solution may be stored at -20 °C
- Place 100 µl of 2 mg/ml of concanavalin A solution on the glass surface of a glass-bottomed dish, and keep at room temperature for 5 min
- Aspirate concanavalin A and wash the glass surface with 100 µl of sterilized deionized water three times
- Dry the glass surface for 10 min before use. The dish may be stored at 4 °C
- Glass-bottomed dish coated with soybean lectin
- Dissolve soybean lectin with sterilized deionized water at 2 mg/ml and aliquot. The solution may be stored at 4 °C
- Place 5 µl of 2 mg/ml lectin solution on the glass surface of a glass-bottomed dish, and spread the solution using the pipette tip
- Dry the glass surface for 10 min before use. The dish may be stored at 4 °C
Acknowledgments
This work has been supported by JSPS grants to the author (15K18525, 16H04781, and 19K06654). This work was also supported by a joint research program of the Institute for Molecular and Cellular Regulation, Gunma University, Japan. The protocol has been adapted from Takaine et al. (2019).
Competing interests
The author declares no competing interests.
References
- Borodina, I. and Nielsen, J. (2014). Advances in metabolic engineering of yeast Saccharomyces cerevisiae for production of chemicals. Biotechnol J 9(5): 609-620.
- Diaz-Ruiz, R., Rigoulet, M. and Devin, A. (2011). The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim Biophys Acta 1807(6): 568-576.
- Dong, J. and Zhao, M. (2016). In-vivo fluorescence imaging of adenosine 5’-triphosphate. TrAc Trends Anal Chem 80: 190-203.
- Hanscho, M., Ruckerbauer, D. E., Chauhan, N., Hofbauer, H. F., Krahulec, S., Nidetzky, B., Kohlwein, S. D., Zanghellini, J. and Natter, K. (2012). Nutritional requirements of the BY series of Saccharomyces cerevisiae strains for optimum growth. FEMS Yeast Res 12(7): 796-808.
- Imamura, H., Nhat, K. P., Togawa, H., Saito, K., Iino, R., Kato-Yamada, Y., Nagai, T. and Noji, H. (2009). Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci U S A 106(37): 15651-15656.
- Ito, H., Sugawara, T., Shinkai, S., Mizukawa, S., Kondo, A., Senda, H., Sawai, K., Ito, K., Suzuki, S., Takaine, M., Yoshida, S., Imamura, H., Kitamura, K., Namba, T., Tate, S. I. and Ueno, M. (2019). Spindle pole body movement is affected by glucose and ammonium chloride in fission yeast. Biochem Biophys Res Commun 511(4): 820-825.
- Lehninger, A., Nelson, D. and Cox, M. (2010). Principles of Biochemistry. CBS Publisher and Distributors. Delhi.
- Mortensen, S. P., Thaning, P., Nyberg, M., Saltin, B. and Hellsten, Y. (2011). Local release of ATP into the arterial inflow and venous drainage of human skeletal muscle: insight from ATP determination with the intravascular microdialysis technique. J Physiol 589(Pt 7): 1847-1857.
- Patel, A., Malinovska, L., Saha, S., Wang, J., Alberti, S., Krishnan, Y. and Hyman, A. A. (2017). ATP as a biological hydrotrope. Science 356(6339): 753-756.
- Pu, Y., Li, Y., Jin, X., Tian, T., Ma, Q., Zhao, Z., Lin, S. Y., Chen, Z., Li, B., Yao, G., Leake, M. C., Lo, C. J. and Bai, F. (2019). ATP-dependent dynamic protein aggregation regulates bacterial dormancy depth critical for antibiotic tolerance. Mol Cell 73(1): 143-156 e144.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Serrano, R. (1977). Energy Requirements for Maltose Transport in Yeast. European Journal of Biochemistry 80(1): 97-102.
- Sridharan, S., Kurzawa, N., Werner, T., Gunthner, I., Helm, D., Huber, W., Bantscheff, M. and Savitski, M. M. (2019). Proteome-wide solubility and thermal stability profiling reveals distinct regulatory roles for ATP. Nat Commun 10(1): 1155.
- Takaine, M., Ueno, M., Kitamura, K., Imamura, H. and Yoshida, S. (2019). Reliable imaging of ATP in living budding and fission yeast. J Cell Sci 132(8). doi: 10.1242/jcs.230649.
- Xu, L. and Bretscher, A. (2014). Rapid glucose depletion immobilizes active myosin V on stabilized actin cables. Curr Biol 24(20): 2471-2479.
- Yaginuma, H., Kawai, S., Tabata, K. V., Tomiyama, K., Kakizuka, A., Komatsuzaki, T., Noji, H. and Imamura, H. (2014). Diversity in ATP concentrations in a single bacterial cell population revealed by quantitative single-cell imaging. Sci Rep 4: 6522.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Takaine, M. (2019). QUEEN-based Spatiotemporal ATP Imaging in Budding and Fission Yeast. Bio-protocol 9(15): e3320. DOI: 10.21769/BioProtoc.3320.
Category
Cell Biology > Cell imaging > Fluorescence
Microbiology > in vivo model > Fungi
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.