- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Axon-seq for in Depth Analysis of the RNA Content of Neuronal Processes


Published: Vol 9, Iss 14, Jul 20, 2019 DOI: 10.21769/BioProtoc.3312 Views: 12448
Reviewed by: Gal HaimovichShyam SolankiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Neuronal processes have an RNA composition that is distinct from the cell body. Therefore, to fully understand neuronal biology in health and disease we need to study both somas, dendrites and axons. Here we describe a detailed protocol of a newly refined method, Axon-seq, for RNA sequencing of axons (and dendrites) grown in isolation using single microfluidic devices. We also detail how to generate motor neurons from mouse and human pluripotent stem cells for sequencing, but Axon-seq is applicable to any neuronal cell. In Axon-seq, the axons are recruited through a growth factor gradient, lysed and directly processed to cDNA without RNA isolation. A careful bioinformatic step ensures that any soma-contaminated samples are easily identified and removed.
Keywords: RNA sequencingBackground
Neurons are highly polarized cells. Their processes, both dendrites and axons, need to be able to respond to changes in the microenvironment in a manner independent of the soma (Holt and Schuman, 2013). To fully understand neuron biology, it is therefore important to be able to study neurites in isolation, in addition to conducting analysis of the cell bodies. This appears particularly important for neurons where the axon and dendrites constitute the majority of the cellular volume, as is the case for spinal motor neurons where we estimate that they comprise approximately 99% of the cellular volume. To isolate neurites, there are excellent tools such as Campenot Chambers (Boyden, 1962; Campenot, 1977) or microfluidic chambers (Taylor et al., 2005).
However, while neurites are separated out in such devices, it is important to bear in mind that cross-contamination between compartments can still occur. Thus, RNA sequencing of isolated axonal compartments (Minis et al., 2014; Saal et al., 2014; Briese et al., 2016; Rotem et al., 2017) can lead to incorrect results/conclusions when the purity and exclusion of somas is insufficiently examined. To ensure detailed and accurate investigation of motor axonal mRNA composition and its modulation in ALS we developed Axon-seq (Nijssen et al., 2018). This is an application of our LCM-seq method for single-cell spatial RNA-sequencing (Nichterwitz et al., 2016).
In Axon-seq, we use microfluidic devices to separate axons from stem cell-derived motor neurons (mouse and human) from their somas. As motor neurons have axons that traverse far longer distances than their dendrites, we are able to analyze axons alone. This may not be possible for all neuronal subtypes, but the method still allows for analysis of neurites as an entity.
In contrast to previous methods, Axon-seq does not require an RNA isolation step, and it allows for high sensitivity and cost-efficient sequencing from a single microfluidic device.
Notably, Axon-seq effectively eliminates all samples with any somatic cross-contamination, as it uses a highly stringent and sensitive bioinformatic quality control step that identifies axonal samples containing trace levels of mRNA from undesired cell somas. Here we provide a detailed protocol for Axon-seq which can be applied to any cell containing longer processes.
Materials and Reagents
- Materials
- Sterile 6 and 10 cm bacterial dishes (non-adhesive plastic) (Corning, catalog numbers: 351007 and 351029)
- Pipette tips (0.5-10 μl, 10-100 μl, 100-1,000 μl)
- Sterile 15 and 50 ml Falcon tubes
- Parafilm
- 70 μm cell-strainer filter (VWR, catalog number: 732-2758P)
- 24 x 32 mm cover glasses (VWR, Menzel Gläser, catalog number: 630-2873)
- Soft pencil-type brush (Any soft brush type that does not have too strong hairs that could pierce/damage the PDMS-surface of which the microfluidic devices are made. Brushes, from a typical hardware/convenience store, with a diameter of 2-5 mm work best, laboratory grade is not required. Nylon/synthetic hairs are not an issue.)
- Waterproof marker with fine (1 mm) or very fine (0.3 mm) tip
- Biological material
- Motor neurons from mouse embryonic stem cells
- Motor neurons from human pluripotent stem cells
- Reagents
- 70% ethanol
- Triton X-100 (Sigma-Aldrich, catalog number: X100)
- PBS (Thermo Fisher, catalog number: 14190136)
- Poly-L-ornithine, 0.01% solution (Sigma-Aldrich, catalog number: A-004-C)
- Fibronectin, 1 mg/ml solution (Sigma-Aldrich, catalog number: F1056)
- Laminin, 1 mg/ml solution (Sigma-Aldrich, catalog number: L2020)
- TrypLe Express (Thermo Fisher, catalog number: 12604021)
- Neurobasal (Thermo Fisher, catalog number: 21103049)
- DMEM/F12 with GlutaMAX (Thermo Fisher, catalog number: 31331028)
- B27 supplement, custom (Thermo Fisher, catalog number: 0080085SA)
- Penicillin/Streptomycin (Thermo Fisher, catalog number: 15140122)
- GlutaMAX supplement 100x (Thermo Fisher, catalog number: 35050061)
- 2-Mercaptoethanol (Sigma-Aldrich, catalog number: M6250)
- Ascorbic acid, 100 mg powder (Sigma-Aldrich, catalog number: A4403), prepare as 200 mM stock solution by dissolving the powder in 2.84 ml of nuclease-free dH2O
- Y-27632, 10 mg powder (Tocris, catalog number: 1254), prepare as 10 mM stock solution by dissolving the powder in 3.12 ml nuclease-free dH2O
- LDN-193189 dihydrochloride, 10 mg powder (Tocris, catalog number: 6053), prepare as 10 mM stock solution by dissolving the powder in 2.09 ml DMSO, then prepare 1 mM working stocks (1:10 diluted) in DMSO
- SB-431542, 10 mg powder (Tocris, catalog number: 1614), prepare as 10 mM stock solution by dissolving the powder in 2.60 ml DMSO
- CHIR-99021, 10 mg powder (Tocris, catalog number: 4423), prepare as 3 mM stock solution by dissolving the powder in 7.16 ml DMSO
- DAPT, 10 mg powder (Tocris, catalog number: 2634), prepare as 10 mM stock solution by dissolving the powder in 2.31 ml DMSO
- SAG, 1 mg powder (Tocris, catalog number: 4366), prepare as 500 μM stock solution by dissolving the powder in 4.08 ml DMSO
- All-trans retinoic acid, 50 mg powder (Sigma-Aldrich, catalog number: R2625), prepare as 100 mM stock solution by dissolving the powder in 1.66 ml DMSO, then prepare 1 mM working stocks (1:100 diluted) in DMSO
- Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, catalog number: D2650)
- Bovine serum albumin (BSA), 7.5% solution in dPBS (Sigma-Aldrich, catalog number: A8412)
- Glial-derived neurotrophic factor (GDNF), 10 μg powder (Peprotech, catalog number: 450-10), prepare as 10 μg/ml stock solution by dissolving the powder in 1 ml dPBS with 0.1% BSA
- Brain-derived neurotrophic factor (BDNF), 10 μg powder (Peprotech, catalog number: 450-02), prepare as 10 μg/ml stock solution by dissolving the powder in 1 ml dPBS with 0.1% BSA
- RNase-inhibitor (Takara, catalog number: 2313A)
- DTT (part of SuperScript II Reverse Transcriptase kit, Thermo Fisher, catalog number: 18064071)
- Nuclease-free dH2O (Thermo Fisher, catalog number: 10977-035)
- 37% formaldehyde solution (Merck Millipore, catalog number: 818708)
- Mouse MN medium (see Recipes)
- N2 medium (see Recipes)
- B27 medium (see Recipes)
- Human EB medium (see Recipes)
- Lysis/harvesting solution (see Recipes)
Equipment
- Glass beaker (500 ml)
- Sterile forceps (Dumont, catalog number: 11251-20, or other forceps of similar standard size)
- Pipettes (0.5-10 μl, 10-100 μl, 100-1,000 μl)
- Magnetic stirrer
- Sterile laminar flow hood
- Humidified incubator (37 °C, 5% CO2)
- Suction aspirator
- Standard Neuron Device (Xona MicrofluidicsTM, SND75/150/450/900)
- Low-speed orbital shaker (Elmi-tech, DOS-20S)
- Inverted bright field microscope (> 20x magnification is required to reliably see axons exiting the microgrooves. Examples are Laxco LMI-3000 or Carl Zeiss PrimoVert. Any similar setup is fine.)
Software
- R (https://www.r-project.org)
- RStudio open source edition, Boston, MA, USA (https://www.rstudio.com)
Procedure
- Storage and washing of microfluidic devices
Commercial microfluidic devices from Xona MicrofluidicsTM (Standard Neuron Devices) with any groove length can be stored in dH2O and used multiple times.
Note: The groove length of the devices could affect the proportion of axons versus dendrites that can cross over to the opposite chamber, as could the length of time of culture in the devices. For mouse motor neurons, we used devices with 150 μm length grooves and saw a clear majority of axons being recruited during the 7 days of culture. For human motor axons, we used devices with 450 μm grooves.
- Place devices in a glass beaker with 500 ml of 1% Triton X-100 in dH2O for 2 h and use a magnetic stirrer to keep devices agitated.
- Use a soft brush to gently clean the microgroove surfaces and the chambers of the devices. Transfer devices into dH2O afterward.
- Wash 3 x 10 min in fresh dH2O each time to remove any residues of Triton X-100. Then, sterilize devices in 70% ethanol for a minimum of 10 min.
Note: The washing procedures can be done at any time and devices can be stored in 70% ethanol to be ready when needed. Start the device attachment (below) the day that the coating procedure is to be initiated.
- Attachment of microfluidic devices to cell culture surface
- Remove microfluidic devices from 70% ethanol using sterile forceps and allow the devices to air-dry in a sterile laminar flow hood for 20 min. Use a suction aspirator to remove any residual ethanol.
Note: It is very important that devices are entirely dry at this stage, as any remaining liquid will negatively affect device attachment and can cause leakage in later stages. - Place 4 drops of 4 μl dH2O in a sterile 10 cm bacterial dish (Figure 1A).
- Place sterile glass cover glasses (32 x 24 mm, thickness #1 or #1.5) on top of the water drops to ensure that the cover glasses do not move around within the dish (Figure 1B).
- Use forceps to pick up one device at a time and gently press it, feature side down, onto the glass coverslip (Figure 1C). Apply gentle force, using the back of the forceps, to the center of the device (above the grooves), and all four corners. A microscope can be used to assess if all the grooves are in the same focal plane and thus attached to the coverslip. Devices attached in this way are stable for at least four weeks (and likely longer). Label the glass coverslips with a waterproof marker in one corner to define the future left-right orientation of the devices. The device forms the ‘ceiling’ for the cells and creates grooves for the axons to grow through. Cells will attach to the glass surface of the coverslip.
- The final setup of devices (Step B4) is a suggestion. Devices can also be placed directly in tissue culture-grade plastic dishes (i.e., directly onto the plastic, without cover glasses). For imaging purposes, however, placing them on glass coverslips is beneficial.
- In addition to the PDMS devices described here, Xona MicrofluidicsTM also offers pre-bonded devices for cell culture use (XonaChips). While these cost the same as regular devices, they are not optimal for reusing, as can be done with the regular devices (SND150). In the case of pre-bonded devices, the attachment step (Procedure B) can be skipped.
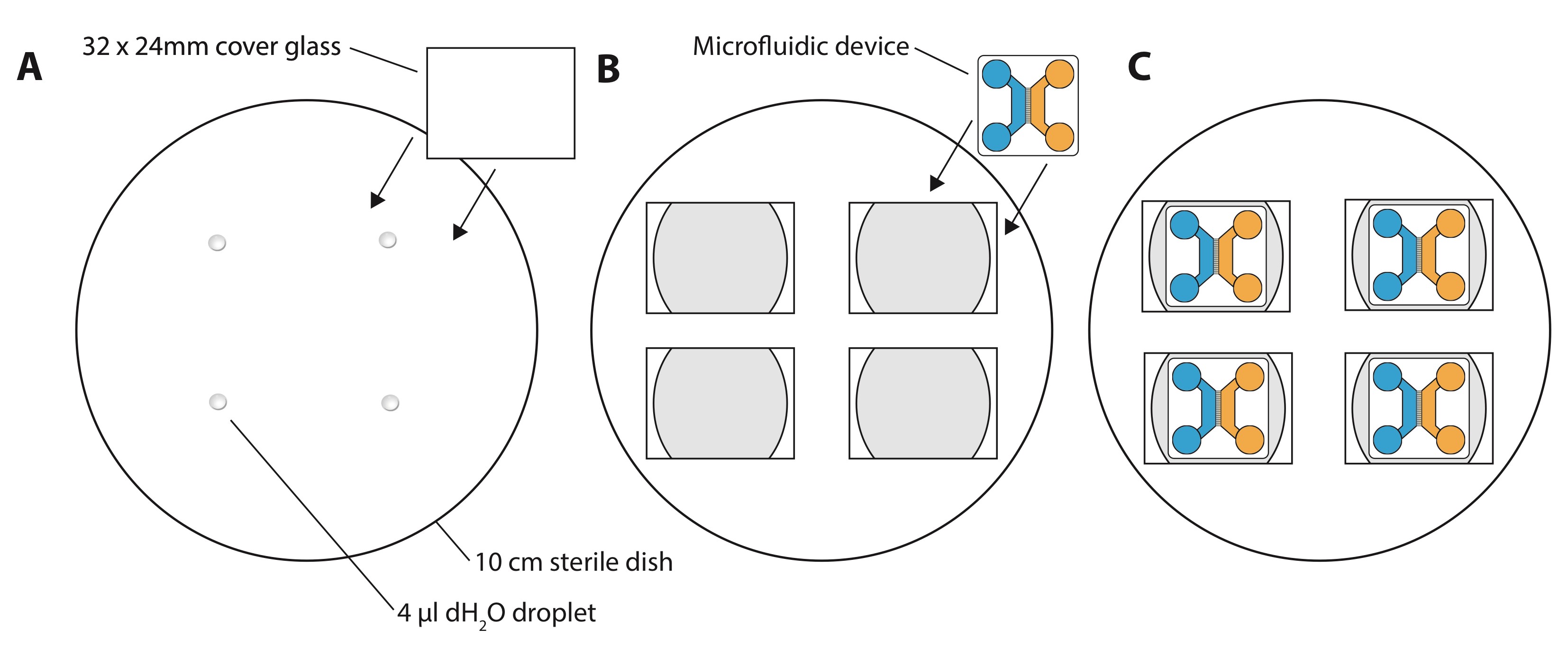
Figure 1. Setup of 4 microfluidic devices in a 10 cm dish. A. First, place 4 drops of 4 μl dH2O spaced apart evenly in the 10 cm dish. Then place glass cover glasses onto the drops to create a water seal between cover glasses and the plastic dish. B. When cover glasses are attached, place one microfluidic device with feature-side down on each cover glass. Press gently on the top of the device to ensure attachment. C. Final setup of four devices in one 10 cm dish. Other arrangements of devices (in terms of dish size) can be made if desired.
- Remove microfluidic devices from 70% ethanol using sterile forceps and allow the devices to air-dry in a sterile laminar flow hood for 20 min. Use a suction aspirator to remove any residual ethanol.
- Coating of surfaces in microfluidic devices
Day 1:- The primary surface coating consists of 0.001% poly-L-ornithine in dH2O. Prepare 0.5 ml solution per device.
- Add 75 μl solution into the two bottom wells of each device. Typically, the liquid will quickly fill the chambers through capillary forces. If this does not happen, use the aspirator with a mounted 1 ml pipette tip to gently suck the liquid through from the top. Do not aspirate all the liquid.
- Once the liquid has flown through, add 150 μl to the top left and 200 μl to the top right compartment. Adding different volumes between the left and right chamber ensures that the coating is forced into the microgrooves over time.
- Leave the coating on overnight at room temperature and shield it from light (e.g., a drawer works well).
- Wash the devices twice using 1x PBS. To wash, aspirate the coating from the wells, then add PBS into the wells, 75 μl in the bottom, then 150 μl in the top.
- The second coating consists of a solution of 2 μg/ml fibronectin and 5 μg/ml laminin in PBS. Add this last coating in a similar fashion to the first–see Steps C2-C4.
- Specification of motor neurons from mouse embryonic stem cells
Typically, a good starting point for differentiation is a 10 cm dish of stem cells at ~70-80% confluency. Cells are always maintained in a humidified incubator at 37 °C and 5% CO2.
Day 1:- Prewarm TrypLE Express to be used for dissociation at 37 °C (i.e., 2 ml for a 6 cm dish, 4 ml for 10 cm dish) in a tube. In addition, pre-warm a tube with 8 ml DMEM/F12-GlutaMAX (or any other plain MEM-based media).
- Add the TrypLE Express to initiate the dissociation.
- When cells have rounded up and are coming off the culture dish, use a pipette to dissociate them fully in the TrypLE Express, then transfer everything into the tube with pre-warmed DMEM.
- Spin down for 4 min at 200 x g, at room temperature.
- Remove supernatant and resuspend cells in 1 ml of mouse MN media (Recipe 1) and count.
- Place cells in bacterial (non-adherent) dishes at a density of 0.5-1.5 million cells per ml (the number of cells is line dependent and needs to be tested out), and place dishes on a low-speed orbital shaker at 30 rpm overnight in the incubator.
- Remove dishes from the shaker and change to fresh mouse MN media. To change media on EBs, pipette the entire volume of media (including EBs) into a tube. Allow the EBs to sink for ~2 min and remove the supernatant. Resuspend EBs in 1 ml fresh media and place into a new culture dish.
- Change to fresh mouse MN media, supplemented with 100 nM all-trans-retinoic acid (RA) and 500 nM smoothened agonist (SAG). Repeat for 4 successive days. See Table 1.
Table 1. Factors for motor neuron differentiation from mouse pluripotent stem cells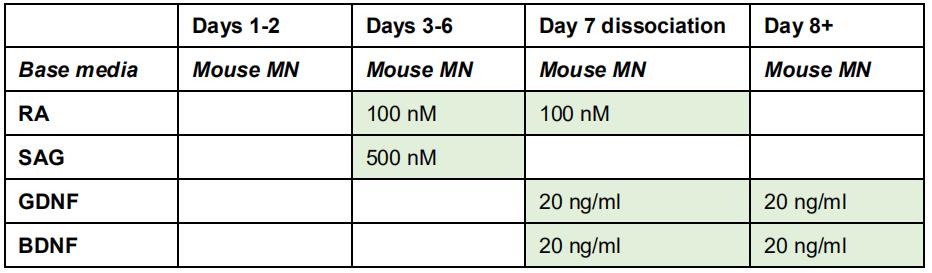
- After 4 consecutive days of patterning, cells are ready to be dissociated and seeded into microfluidic devices (see below).
- Specification of motor neurons from human pluripotent stem cells
Typically, a good starting point for differentiation is a 10 cm dish of stem cells at ~70-80% confluency. Cells are always maintained in a humidified incubator at 37 °C and 5% CO2.
Day 1:
The procedures on the first day (EB formation) are the same as described above for mouse ES cells. However, in the end, resuspend the cells in human EB media at a density of 0.5 M-0.75 M cells per ml (this density may require a degree of optimization between cell lines). Supplement the Day 1 human EB media with the required factors for Day 1 (for overview, see Table 2):
5 μM Y-27632 (ROCK-inhibitor)
200 nM LDN-193189
40 μM SB-431542
3 μM CHIR-99021
200 μM ascorbic acid
Table 2. Factors for motor neuron differentiation from human pluripotent stem cells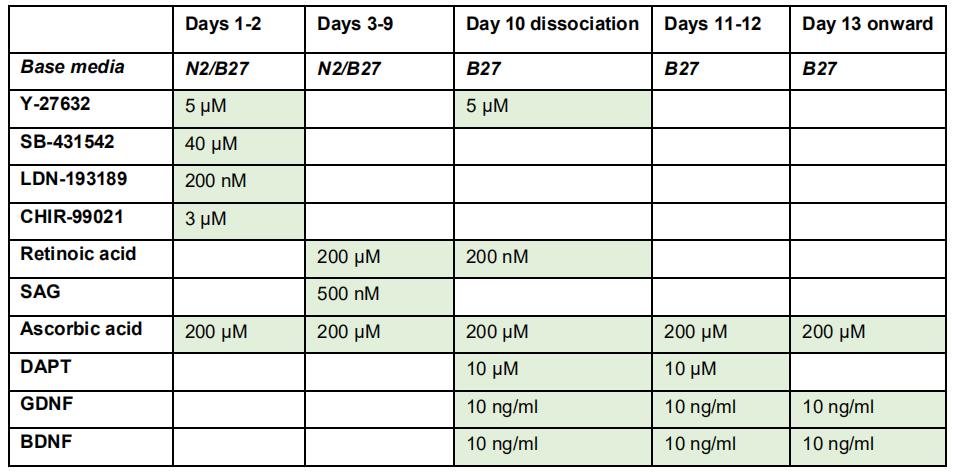
Days 2-10:
Perform daily media changes at approximate 24 h intervals with the corresponding factors for each day (Table 2).
Note: During the RA-SAG stage of the protocol (days 3-10), it is not absolutely required that media changes follow 24 h intervals, and if necessary (but not recommended) one day of media changes can be omitted. However, it is critical that the dual-SMAD inhibition (using SB-431542 and LDN-193189) during the first two days is 48 h and not shorter.
Day 10:
Cells are ready to be dissociated and seeded into microfluidic devices (see below). - Cell dissociation and preparation for seeding
- Transfer EBs into a sterile 15 ml Falcon tube. Allow the EBs to sink to the bottom for 1-5 min and aspirate the remaining media.
- Add 1 ml pre-warmed (37 °C) TrypLE Express to the Falcon tube with EBs. The dissociation takes 10-20 min, depending on the size of the EBs.
- Gently agitate the EBs every 5 min by tapping the tube or gently pipetting up and down using a 1 ml pipette.
Note: If you resuspend EBs by pipette for the first time after 5 min in TrypLE Express, do not pipette them up into the pipette tip as they tend to stick on the inside. Instead, just pipette the liquid and ensure the EBs consequently are agitated slightly in the suspension. - Once the EBs are beginning to break apart, pipette them up and down rather vigorously approximately 10 times to finalize the dissociation. Add 9 ml of pre-warmed DMEM to dilute the 1 ml of TrypLE Express.
- Filter the resulting suspension through a 70 μm cell-strainer filter to remove undissociated clumps/debris, then spin at 200 x g for 4 min, at room temperature.
- Resuspend the pellet in 1 ml of DMEM/F12-GlutaMAX (or any other plain MEM-based media). Count the cells.
- Adjust the density to a final concentration of 2.5 x 104 cells per μl (this will ensure that a total of 1 x 105 cells are seeded per microfluidic device later on). To do this, transfer 1 ml of the cell suspension into an Eppendorf tube and spin once more at 200 x g for 4 min. Resuspend cells in the required amount of media (i.e., 1 μl for every 2.5 x 104 cells). At this stage, the cells should be resuspended in the final culturing media, for human cells this is B27 media with the Day 10 factors (see Table 2), for mouse cells this is mouse MN media with Day 7 factors (see Table 1).
Note: The exact composition of the final media can vary based on the type of neurons in culture. However, the addition of ascorbic acid and trophic factors (and ROCK-inhibitor for human cells) is highly recommended to improve survival upon plating in the confined environment of the microfluidic device. - Prepare sufficient final media to fill all devices (0.5 ml per device). In the end, the device will hold media volumes as shown in Figure 2.
- Take the required amount of media (see Figure 2) for the axonal compartment (top and bottom well) and transfer to a new tube. Add trophic factors to increase the concentration to 50 ng/ml. The higher concentration of trophic factors on the “axonal” side will improve axonal recruitment across the microgrooves.
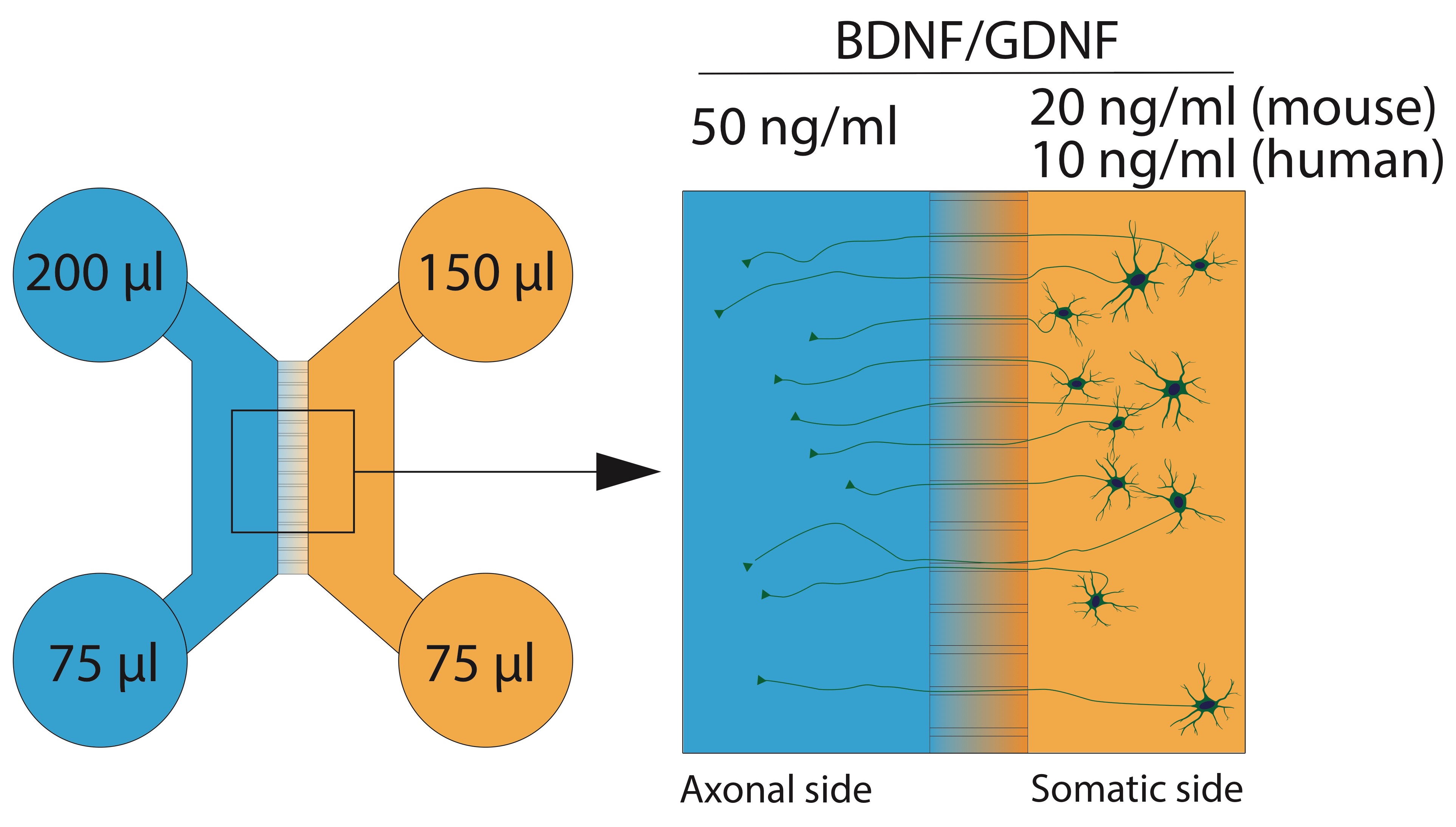
Figure 2. Media volumes and trophic factor concentrations in the microfluidic device at seeding. Note that somatic concentrations of the neurotrophic factors differ between human and mouse MNs. After axons have crossed the microgrooves into the other compartment, trophic factor concentrations can be equalized to 10 ng/ml on both sides, both for mouse and human.
- Cell seeding
- Start removing coating from the devices by aspiration (from one 10 cm dish with four devices at a time).
- Add 4 μl of the axonal media (containing 50 ng/ml BDNF/GDNF) into the chamber on the axonal side. Pipette close to the chamber entrance on the top-left (in the orientation of Figure 2, see Figure 3A for photograph) and allow capillary forces to pull the liquid into the chamber.
- Add the cells in the same manner to the other compartment. Once cells are seeded in the chamber, place devices in the incubator and allow the cells to attach for approximately 15-20 min.
- Start checking devices from 10 min after seeding the cells and onward to avoid excessive evaporation of the media. Once cells appear attached, add the final media amounts to the wells as indicated in Figure 2. Check if the cells remain attached and then place devices in the incubator overnight.
Notes:
- Occasionally cells do not attach well even after 15 min, maybe in part due to the flow between the compartments. If this occurs, we recommend adding only 40 μl of media to all wells (keeping the right media types for each side), which will avoid excessive flow between compartments. This way, there is minimal flow and evaporation will not be an issue. Then keep the cells like this for an additional hour and finally add media to the recommended amounts in Figure 2.
- For human cells, the dissociation takes place on Day 10 of the protocol. In addition to everything described here, maintain the corresponding factors in the media for each day as described in Table 2. The trophic factor (BDNF/GDNF) concentrations described there are for the somatic compartment, the axonal compartments receive the trophic factor gradient for the first few days (see below).

Figure 3. Seeding cells into microfluidic chambers and representative micrographs. A. Example photographs of the angle at which media/cells can be added into the chambers of the microfluidic device. Dashed lines show the location of the chambers, and chamber entrances. B. Micrographs of a good culture in the axonal compartment and the somatic compartment, where clusters of neuron somas are visible.
- Culturing devices
- After seeding neurons into microfluidic devices, change media daily to ensure that the trophic factor gradient stays intact. A bright field microscope can be used to keep track of the axonal growth.
Note: When changing media, do NOT aspirate media out of the chambers as this will forcibly remove all cells/axons, only change media in the wells. - Once axons can be seen crossing several microgrooves into the axonal compartment (i.e. > 10 grooves with axons), the trophic factor gradient can be removed, and both compartments instead receive 10 ng/ml of GDNF and BDNF. At this point, media can be exchanged every other day. See Figure 3B for a successful micrograph of a microfluidic device with growing axons.
- After seeding neurons into microfluidic devices, change media daily to ensure that the trophic factor gradient stays intact. A bright field microscope can be used to keep track of the axonal growth.
- Compartmental harvesting for RNA sequencing
- Prepare the lysis/harvesting solution. Prepare ~70 μl solution per microfluidic device, or less if only one compartment is harvested (final recommended lysis volumes are 50 μl for the somatic compartment and 10 μl for the axonal compartment). The lower volume for axons ensures that a higher concentration of RNA is present in the final solution, which aids in downstream applications.
- The lysis solution (see Recipes) is made up as a 2% Triton X-100 solution in nuclease-free dH2O. First, cool this solution on ice, then supplement with:
0.5 μl RNase-inhibitor per 100 μl (final concentration of 0.2 U/μl)
1 mM dithiothreitol (DTT), required for RNase-inhibitor enzymatic activity
Note: Always keep lysis solution on ice during preparation and use. - Prepare nuclease-free Eppendorf tubes. Label them according to the samples that are being collected, and pre-cool them on ice.
- Have dry ice ready to snap-freeze harvested samples immediately following the collection.
- Optionally, before starting the harvesting, take bright field images along the grooves. If somatic contamination is detected in the downstream bioinformatics analysis of axon samples, these images can be scrutinized for evidence of any cross-contaminating somas.
- Harvest one device at a time. Begin by washing both compartments once with pre-warmed (37 °C) PBS. Use at least 100 μl PBS.
- Remove PBS from the axonal wells and add 10 μl lysis solution to the top of the chamber, as if seeding cells (the vastly higher volume of PBS on the somatic side prohibits any lysis solution from flowing through the grooves and accidentally lyse somas).
- Wait for 10 s, then pipette up and down ~5 x in the bottom well, and extract as much solution as possible from the bottom of the chamber (can be > 10 μl). Transfer solution into a prepared Eppendorf tube and snap-freeze on dry ice.
- Add at least 100 μl PBS to the axonal side again.
- Repeat Steps I7 and I8 above for the somatic side, but instead use 50 μl lysis solution. When extracting the solution, try to extract as much as possible (> 50 μl). Snap-freeze on dry ice.
- After collection and snap-freezing, samples for RNA sequencing can be stored at -80 °C until further used.
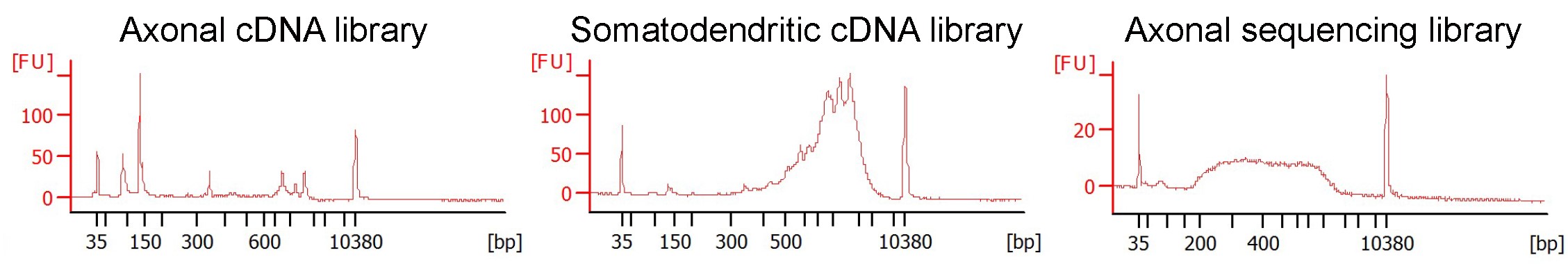
Figure 4. Typical bioanalyzer profiles for RNA sequencing using Axon-seq. Axonal cDNA samples have a distinct profile compared to somatodendritic samples. An example of ready to sequence axonal sequencing library is also shown after tagmentation reaction and barcoding. - Fixation of cells in devices for staining purposes
- Remove all media from the wells and wash both compartments once with PBS.
- Add a solution of 4% formaldehyde in PBS. When adding the formaldehyde, maintain similar volumes in all wells as used during media changes (see Figure 2) to ensure a flow both through the compartments and across the microgrooves.
- Leave the devices to fix at room temperature for 30 min.
- Wash 3 times with PBS, dispose of formaldehyde waste.
- Store devices in PBS (~200 μl in all wells) until further use. If not used within a few days, wrap the 10 cm dish with parafilm.
Note: Either before or after performing the desired staining method (immunocytochemistry/FISH/RNAscope), devices need to be removed along with coverslips from the 10 cm plastic dish. - Using a 1 ml pipette, add a thin line of H2O around the edges of the cover glass. This will loosen it from the underlying surface and cause it to float after a few seconds/minutes.
Data analysis
Bioinformatic quality control
- After RNA sequencing has been performed, a bioinformatic quality control (QC) step should be performed to exclude axonal samples with (traces of) soma contamination.
Note: The following is a simplified version of the bioinformatic QC that is conducted using the programming language R (R Core Team, 2018). More in-depth knowledge of bioinformatics and R will be required to perform these analyses, but what follows is a general guide to exclude any soma-contaminated samples.
- Ensure that the RNA sequencing data is processed to both a count table (raw read counts) and an RPKM table with normalized values (reads per kilobase per million mapped reads).
- As a general QC guideline, samples with too few mapped reads or detected genes should be excluded. To perform this, extract the total number of counts (= mapped reads) per sample from the count table, as well as the total number of genes with counts.
- Exclude samples with fewer than 2.5 x 105 mapped reads.
- Exclude samples that have fewer than 2,500 genes with any counts.
- After this general QC, the data can be inspected for soma-contamination of axonal samples. Briefly, there are four indicators to investigate:
The number of detected genes in axonal samples
2D visualization plots (PCA/umap)
Sample correlations in a correlation heatmap
Hierarchical clustering based on all expressed genes (or a subset)- Obtain the number of detected genes in the axonal samples, and plot these as a dot plot. As a rough guideline, axon samples contain fewer than ~8,000 genes. Investigate any outliers in this distribution. Soma-contaminated samples will have a strong increase in the number of detected genes.
- Using the base R function prcomp, or alternatively the package umap (Konopka, 2018), plot a 2D visualization of your samples (first 2 principal components for prcomp, or n_components = 2 for umap). Investigate the plot. Typically, axon and soma samples will separate strongly in 2D space. Contaminated axon samples will be significantly shifted towards the somatic samples (Figure 5).
- Using the base R function corr, calculate the correlation between samples based on all expressed genes. This correlation matrix can be plotted as a heatmap using the function pheatmap from the package carrying the same name. Typically, axonal samples have a low correlation to somatic samples. However, contaminated samples will have an increased overall correlation to all somatic samples.
- Using pheatmap (Kolde, 2019), a heatmap with corresponding hierarchical tree clustering can be generated based on all expressed genes in the dataset. Where axon and soma samples normally cluster in distinct branches, contaminated samples can cluster together with soma samples.
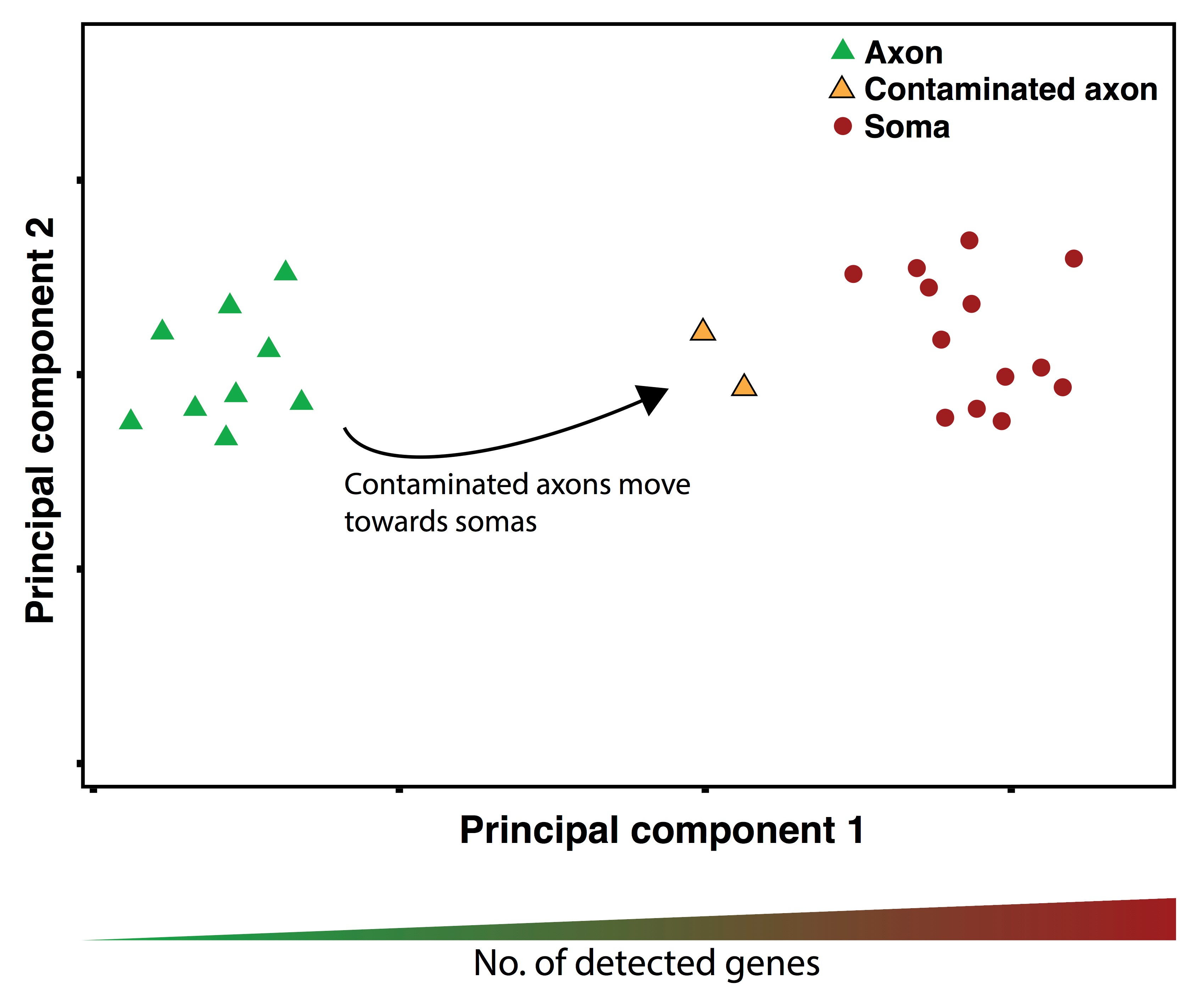
Figure 5. Example PCA-plot showing the shift of contaminated axon samples towards the somatic samples. They additionally have an increase in the number of detected genes per sample.
- Using these four measures, one can obtain an idea of samples that are possibly soma contaminated. Note that cutoffs need to be set for each of the measures, and the decision to exclude samples depends on the severity of the contamination.
Recipes
- Mouse MN medium (500 ml)
240 ml Neurobasal
240 ml DMEM/F12-GlutaMAX
10 ml B27 supplement, custom (stock 50x, add the full supplement bottle)
5 ml Penicillin/Streptomycin
5 ml GlutaMAX supplement
0.91 ml 2-Mercaptoethanol - N2 medium
1 bottle (500 ml) of DMEM/F12-GlutaMax
5 ml N2 supplement (stock 100x, add the full supplement bottle)
5 ml Penicillin/Streptomycin - B27 medium
1 bottle (500 ml) of Neurobasal
10 ml B27 supplement, custom (stock 50x, add the full supplement bottle)
5 ml Penicillin/Streptomycin - Human EB medium
50/50 mixture of N2 medium and B27 medium (above) - Lysis/harvesting solution (~70 μl per microfluidic device)
2% Triton X-100 in nuclease-free dH2O. Cool on ice, then supplement with:
0.5 μl RNase-inhibitor per 100 μl (final concentration of 0.2 U/μl)
1 mM dithiothreitol (DTT), required for RNase-inhibitor enzymatic activity
Acknowledgments
This work was supported by grants from the Swedish Research Council [2016-02112 and 2018-03255]; EU Joint Programme for Neurodegenerative Disease (JPND) [529-2014-7500]; the Strategic Research Programme in Neuroscience (StratNeuro); Karolinska Institutet; Birgit Backmark’s Donation to ALS Research at Karolinska Institutet in memory of Nils and Hans Backmark; Åhlén-stiftelsen (mA1/h17 and mA1/h18); Ulla-Carin Lindquists stiftelse för ALS forskning; and Magnus Bergvalls stiftelse [2015-00783 and 2016-01531] to E.H. J.A.B. is supported by a postdoctoral fellowship from the Swedish Society for Medical Research (SSMF).
This protocol is based on our previous study published in Stem Cell Reports in 2018: Nijssen et al. (2018).
Competing interests
The authors declare that there are no financial or non-financial competing interests.
Ethics
All work was carried out according to the Code of Ethics of the World Medical Association (Declaration of Helsinki) and with national legislation and institutional guidelines. The use of human stem cell lines was approved by the regional ethical review board in Stockholm, Sweden (Regionala Etikprövningsnämnden, Stockholm, EPN).
References
- Boyden, S. (1962). The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J Exp Med 115: 453-466.
- Briese, M., Saal, L., Appenzeller, S., Moradi, M., Baluapuri, A. and Sendtner, M. (2016). Whole transcriptome profiling reveals the RNA content of motor axons. Nucleic Acids Res 44(4): e33.
- Campenot, R. B. (1977). Local control of neurite development by nerve growth factor. Proc Natl Acad Sci U S A 74(10): 4516-4519.
- Holt, C. E. and Schuman, E. M. (2013). The central dogma decentralized: new perspectives on RNA function and local translation in neurons. Neuron 80(3): 648-657.
- Kolde R. (2019). R-package: pheatmap. Pretty Heatmaps.
- Konopka T. (2018). R-package: umap. Uniform Manifold Approximation and Projection.
- Nichterwitz, S., Chen, G., Aguila Benitez, J., Yilmaz, M., Storvall, H., Cao, M., Sandberg, R., Deng, Q. and Hedlund, E. (2016). Laser capture microscopy coupled with Smart-seq2 for precise spatial transcriptomic profiling. Nat Commun 7: 12139.
- Nichterwitz, S., Benitez, J. A., Hoogstraaten, R., Deng, Q. and Hedlund, E. (2018). LCM-Seq: A Method for Spatial Transcriptomic Profiling Using Laser Capture Microdissection Coupled with PolyA-Based RNA Sequencing. Methods Mol Biol 1649: 95-110.
- Nijssen, J., Aguila, J., Hoogstraaten, R., Kee, N. and Hedlund, E. (2018). Axon-Seq Decodes the Motor Axon Transcriptome and Its Modulation in Response to ALS. Stem Cell Reports 11(6): 1565-1578.
- R Core Team. (2018). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/.
- Rotem, N., Magen, I., Ionescu, A., Gershoni-Emek, N., Altman, T., Costa, C. J., Gradus, T., Pasmanik-Chor, M., Willis, D. E., Ben-Dov, I. Z., Hornstein, E. and Perlson, E. (2017). ALS along the axons–expression of coding and noncoding RNA differs in axons of ALS models. Sci Rep 7: 44500.
- Saal, L., Briese, M., Kneitz, S., Glinka, M. and Sendtner, M. (2014). Subcellular transcriptome alterations in a cell culture model of spinal muscular atrophy point to widespread defects in axonal growth and presynaptic differentiation. RNA 20(11): 1789-1802.
- Taylor, A. M., Blurton-Jones, M., Rhee, S. W., Cribbs, D. H., Cotman, C. W. and Jeon, N. L. (2005). A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods 2(8): 599-605.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Nijssen, J., Aguila, J. and Hedlund, E. (2019). Axon-seq for in Depth Analysis of the RNA Content of Neuronal Processes. Bio-protocol 9(14): e3312. DOI: 10.21769/BioProtoc.3312.
Category
Neuroscience > Cellular mechanisms > RNA Localization
Stem Cell > Pluripotent stem cell > Cell-based analysis
Molecular Biology > RNA > RNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.