- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Lipid-exchange Rate Assay for Lipid Droplet Fusion in Live Cells


Published: Vol 9, Iss 14, Jul 20, 2019 DOI: 10.21769/BioProtoc.3309 Views: 7365
Reviewed by: David PaulSijie WeiSurabhi Sonam
Original research article
The authors used this protocol in:

Dec 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Lipid droplets (LDs) are central organelles in maintaining lipid homeostasis. Defective LD growth often results in the development of metabolic disorders. LD fusion and growth mediated by cell death–inducing DNA fragmentation factor alpha (DFFA)-like effector (CIDE) family proteins are crucial for various biological processes including unilocular LD formation in the adipocytes, lipid storage in the liver, milk lipid secretion in the mammary epithelia cells, and lipid secretion in the skin sebocytes. Previous methodology by Gong et al. (2011) first reported a lipid-exchange rate assay to evaluate the fusion ability of each LD pair in the cells mediated by CIDE family proteins and their regulators, but photobleaching issue remains a problem and a detailed procedure was not provided. Here, we provide an improved and detailed protocol for the lipid-exchange rate measurement. The three key steps for this assay are cell preparation, image acquisition, and data analysis. The images of the fluorescence recovery are acquired after photobleaching followed by the measurement of the intensity changes in the LD pair. The difference in fluorescent intensity is used to obtain the lipid exchange rate between the LDs. The accuracy and repetitiveness of the calculated exchange rates are assured with three-cycle of photobleaching process and the linear criteria in data fitting. With this quantitative assay, we are able to identify the functional roles of the key proteins and the effects of their mutants on LD fusion.
Keywords: Lipid metabolismBackground
Lipid droplets (LDs) play a key role in maintaining lipid homeostasis (Farese and Walther, 2009; Yang et al., 2012). Defective LD maturation and growth are closely associated with the development of metabolic diseases such as obesity, fatty liver disease, cardiovascular disease, and type II diabetes (Krahmer et al., 2013; Rosen and Spiegelman, 2014; Gluchowski et al., 2017). LDs are dynamic organelles which budded from the endoplasmic reticulum (ER) (Gross et al., 2011; Choudhary et al., 2015) and continue to grow via triglyceride synthesis and lipid transfer from the ER (Fujimoto et al., 2007; Wilfling et al., 2013; Xu et al., 2018) or LD fusion (Gong et al., 2011; Somwar et al., 2011). LD-associated cell death–inducing DNA fragmentation factor alpha-like effector (CIDE) family proteins including CIDEA, CIDEB, and CIDEC/Fsp27 (Gao et al., 2017) are crucial regulators in the lipid homeostasis by governing atypical LD fusion and growth for lipid storage. Previously, we reported that CIDEC mediates LD fusion through directional lipid transfer from small (donor) to large (acceptor) LDs (Gong et al., 2011; Somwar et al.). The enrichment of CIDEC at the LD-LD contact site (LDCS) and the formation of the fusion pore are two essential steps for lipid exchange and transfer to occur. To evaluate the fusion ability of each LD pair in cells mediated by the CIDE family proteins and their regulators such as Perilipin1 (Sun et al., 2013a) or Rab8a (Wu et al., 2014), lipid-exchange rate assay was first proposed and performed as previously described (Gong et al., 2011; Sun et al., 2013b). However, the analytic process reported in the previous methodology neither eliminated the photobleaching effect upon laser exposure nor provided a detailed procedure to ensure experimental reproducibility and accuracy.
Here, we detailed the protocol of our renewed lipid-exchange rate assay used in Wang et al. (2018). The three key steps in the assay including cell preparation, fluorescence recovery after photobleaching (FRAP) image acquisition, and data analysis using mean of intensities (MOI) and BODIPY-C12-stained size measurement of LDs were reported. Upon performing a three-cycle photobleaching process and the pre-estimation of exchange rate based on the linear criteria in the data analysis, we can ensure the accuracy and repetitiveness of the fitted exchange rates by using our proposed equation underlying molecular thermodynamics in the Theory section. This lipid-exchange rate assay is also applicable for the evaluation of a nanometer size channel connecting two vesicles when the micrometer volumes of the vesicles are measured in advance.
Materials and Reagents
- Materials
- Pipette tips (Corning, Axygen, catalog numbers: T-1000-B, T-200-Y, T-300)
- 100 mm culture dish (Thermo Fisher, Nunc, catalog number: 150462)
- 35 mm glass bottom culture dish (Thermo Fisher, catalog number: 150682)
- 1.5 ml Eppendorf tube (Corning, Axygen, catalog number: MCT-150-C)
- 50 ml, 15 ml centrifuge tubes (Thermo Fisher, catalog numbers: 339652, 339650)
- Gene Pulser (Bio-Rad, catalog number: 165-2086)
- Biological materials
- 3T3-L1 pre-adipocyte (ATCC, catalog number: CL-173)
- Cidec-GFPN1 plasmid (Wang et al., 2018; available from the corresponding author upon request)
- Reagents
- BODIPY 558/568 C12 (Thermo Fisher, Molecular Probes, catalog number: D3835)
- Sodium oleate (Sigma-Aldrich, catalog number: O7501)
- Dulbecco’s modified Eagle’s medium (DMEM), high glucose, with L-Glutamine and Phenol Red (Gibco, catalog number: 11965084)
- Fetal bovine serum (FBS) (Gibco, catalog number: 16140071)
- Penicillin-streptomycin Mixed Solution (P/S) (Gibco, catalog number: 15140122)
- Electroporation buffer (Bio-Rad, catalog number: 1652676)
- Trypsin-EDTA (Thermo Fisher, Life Technologies, catalog number: 25200-072)
- NaCl (Sigma-Aldrich, catalog number: S3014)
- KCl (Sigma-Aldrich, catalog number: P9541)
- Na2HPO4 (Sigma-Aldrich, catalog number: S5136)
- KH2PO4 (Sigma-Aldrich, catalog number: P9791)
- CaCl2 (Sigma-Aldrich, catalog number: C5670)
- MgCl2 (Sigma-Aldrich, catalog number: M4880)
- Phosphate-buffered saline (PBS) (see Recipes)
Equipment
- Pipettes (Gilson, models: PIPETMAN P10, P20, P200, P1000)
- Cell counting chamber (Easybio, catalog number: BE6138)
- 37 °C, 5% CO2 cell culture incubator (NuAire, model: NU-49SOE)
- Biosafety cabinet (NuAire, model: NU-425-400E)
- Centrifuge (Cence, China, model: TDZ5-WS)
- Electroporation (Lonza, model: Amaxa Nucleofactor II)
- Confocal fluorescent microscope (Nikon Instruments, model: Nikon A1+ Confocal Microscope)
- Live cell station (Oko laboratory, model: A1 Confocal)
- Computer (Lenovo, model: ThinkStation P510)
Software
- NIS-element analysis (Nikon, https://www.microscope.healthcare.nikon.com/products/software)
- Fiji (NIH software, http://fiji.sc/Fiji)
- Open-source plugin-based image analysis software based on ImageJ (https://imagej.nih.gov/ij/)
- LabVIEW 8.5 with a plug-in installation of NI Vision 8.6 module (National Instruments, http://www.ni.com/en-us/support/downloads/software-products/download.labview.html)
- Custom-made LabVIEW modules:
- 1_Exchange rate assay.llb (applied in Step B of Data analysis)
- 2_Check a fitting region.vi (applied in Step C of Data analysis)
- 3_Calculation of exchange rate.vi (applied in Step D of Data analysis)
- Prism 5 (GraphPad Inc.)
Procedure
Theory: For the estimation of LD fusion ability, we measure a lipid-exchange rate as an alternative indicator of potential pore size between two contacted LDs. During LD fusion, the lipids constantly exchange between the two LDs. For the measurement of the lipid-exchange rates between the LDs in live cells, the small (donor) LD of each LD pair pre-stained with BODIPY-C12 was photobleached. The fluorescence-labeled lipids in the large (acceptor) LD diffused into the small LD, and the photobleached lipids went into the large LD. The images of the small and large (acceptor) LDs, before and after photobleaching, were recorded and their fluorescent intensities were normalized to the initial fluorescent intensity of the large LD at time 0 s after photobleaching, giving rise to the initial intensity ratio of 1 for the large LD. The ratio of fluorescent intensity of the photobleached small LD to the large LD at time 0 s is denoted as G0. At any time after photobleaching, the time-dependent ratios of the fluorescent intensities of the large and small LDs are denoted as G1 (t) and G2 (t), respectively. Therefore, a time-invariable exchange rate (∅e) of the neutral lipid molecules between an LD pair is calculated from the measurement of the fluorescence recovering rate in the LD pair according to the ordinary differential equation as follows: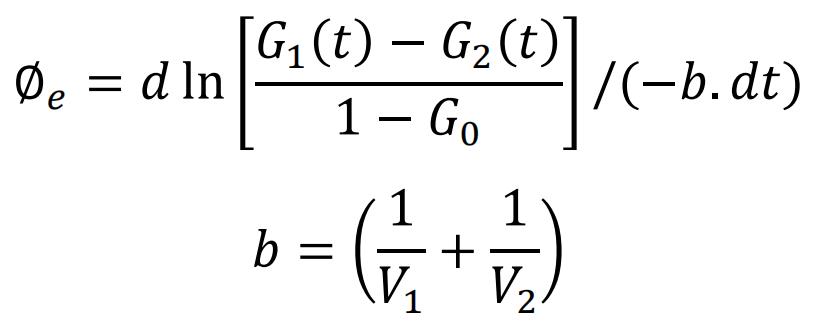
where V1 and V2 represent the volumes of the large and small LDs, respectively. The unit of the lipid-exchange rate is μm3/s. The detailed equation derivation is shown in Supplementary Information.
- Overall procedure
The procedure for the lipid-exchange rate assay of LDs in live cells is shown in Figure 1.
Figure 1. Schematics of LD lipid-exchange rate measurement in live cells - Cell preparation
- Culture 3T3-L1 pre-adipocytes in 10 cm plastic dish in 10 ml DMEM supplemented with 10% FBS, 100 μg/ml streptomycin and 100 U/ml penicillin.
- At 80% confluence, trypsinize the cells using 2 ml 0.25% trypsin solution. Terminate the process using 5 ml DMEM. Transfer the cells with DMEM into a 15 ml tube and centrifuge at 150 x g for 3 min, discard the supernatant.
- Resuspend the cell using 1 ml PBS.
- Count cells using a cell counting chamber.
- Centrifuge at 150 x g for 3 min, discard the supernatant.
- Resuspend the cells with appropriate electroporation buffer to reach a cell density of 2-3 x 106/ml.
- Aspirate 100 μl of cells, mix with 0.5-1 μg plasmid (Cidec-GFP) in a 1.5 ml Eppendorf tube.
- Transfer the cell-DNA mix into a genepulser, place the genepulser into Nucleofector II, choose the program A-033, which is optimized for 3T3-L1 pre-adipocytes transfection, execute the electroporation step.
- Add 100 μl of DMEM into the genepulser, aspirate out the mixture into a 15 ml centrifuge tube with 2 ml DMEM medium.
- Add BODIPY-C12 558/568 into the medium to a final concentration of 1 μg/ml.
- Add sodium oleate solution into the medium to a final concentration of 200 μM to provide lipids for LD formation and growth in cells.
- After the addition of the above reagents, aspirate the mixture into a 35 mm glass bottom culture dish and incubate for 18-20 h in an incubator at 37 °C, 5% CO2.
- Change the culture medium with 2 ml fresh DMEM for two reasons: 1) for the removal of BODIPY-C12 dye from the medium to eliminate background fluorescence; 2) for the removal of sodium oleate to reduce the influence of triglycerides synthesis on LD fusion. Next, place the culture dish with the cells in the incubator for 1 h before FRAP experiments to help the cells accommodate to the changed medium.
- Fluorescence recovery after photobleaching (FRAP) image acquisition
The module of FRAP is conventional in most commercial confocal microscopes. Here, we used a Nikon A1 confocal microscope to perform FRAP measurements as an example. This following procedures can also be used for FRAP experiments when using the other confocal microscopes.- Turn on the live cell station, set the temperature to 37 °C, switch on the 5% CO2, add an appropriate amount of double distilled water in a heating trough.
- Turn on the confocal microscope, laser generator, microscope controller, mercury lamp, electric automatic stage, z-axis piezoelectric stage, and computer workstation. Turn on the perfect focus system (PFS). Launch the Nis-element software. Choose the 100x oil-immersion objective (Numerical aperture 1.45). Add objective oil dropwise onto the objective, place the 35 mm glass bottom culture dish onto the sample holder, and adjust the height of the objective to immerse the culture dish in the oil. Allow the cells to accommodate for 1 h if possible, or at least 0.5 h to ensure the stability of the following measurement.
- Set the parameters of the microscope as shown in Figure 2. Click on the channel button, select “Fluorescein isothiocyanate (FITC)” and “Tetramethylrhodamine (TRITC)” channels. Choose “none” in Channel series. To capture the fluorescent images, fine settings of the optimized laser power and the gain and sensitivity of detector are critical. There are several commercial microscopes but their definitions of the setting parameters are different. Here, we used Nikon A1 confocal microscope as an example to show our settings, in which “Laser power” indicates the level of real laser power, “HV” indicates the gain of detector (brightness of images), and “Offset” indicates the sensitivity of detector (contrast of images). Thus, set the FITC laser power to “6.0”, HV “60-120”, off set “0”; the TRITC laser “0.3”, HV “60-120”, off set “0”. Set pinhole as home under the wavelength of 561 nm. Set scan size to “512 x 512 pixels”, scan speed “1 frame/s”, zoom “3.0”, and line average “none”.
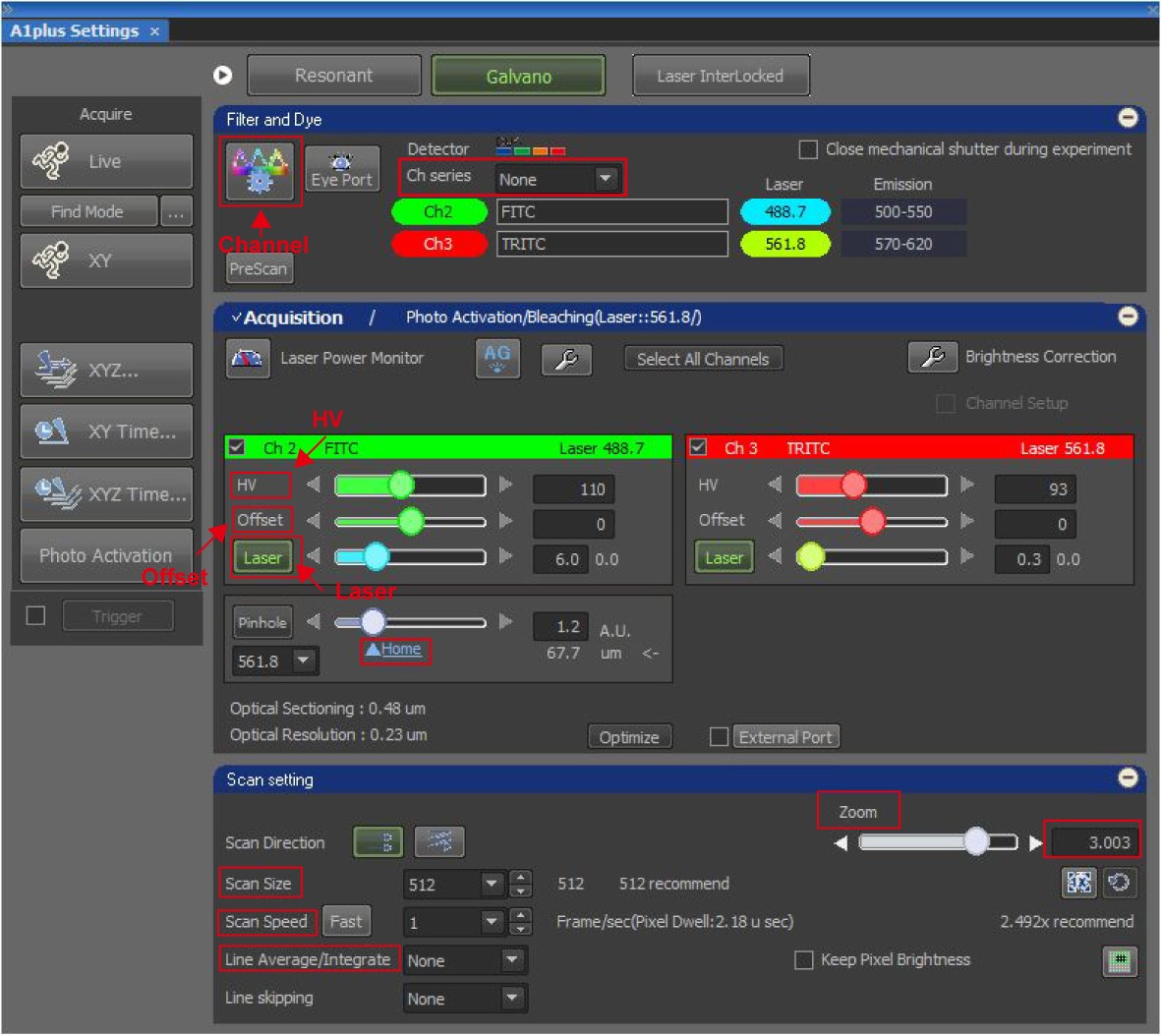
Figure 2. NIS-element settings of images acquisition. The image shows the settings of the laser channel, laser power, HV, Offset, pinhole size, scan size, scan speed, and other detailed parameters for image acquisition. - Set the photobleaching parameters:
- Switch on the photoactivation interface as shown in Figure 3. Set the photobleaching laser as the wavelength of 561 nm, power of 100%. Set the stimulation scanning speed as 0.25 sec/frame.
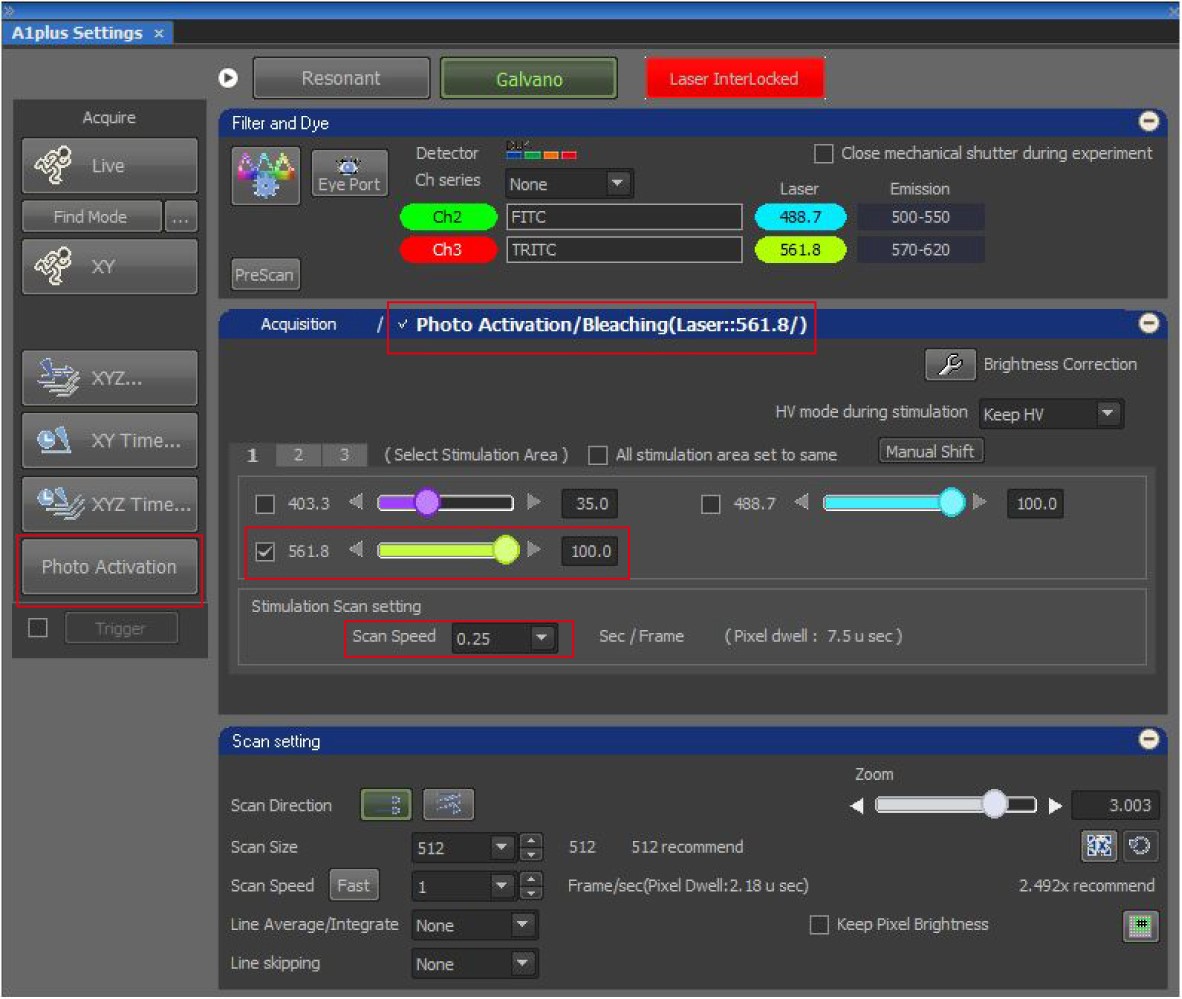
Figure 3. NIS-element settings of photobleaching. The image shows the settings of laser power and scan speed for photobleaching. - Go to the N-dimensional (ND) Stimulation Interface as shown in Figure 4, in which we could set the XYZ locations of a few images captured and the time schedule options. Create three phases corresponding to the three-steps in time-sequential operations. In Phase 1, choose “Acquisition” in Acq/Stim, “No delay” in Interval, a duration of 1.06 s and 1 loop to capture one image, which shows the initial fluorescent intensities of an LD pair before photobleaching. In Phase 2 and Phase 3, set 3x in Group option. In addition, in Phase 2, set “Stimulation” in Acq/Sim, “S1” in ROIs and 1 loop to perform photobleaching. The stimulation duration will be automatically set according to the setting of stimulation scanning speed in Figure 3. In Phase 3, choose “Acquisition” in Acq/Stim, “1.06 s” in time Interval, and 30 (fast exchange rate) or 50 (slow exchange rate) loops to capture time-lapse images with a time interval of 1.06 s, which record the change in the fluorescent intensities of the LD pair after photobleaching. Subsequently, the duration is automatically calculated. Choose “Perform Time Measurement”. Check “Save to File”, input the path and name the file.
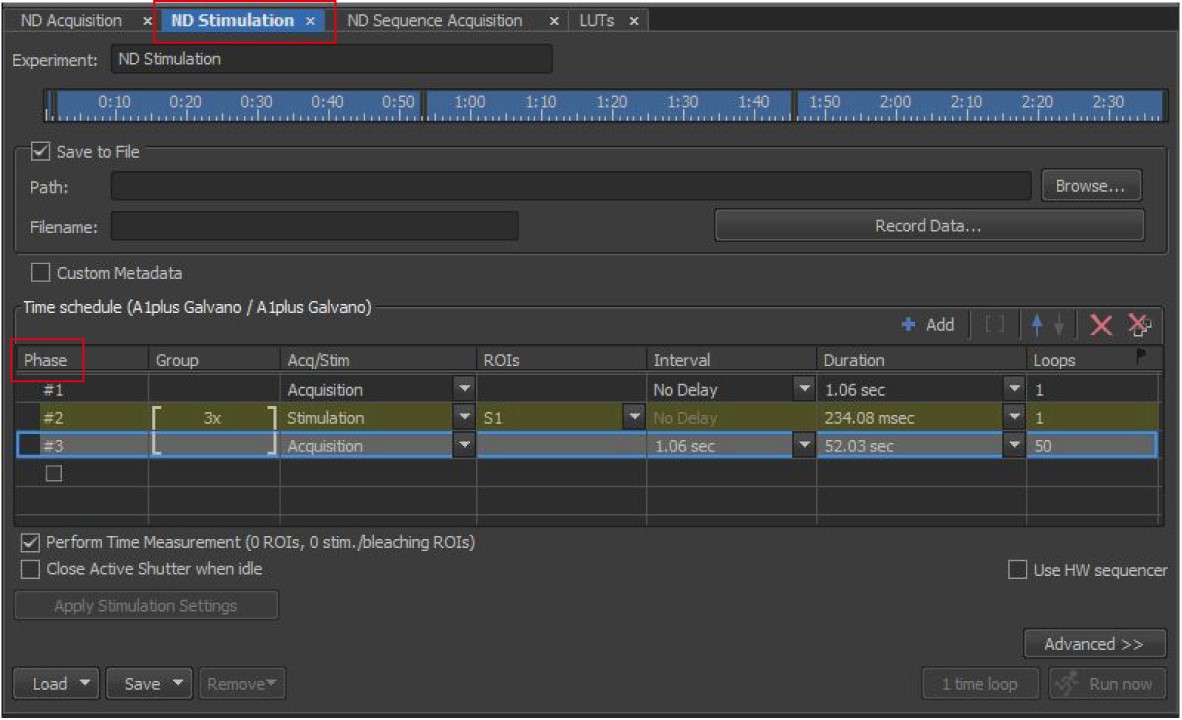
Figure 4. NIS-element settings of ND Stimulation. The image shows three phases corresponding to the three-step time-sequential operations in the ND stimulation setting. - Click on the button “Time measurement” in the menu to stick the time measurement interface in front of all interfaces. The interface is used to monitor the change in fluorescent signals of the selected regions-of-interest over time.
- Switch on the photoactivation interface as shown in Figure 3. Set the photobleaching laser as the wavelength of 561 nm, power of 100%. Set the stimulation scanning speed as 0.25 sec/frame.
- Find target LD pairs:
- Click on the FITC or GFP button in the interface of the NIS-element software and switch to the microscope control mode. Using the mercury lamp illumination, select an LD pair with GFP signal enriched at the LD-LD contact site (LDCS) as shown in Figure 5. Adjust the focus plane of the objective to get a clear image of the LD pair. The diameters of the selected donor LDs are preferred to be in the range of 3-6 μm to reduce variation.
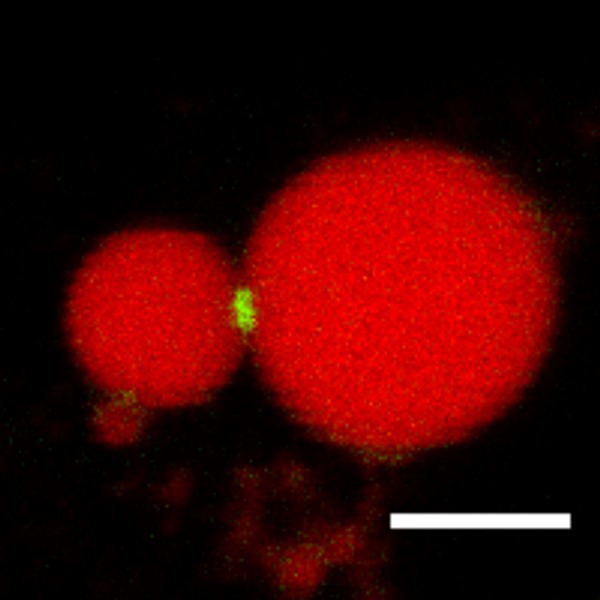
Figure 5. An example image of a suitable LDCS. The image shows the red signal of BODIPY-C12 stained LD pairs and the green signal of Cidec-GFP enriched at the LDCS. Scale bar, 5 μm. - Switch to the computer control mode by clicking on the button “A1”. Click on the button “Laser interlocked” to stimulate the laser. Click on the button “Live” to preview the LD pair. Move the cursor on the interface of the software to the LDCS, right click on the mouse, choose “move the point to the center of the image” to place the LD pair at the center of the vision. Scroll the mouse to adjust the focus plane of the objective once the LD pair went out of focus. Adjust HV in GFP and TRITC channels to achieve the highest fluorescent intensity without overexposure as the photograph taken by detector. Press the button “PFS” on the microscope front panel to lock the imaging plane during the acquisition process.
- Click on the ROI button as shown in Figure 6, select the round circle, draw a circle covering about 70% area of the donor LD at its center, and denote as S1 (stimulation ROI). The reason for photobleaching donor LDs is described in the Notes section below. Next, draw another circle at the center of the donor LD that covers about 70% area of the donor LD; Draw a third circle at the center of the acceptor LD that covers about 70% area of the acceptor LD. The reason for selecting 70% area is dependent on our experience with the optimal HV value and bleaching laser power used. It is possible to adjust the percentage area on case by case basis. In addition, the ROIs selection here was only used to preview the real-time fluorescent intensities of the BODIPY-C12 dyes in LDs and to pre-check the fluorescence recovery data.
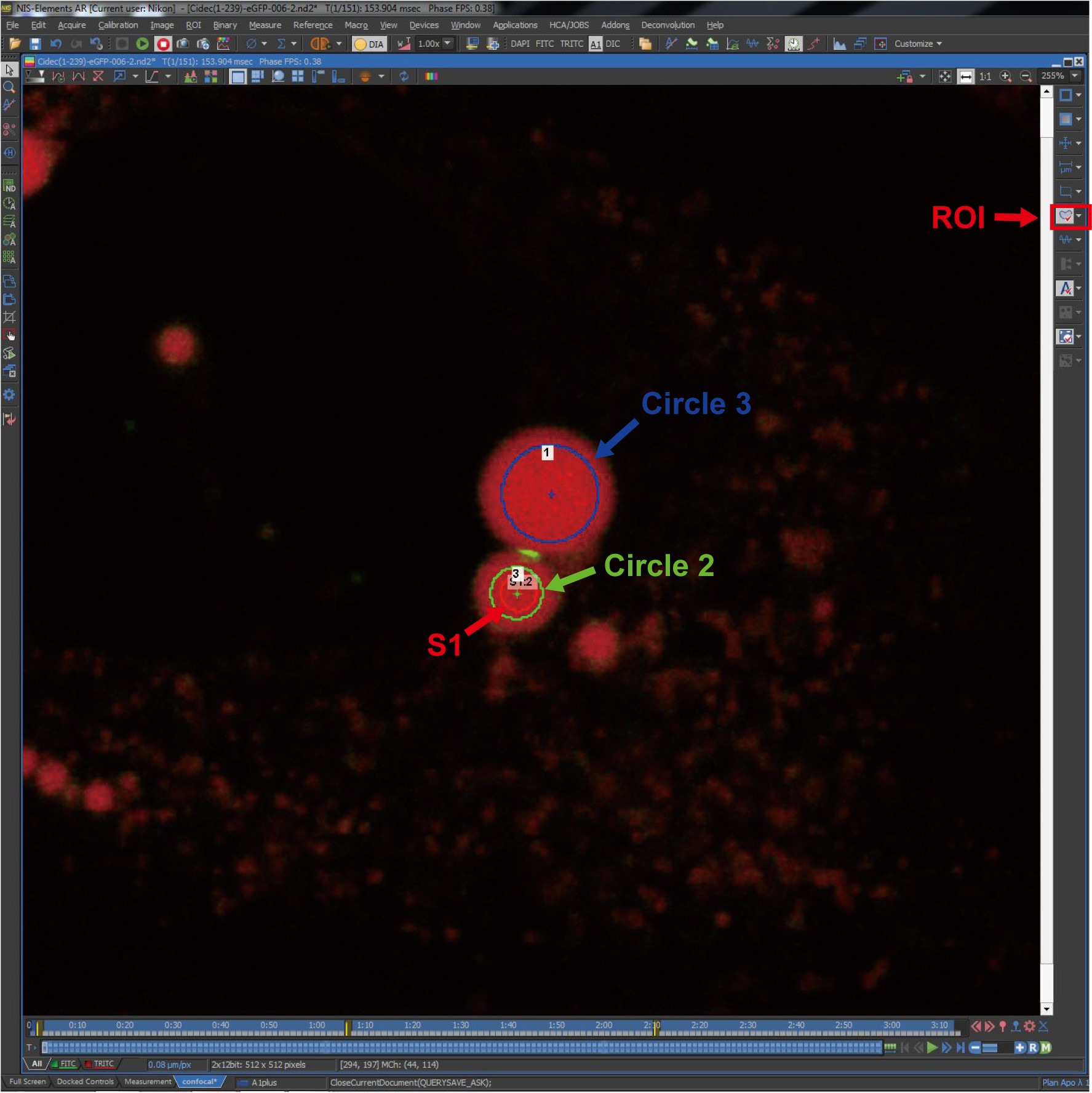
Figure 6. ROI settings. The image shows the ROIs and the schematic diagram of circles on LDs. S1, a photobleaching region on the donor LD of an LD pair; Circle 2, the monitoring ROI on the photobleaching donor LD; Circle 3, the monitoring ROI on the acceptor LD.
- Click on the FITC or GFP button in the interface of the NIS-element software and switch to the microscope control mode. Using the mercury lamp illumination, select an LD pair with GFP signal enriched at the LD-LD contact site (LDCS) as shown in Figure 5. Adjust the focus plane of the objective to get a clear image of the LD pair. The diameters of the selected donor LDs are preferred to be in the range of 3-6 μm to reduce variation.
- Execute the photobleaching process. Before the process, make sure the LD pair is ready on the imaging plane and all the parameters are set correctly. Click on the button “run now” in the ND stimulation interface and allow the program to complete.
- Look for next LD pairs and repeat Steps C5 and C6 to acquire FRAP data for a series of LD pairs.
Data analysis
Below, we will demonstrate two alternative methods to analyze the FRAP image data. Specifically, the first method is a partial manual measurement, using Fuji software. The second method is a totally automatic measurement, using custom-built codes in LabVIEW program. After testing on cases of LD pairs ranged from 3 to 6 μm in diameter, the final FRAP results measured using both the above methods are almost alike. But, in view of the other to-be-measured LD pairs maybe of different sizes and fluorescent intensities, as well as the potential limitation of the custom-built LabVIEW program to handle any condition of FRAP data, we also provide the manual method to process the most difficult cases. For example, for donor LDs with a diameter smaller than 1 μm. However, if possible, we recommend the automatic method.
- Mean of intensity (MOI) and size measurement of BODIPY-C12 stained LDs by manual
- Open the Fiji software.
- Go to “File” → “Import” → “Image Sequence”, to open a series of time-sequential images (8-bit TIF format files, the data in Figure 7 is used as an example).
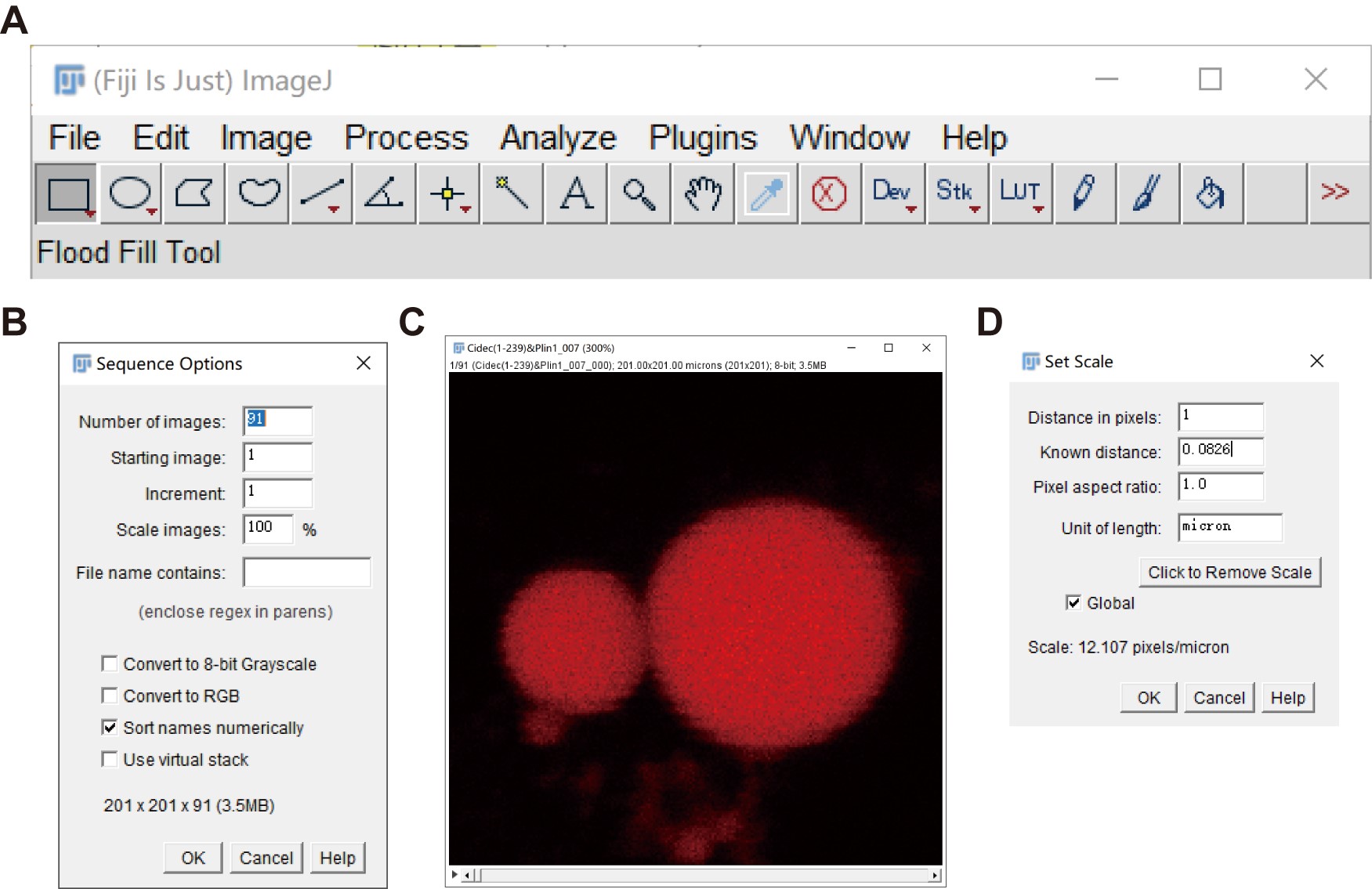
Figure 7. Import a series of 8-bit TIF format images. A. The menu of the Fiji software. B. The sequence options of the imported image sequence. C. A representative set of time-sequential images input. D. The scale of the input images. - To set the scale bar of the images, go to “Analysis” → “Set Scale”. Here the calibrated scale is 12.107 pixels/μm as shown in Figure 7D.
- To obtain a series of “Mean gray values (Mean)” information of a large LD over time.
- Select at least 70% of large LD area with “Oval” tool.
- Go to “Analyze” → “Measure”, to get “Area”, “Mean gray value (Mean)”, “Min gray value (Min)”, and “Max gray value (Max)” information of the large LD.
- Press on the button “►” as shown in the lower right of Figure 7C or “>” on the keyboard to go to the next frame of the sequential images.
- Click the center of the selected circle and move the circle to the center of the large LD in the next frame with drag-and-drop.
- Repeat steps b-d to obtain a series of information of the large LD to the last frame (Figure 8).
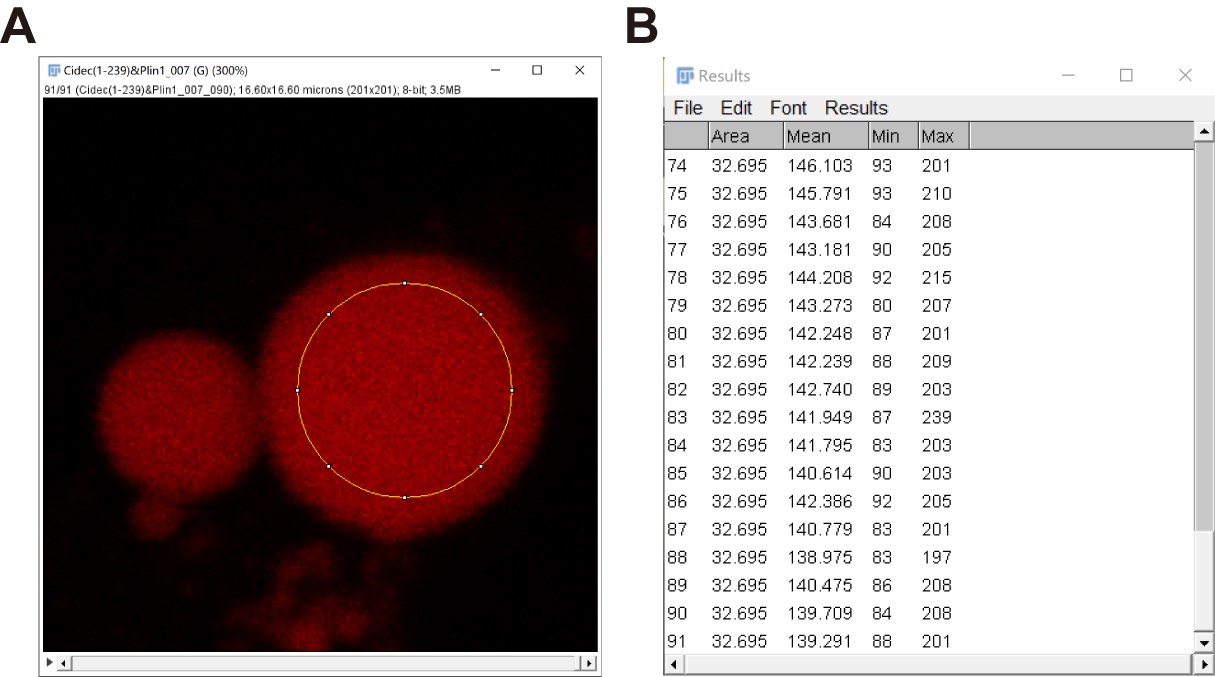
Figure 8. “Mean gray values (Mean)” data of a large LD in a series of time-lapse fluorescent images quantified by Fiji. Here, the total number of frames analyzed in this image is 91 with a time interval of 1.12 s
- Save the data set of the large LD as an Excel file.
- To obtain a series of “Mean” information of a small LD over time.
- Press on the back button “◄” as shown in the lower left of Figure 9A or “<” on the keyboard to return to the first frame of the sequential images (for example press 90 times for the sequential set of 91 images).
- Re-select at least 70% of the small LD area with “Oval” tool.
- Go to “Analyze” → “Measure”, to get “Area”, “Mean gray value (Mean)”, “Min gray value (Min)”, and “Max gray value (Max)” information of the small LD.
- Press on the button “►” or “>” to go to the next frame of the sequential images.
- Click the center of the selected circle and move the circle to the center of the small LD in the next frame with drag-and-drop.
- Repeat steps c-e to get a series of information of the small LD to the last frame (Figure 9).
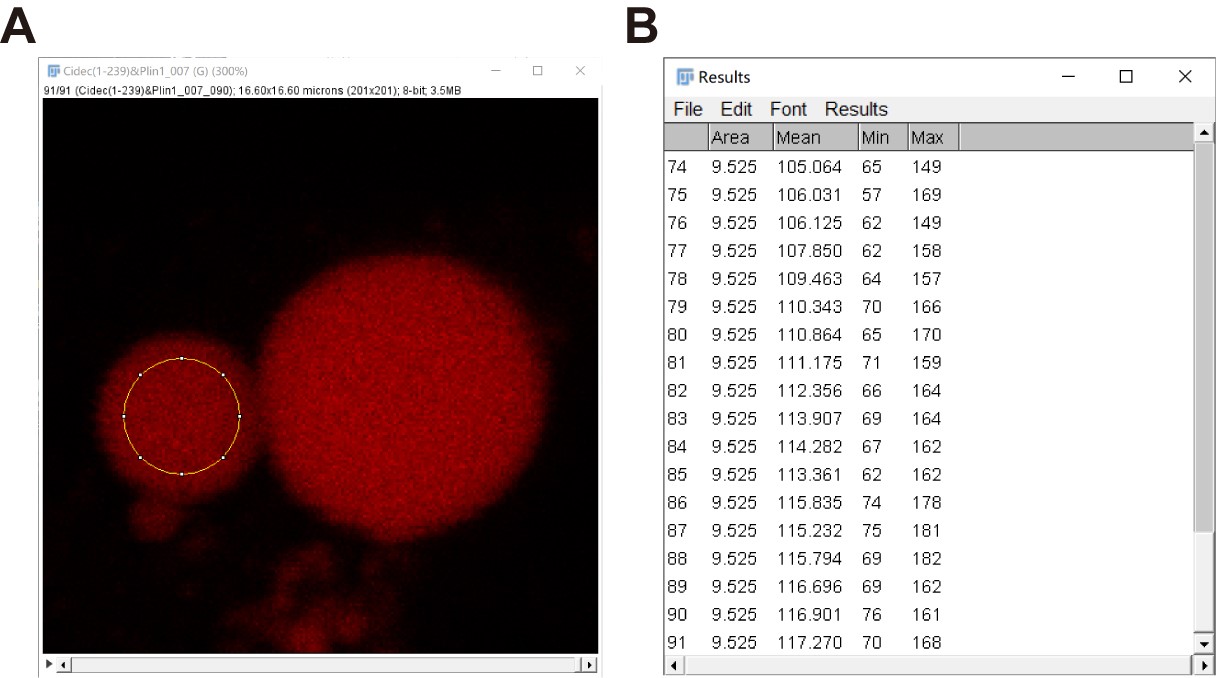
Figure 9. “Mean gray values (Mean)” data of a small LD in a series of time-lapse fluorescent images quantified by Fiji
- Save the data set of the small LD as an Excel file.
- To obtain a series of “Size” information in the region of the large LD over time.
- Press on the back button “◄” or “<” to return to the first frame of the sequential images (for example press 90 times for the sequential set of 91 images).
- Measure the size of the large LD with the “Line” tool.
- Go to “Analyze” → “Measure”, to get “Area”, “Mean gray value (Mean)”, “Min gray value (Min)”, “Max gray value (Max)”, “Angle”, and “Length” information of the large LD. For the following analytical process, only “Length” information is required.
- Press on the button “►” or “>” to go to the next frame of the sequential images.
- Repeat steps b-d to get a series of the size information of the large LD to the last frame (Figure 10).
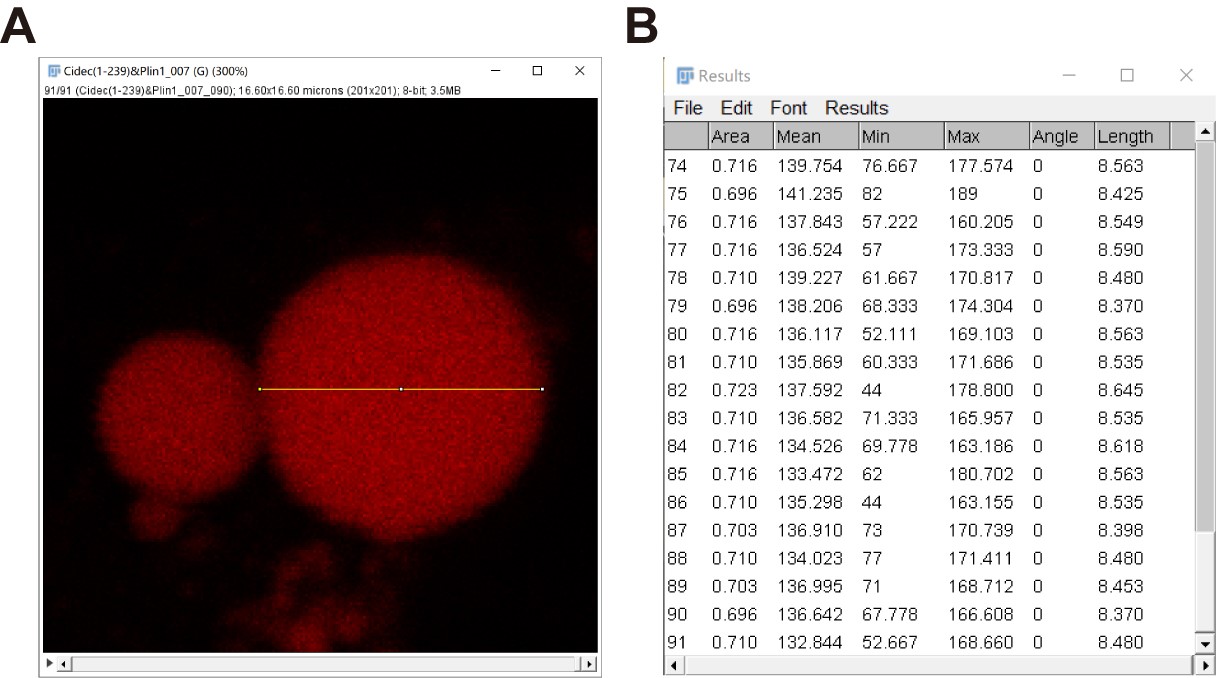
Figure 10. “Size” data of a large LD in a series of time-lapse fluorescent images quantified by Fiji
- Save the data set of the large LD as an Excel file.
- To obtain a series of “Size” information of the small LD over time.
- Press on the back button “◄” or “<” to return to the first frame of the sequential images (for example press 90 times for the sequential set of 91 images).
- Measure the size of the small LD with the “Line” tool to obtain a series of the size information of the small LD until the last frame by repeating the same steps 8b-8e for the large LD measurement (Figure 11).
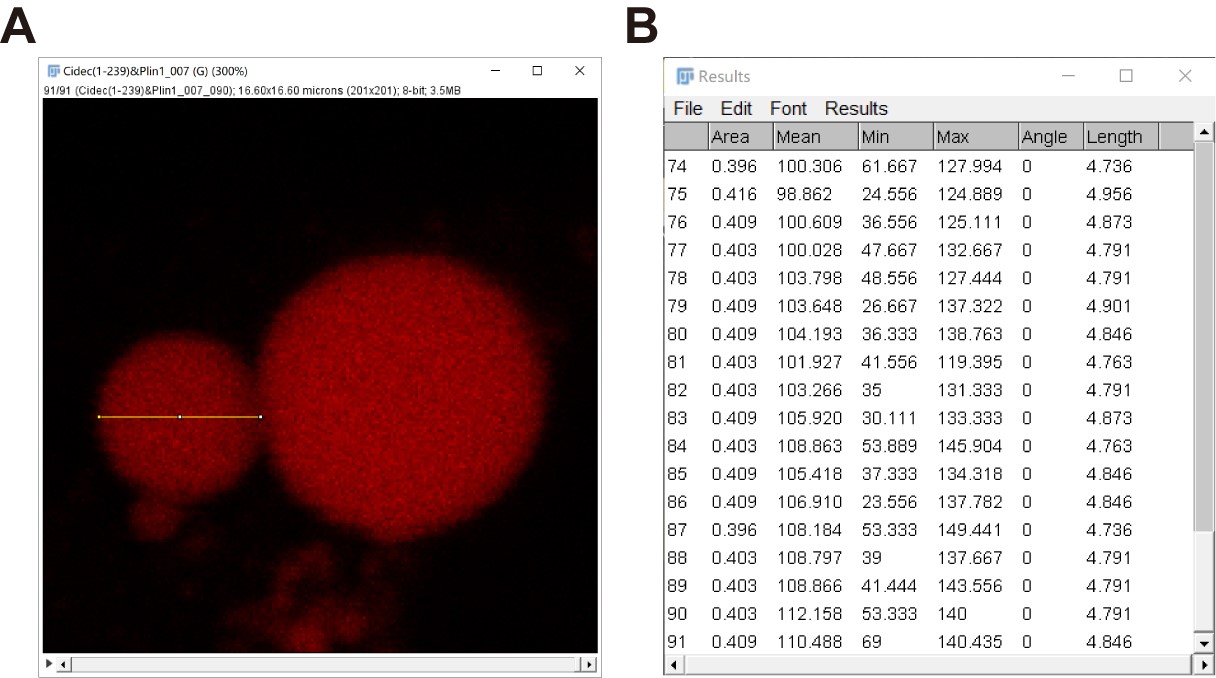
Figure 11. “Size” data of a small LD in a series of time-lapse fluorescent images quantified by Fiji
- Save the data set of the small LD as an Excel file.
- To get the plot of time-sequential MOIs over time, only “Mean” value from the intensity measurement and the “Length” values from the size measurement are required. Next, reformat the data sets as a new Excel file as shown in Figure 12. The data is now ready for the subsequent analysis in Step C of Data analysis.
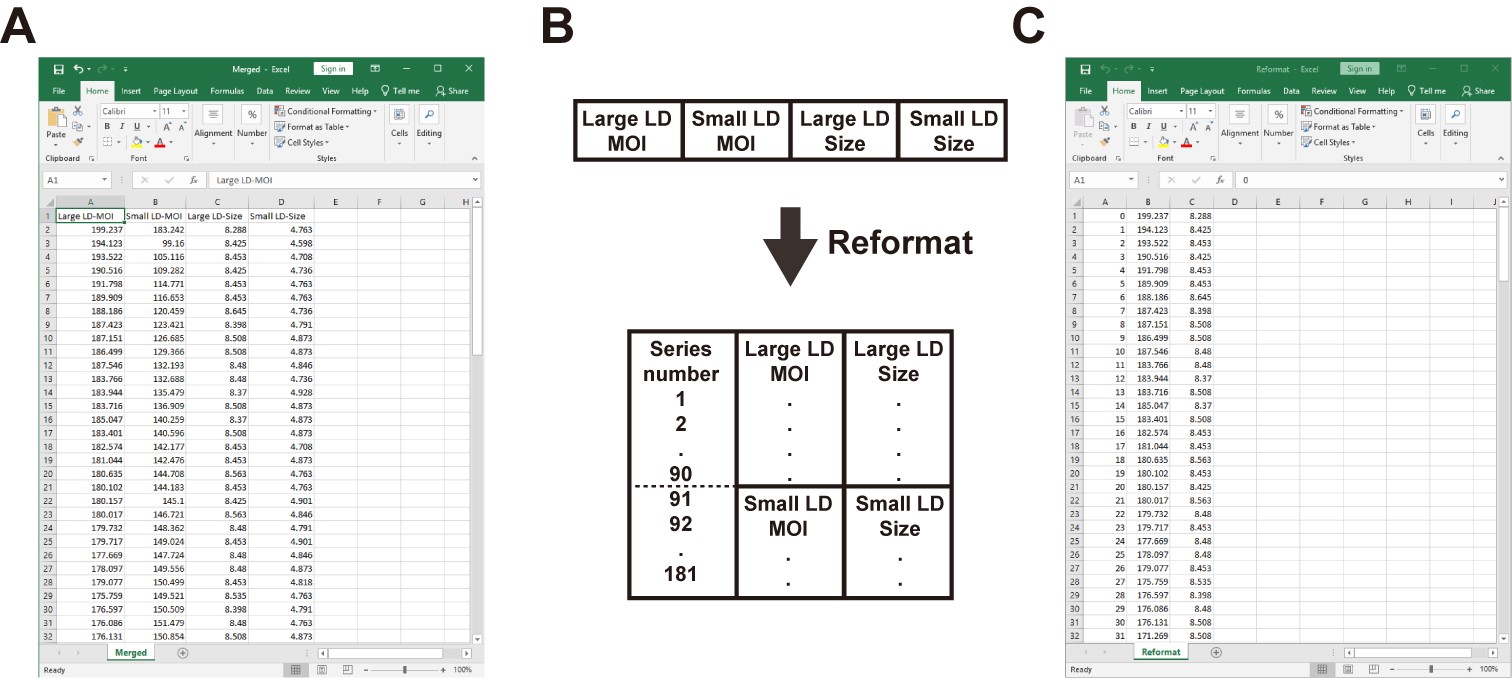
Figure 12. Merge and reformat the data sets in a new Excel file - Plot the sequential MOIs of the two regions-of-interest of the LD pair over time in the Excel software (Figure 13).
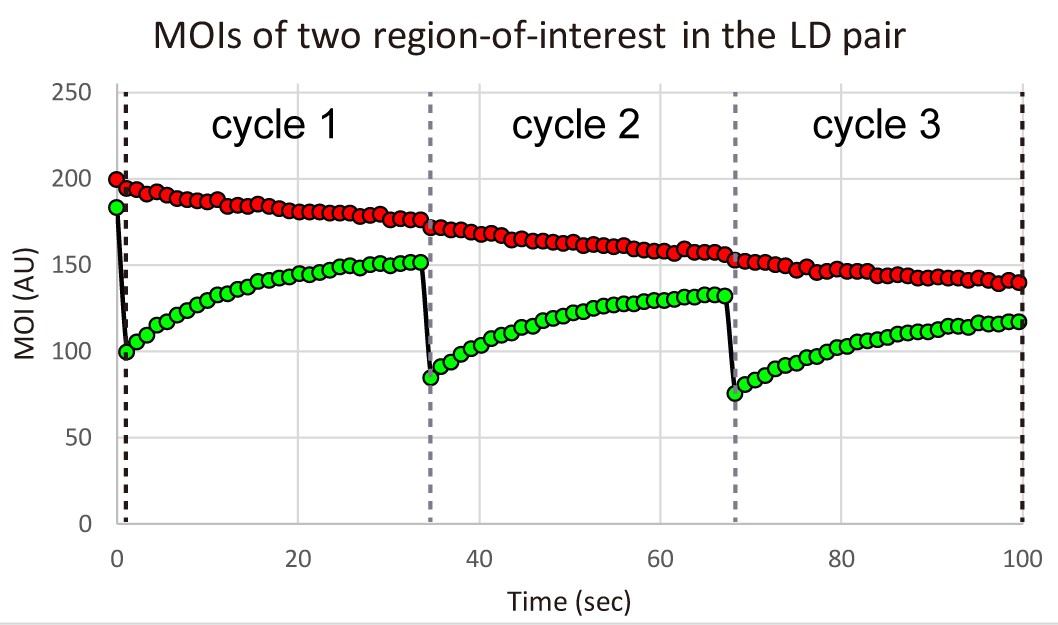
Figure 13. Plot of sequential MOIs over time
- MOI and size measurements of BODIPY-C12 stained LDs using an automatic identification program
- Open the LabVIEW software (here using the version of LabVIEW 8.5 with a plug-in installation of NI Vision 8.6 module).
- Double-click the LabVIEW sub-llb file “1_Exchange rate assay.llb” to open the main module “Main program.vi” as shown in Figure 14. The interface of the sub-VI program is shown in Figure 15.
Figure 14. The dialog box of LLB manager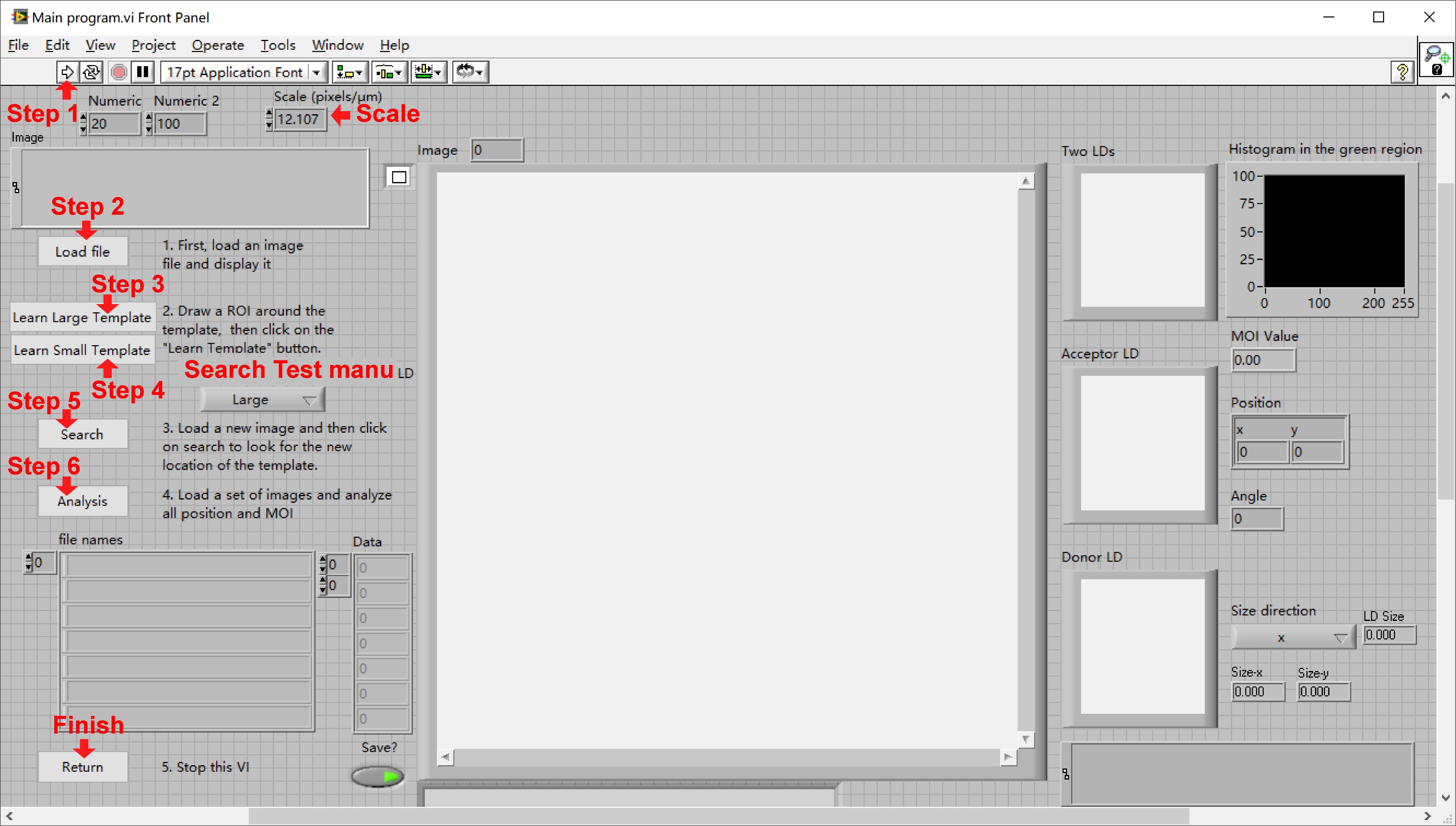
Figure 15. The interface of the main module “Main program.vi”. Red asterisks indicate a set of regular steps in a standard procedure. Follow the steps to obtain the series of information of each LD pair over time. - To obtain a series of “Mean” information of the large and small LDs over time.
- Set the scale of input images marked by “Scale” in Figure 15 (here using the value of 12.107 pixels/μm).
- Run the sub-VI program by pressing on the button marked by “Step 1” in Figure 15 or alternatively press on a combination keyboard button “Ctrl + R.”
- Press on “Load file” marked by “Step 2” in Figure 15 to open a series of time-sequential images (8-bit TIF format files, here using the data of Figure 16A as example).
- Select the whole large LD area with a default “Square” tool by drag-and-drop directly on the image as shown in Figure 16B and press on “Learn Large Template” marked by “Step 3” in Figure 15 to record the template image of the large LD.
- Repeat step d for the small LD: select the whole small LD area with “Square” tool as shown in Figure 16C and press on “Learn Small Template” marked by “Step 4” in Figure 15 to record the template image of the small LD.
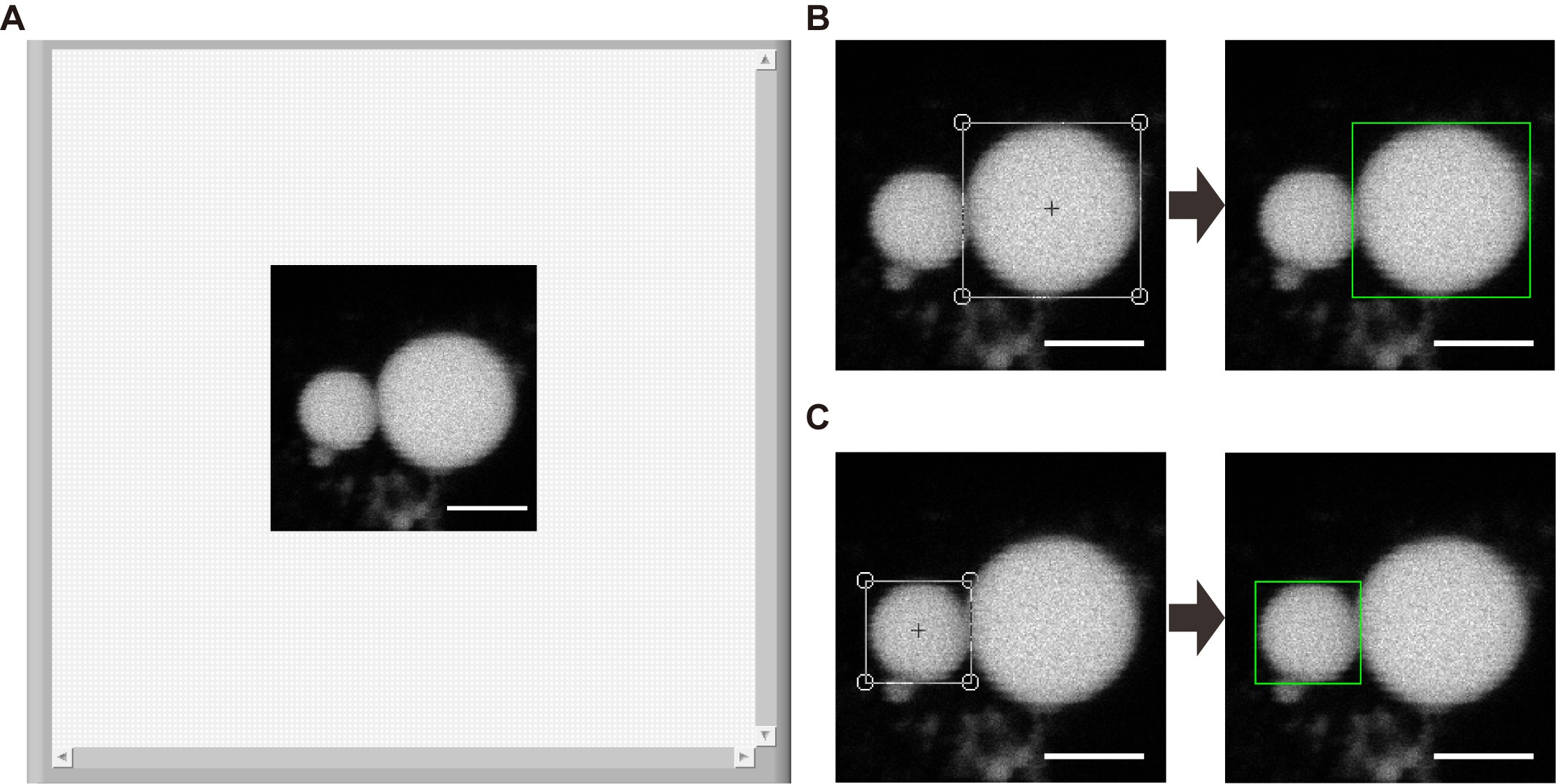
Figure 16. Loading an image set and recording the template images of large and small LDs. A. A set of time-sequential images of an LD pair were loaded into the computer memory and showed accordingly on the interface of the main program. B. A template image of a large LD was selected with “Square” tool and recorded into the computer memory as a reference for the subsequent identification of the large LD in the next frames. C. A template image of a small LD was selected with “Square” tool and recorded into the computer memory as a reference for the subsequent identification of the small LD in the next frames. Scale bars, 5 μm. - Select “Large” or “Small” in the Search Test menu and press on the button “Search” marked by “Step 5” in Figure 15 to confirm the accurate identification of the selected large or small LDs (Here showing Figure 17 as an example).

Figure 17. Confirmation of the identified LD size. Select “Large” (A) or “Small” (C) to check that the identification of the selected large (B) or small (D) LD is accurate. Scale bars, 5 μm. - Select “Large” in the Search Test menu and press on the button “Analysis” marked by “Step 6” in Figure 15 to get a series of “Frame”, “Mean gray value (Mean)”, “Min gray value (Min)”, and “Size” information of the large LD over time automatically.
- Select “Small” in the Search Test menu and press on the button “Analysis” again to get a series of “Frame”, “Mean gray value (Mean)”, “Min gray value (Min)”, and “Size” information of the small LD over time automatically.
- Press on the button “Load file” marked by “Step 2” in Figure 15 to input the next set of time-sequential images and repeat steps d-h to get the series of information of the next LD pairs over time. Or press on the button “Return” marked by “Finish” in Figure 15 after the completion of the MOI measurement of the LD pairs.
Note: After pressing either the “Load file” or “Return” button, the series of Mean and Size information of the larger and smaller LDs will be saved as an Excel file automatically. The generated Excel file is in ready format for the subsequent analysis in Step C of Data analysis.
- Check the data sets of the LD pairs saved previously as an Excel file (here using the two sets of data as example).
- Plot the sequential MOIs of the two regions-of-interest in the individual LD pairs over time in the Excel software (Figure 18).
- Create a folder name “MOI&Size” and copy the Excel data set into the folder for the subsequent exchange rate analysis. Transfer the folder with these data into the same folder where there are LabVIEW sub-VIs “2_Check a fitting region.vi” and “3_Calculation of exchange rates.vi” files.
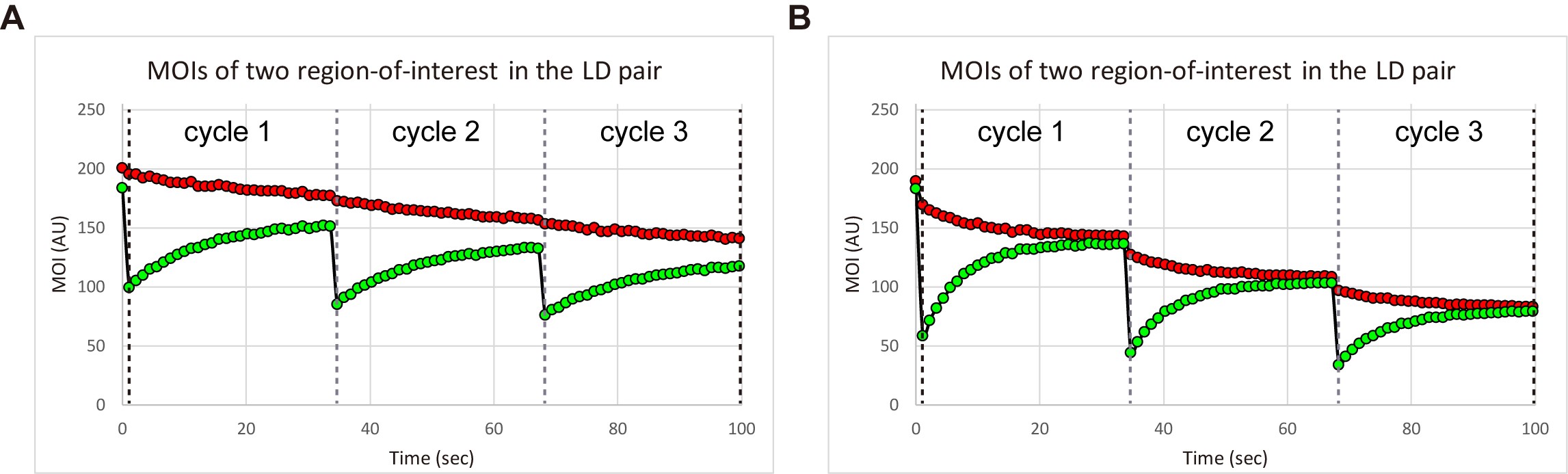
Figure 18. Plot of sequential MOIs over time. Data shown in (A) are the same data shown in Figure 13. B. In contrast, the MOIs were obtained using another set of time-sequential images as mentioned in Step B3i of Data analysis.
- Pre-estimation of the exchange rate underlying linear criteria
After the imaging process and obtaining “MOI” and “Length” information generated in Step A or B of Data analysis, the Excel files are now ready for the following analysis. Here, we show the pre-estimation process of the exchange rate.- Open the LabVIEW software.
- Open the LabVIEW sub-VI “2_Check a fitting region.vi” file. The interface of the sub-VI program is shown in Figure 19.
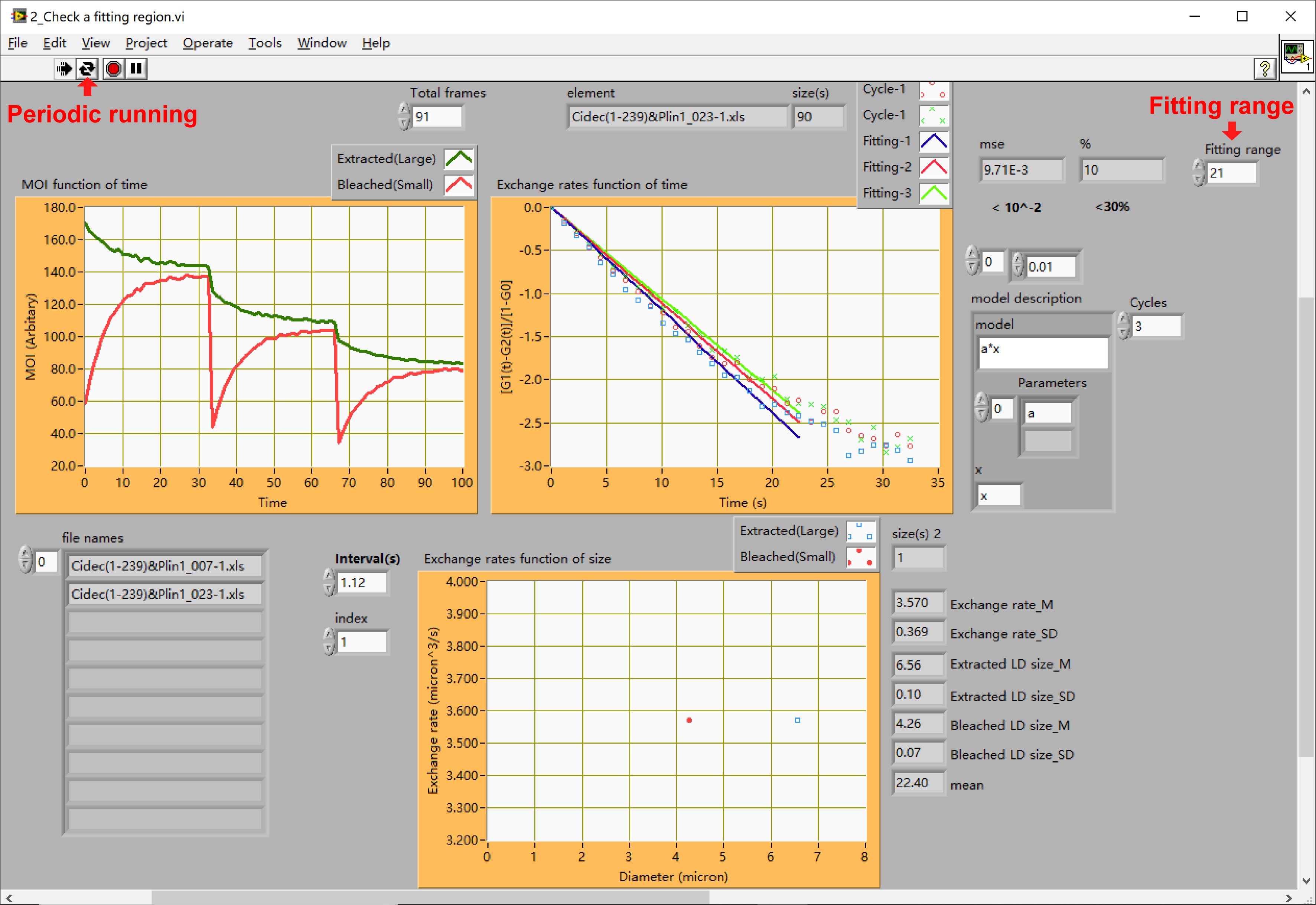
Figure 19. The interface of sub-VI “2_Check a fitting region.vi”. Here using the data from Figure 18B as an example. - Obtain the optimal fitting range of an LD pair’s exchange rate.
- Run the sub-VI program by pressing on the button marked by “Periodic running” in Figure 19.
- Adjust and choose the maximum value of the fitting range marked by “Fitting range” in Figure 19 to ensure the linearity (MSE) and repetitiveness (%) of the adjusted fitted lines satisfy the experienced optimal values (MSE < 0.01 and % < 30%). We define the two upper limit values as linear criteria.
- Record each optimal fitting range for each set of LD pair measured.
- Calculation of the exchange rates between individual fused LD pairs
- Open the LabVIEW sub-VI “3_Calculation of exchange rates.vi” file. The interface of the sub-VI program is shown in Figure 20.
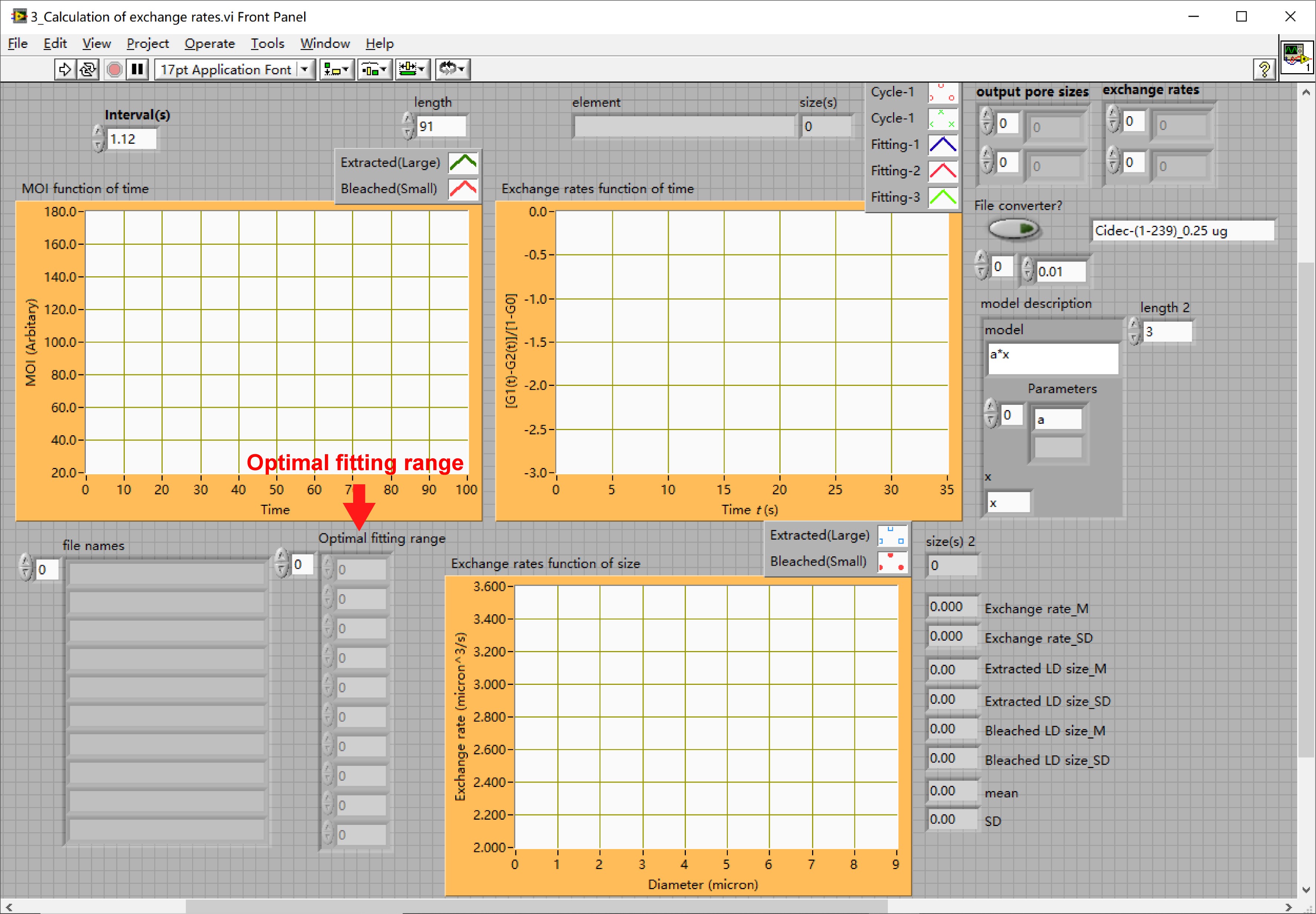
Figure 20. The interface of sub-VI “3_Calculation of exchange rates.vi” - Input all the optimal fitting ranges in the table marked by “Optimal fitting range” as shown in Figure 21A.
- Run the sub-VI program by pressing on “Ctrl + R” to obtain the mean ± SD values of the exchange rates. Exchange rates were plotted against large or small LD sizes as shown in Figure 21B.
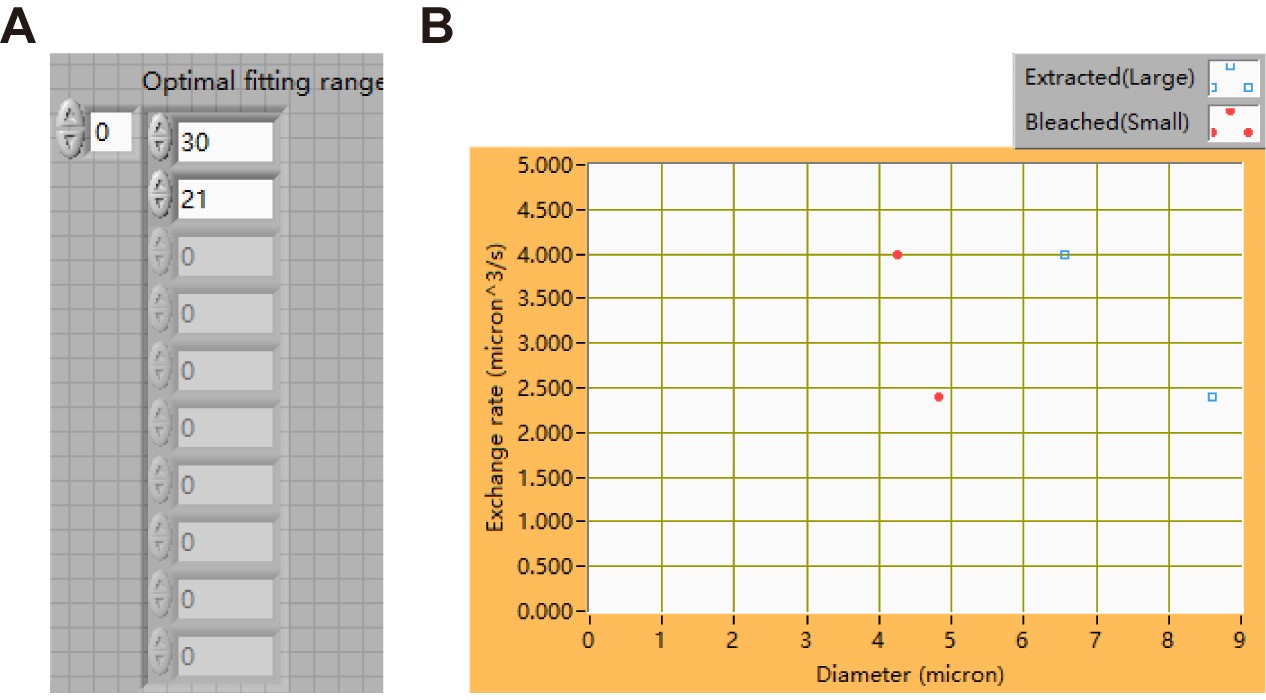
Figure 21. Results of running the sub-VI “3_Calculation of exchange rates.vi”. A. Table of optimal fitting range. B. A plot showing the exchange rates against the sizes of large and small LDs.
- Open the LabVIEW sub-VI “3_Calculation of exchange rates.vi” file. The interface of the sub-VI program is shown in Figure 20.
Notes
- The photobleaching process has to be performed on the smaller (namely donor) LD of an LD pair three times for statistical analyses as described in our original publication (Wang et al., 2018). Briefly, because there are more fluorescent molecules in the larger LDs, photobleaching these large LDs more than twice will result in the rapid decline of the fluorescent intensities in the LD pair and pose difficulty in the fluorescence recovery detection.
- The most critical step in this protocol is to obtain the optimal linearity (mse) and repetitiveness (%) of three fitted lines as described in “Procedure C”. If either the evaluated linearity or the repetitiveness of the fitted lines in the three cycles measured is out of the linear criteria, the data set should be discarded as these indicate that the exchange rate event might be unstable during the acquisition interval. These unstable exchange rates possibly resulted from the physical size change of the LD pair, the height of imaging plane, deformation of LD fusion complex, and serious photobleaching et al. We recommend 50 sets or more of the lipid-exchange rates to be measured. In general, 30-40 events of lipid-exchange is sufficient to satisfy the criteria if acquired under an optimal environmental control condition.
Recipes
- Phosphate-buffered saline (PBS)
- Mix 1.37 M NaCl, 25 mM KCl, 8 mM Na2HPO4, 14.7 mM KH2PO4, 9 mM CaCl2, and 5 mM MgCl2 in 900 ml deionized H2O
- Adjust to a final pH of 7.4
- Add deionized H2O to 1,000 ml
- Autoclave to store (This is 10x PBS stock)
- Dilute PBS stock with deionized H2O to 1x PBS
Acknowledgments
We thank the members of P. Li Laboratory at Tsinghua University for their helpful discussions and Jinyu Wang at SLSTU-Nikon Biological Imaging Center for imaging support. This work was supported by grants from National Basic Research Program Grants 2018YFA0506900 (to P.L.) and National Natural Science Foundation of China Grants 91857103 (to F.J.C), 31430040, 31690103, and 31621063 (to P.L.). This protocol was adapted from our previous publication in the Journal of Biological Chemistry (Wang et al., 2018).
Competing interests
The authors declare no conflicts of interest or competing interests.
References
- Choudhary, V., Ojha, N., Golden, A. and Prinz, W. A. (2015). A conserved family of proteins facilitates nascent lipid droplet budding from the ER. J Cell Biol 211(2): 261-271.
- Farese, R. V., Jr. and Walther, T. C. (2009). Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 139(5): 855-860.
- Fujimoto, Y., Itabe, H., Kinoshita, T., Homma, K. J., Onoduka, J., Mori, M., Yamaguchi, S., Makita, M., Higashi, Y., Yamashita, A. and Takano, T. (2007). Involvement of ACSL in local synthesis of neutral lipids in cytoplasmic lipid droplets in human hepatocyte HuH7. J Lipid Res 48(6): 1280-1292.
- Gao, G., Chen, F. J., Zhou, L., Su, L., Xu, D., Xu, L. and Li, P. (2017). Control of lipid droplet fusion and growth by CIDE family proteins. Biochim Biophys Acta Mol Cell Biol Lipids 1862(10 Pt B): 1197-1204.
- Gluchowski, N. L., Becuwe, M., Walther, T. C. and Farese, R. V., Jr. (2017). Lipid droplets and liver disease: from basic biology to clinical implications. Nat Rev Gastroenterol Hepatol 14(6): 343-355.
- Gong, J., Sun, Z., Wu, L., Xu, W., Schieber, N., Xu, D., Shui, G., Yang, H., Parton, R. G. and Li, P. (2011). Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. J Cell Biol 195(6): 953-963.
- Gross, D. A., Zhan, C. and Silver, D. L. (2011). Direct binding of triglyceride to fat storage-inducing transmembrane proteins 1 and 2 is important for lipid droplet formation. Proc Natl Acad Sci U S A 108(49): 19581-19586.
- Krahmer, N., Farese, R. V., Jr. and Walther, T. C. (2013). Balancing the fat: lipid droplets and human disease. EMBO Mol Med 5(7): 973-983.
- Rosen, E.D. and Spiegelman, B.M. (2014). What we talk about when we talk about fat. Cell 156(1-2): 20-44.
- Sun, Z., Gong, J., Wu, H., Xu, W., Wu, L., Xu, D., Gao, J., Wu, J. W., Yang, H., Yang, M. and Li, P. (2013a). Perilipin1 promotes unilocular lipid droplet formation through the activation of Fsp27 in adipocytes. Nat Commun 4: 1594.
- Sun, Z., Gong, J., Wu, L. and Li, P. (2013b). Imaging lipid droplet fusion and growth. Methods Cell Biol 116: 253-268.
- Wang, J., Yan, C., Xu, C., Chua, B. T., Li, P. and Chen, F. J. (2018). Polybasic RKKR motif in the linker region of lipid droplet (LD)-associated protein CIDEC inhibits LD fusion activity by interacting with acidic phospholipids. J Biol Chem 293(50): 19330-19343.
- Wilfling, F., Wang, H., Haas, J. T., Krahmer, N., Gould, T. J., Uchida, A., Cheng, J. X., Graham, M., Christiano, R., Frohlich, F., Liu, X., Buhman, K. K., Coleman, R. A., Bewersdorf, J., Farese, R. V., Jr. and Walther, T. C. (2013). Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev Cell 24(4): 384-399.
- Wu, L., Xu, D., Zhou, L., Xie, B., Yu, L., Yang, H., Huang, L., Ye, J., Deng, H., Yuan, Y. A., Chen, S. and Li, P. (2014). Rab8a-AS160-MSS4 regulatory circuit controls lipid droplet fusion and growth. Dev Cell 30(4): 378-393.
- Xu, D., Li, Y., Wu, L., Li, Y., Zhao, D., Yu, J., Huang, T., Ferguson, C., Parton, R. G., Yang, H. and Li, P. (2018). Rab18 promotes lipid droplet (LD) growth by tethering the ER to LDs through SNARE and NRZ interactions. J Cell Biol 217(3): 975-995.
- Yang, H., Galea, A., Sytnyk, V. and Crossley, M. (2012). Controlling the size of lipid droplets: lipid and protein factors. Curr Opin Cell Biol 24(4): 509-516.
- Somwar, R., Roberts Jr., C. T. and Varlamov, O. (2011). Live-cell imaging demonstrates rapid cargo exchange between lipid droplets in adipocytes. FEBS Lett 585(12):1946-1950.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Wang, J., Chua, B. T., Li, P. and Chen, F. (2019). Lipid-exchange Rate Assay for Lipid Droplet Fusion in Live Cells. Bio-protocol 9(14): e3309. DOI: 10.21769/BioProtoc.3309.
- Wang, J., Yan, C., Xu, C., Chua, B. T., Li, P. and Chen, F. J. (2018). Polybasic RKKR motif in the linker region of lipid droplet (LD)-associated protein CIDEC inhibits LD fusion activity by interacting with acidic phospholipids. J Biol Chem 293(50): 19330-19343.
Category
Cell Biology > Cell imaging > Fluorescence
Developmental Biology > Cell signaling > Energy homeostasis
Cell Biology > Cell metabolism > Lipid
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.