- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Protocol to Map the Spatial Proteome Using HyperLOPIT in Saccharomyces cerevisiae
Published: Vol 9, Iss 14, Jul 20, 2019 DOI: 10.21769/BioProtoc.3303 Views: 7232
Reviewed by: Alessandro DidonnaJulie WeidnerAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2014
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The correct subcellular localization of proteins is vital for cellular function and the study of this process at the systems level will therefore enrich our understanding of the roles of proteins within the cell. Multiple methods are available for the study of protein subcellular localization, including fluorescence microscopy, organelle cataloging, proximity labeling methods, and whole-cell protein correlation profiling methods. We provide here a protocol for the systems-level study of the subcellular localization of the yeast proteome, using a version of hyperplexed Localization of Organelle Proteins by Isotope Tagging (hyperLOPIT) that has been optimized for use with Saccharomyces cerevisiae. The entire protocol encompasses cell culture, cell lysis by nitrogen cavitation, subcellular fractionation, monitoring of the fractionation using Western blotting, labeling of samples with TMT isobaric tags and mass spectrometric analysis. Also included is a brief explanation of downstream processing of the mass spectrometry data to produce a map of the spatial proteome. If required, the nitrogen cavitation lysis and Western blotting portions of the protocol may be performed independently of the mass spectrometry analysis. The protocol in its entirety, however, enables the unbiased, systems-level and high-resolution analysis of the localizations of thousands of proteins in parallel within a single experiment.
Keywords: Saccharomyces cerevisiaeBackground
Knowledge of the subcellular localization of a protein is crucial in defining its functions within the cell and regulation of the spatial organization of the proteome is essential for cellular homeostasis. Subcellular protein localization allows proteins to act as members of higher-order complexes with binding partners such as proteins, metabolites and co-factors, and contribute to the functions of organelles. In some cases, the sequestration of enzymes within specific subcellular compartments is critical to ensure that intermediates are not exchanged between different metabolic pathways or that potentially detrimental reactions (e.g., proteolysis) are confined to a ‘safe’ space. Protein localization can be influenced by post-translational modification, such as the phosphorylation of proteins within signaling cascades that ultimately allow proteins such as transcription factors (TFs) to localize within the nucleus to exercise their gene regulatory functions, or allowing other proteins to traverse the nuclear membrane and regulate nuclear-localized transcription factors. One example is the case of the osmostress response pathway in yeast, where a signaling cascade is activated that results in the phosphorylation of the protein Hog1p, whereupon it can translocate to the nucleus and phosphorylate the TF Hot1p which, in turn, can recruit RNA polymerase II to the promoters of responsive genes (Alepuz et al., 2003).
A growing body of evidence suggests that differential protein localization, even within the same cell type, may allow the protein to fulfil different roles depending on its respective localization within a cell, sometimes referred to as “moonlighting” (Gancedo et al., 2016). In Saccharomyces cerevisiae, examples of such proteins include Gcn4p, which is the general transcription factor that controls amino acid biosynthesis (Hope and Struhl, 1986) and also acts as a ribonuclease (Nikolaev et al., 2010); as well as Aco1p, which has roles in mitochondrial DNA stability in addition to its more widely known function as tricarboxylic acid cycle enzyme (Chen et al., 2005). Isoforms of the same protein have further been observed with different localizations in the same cell type, suggesting that this differential localization may offer another level of regulation with regard to protein function. An example of this is the case of a DNA methyltransferase (Dnmt1) in murine stem cells, in which one isoform localized to chromatin and the other isoform is found in an alternative nuclear location (Christoforou et al., 2016).
It is clear that there are many levels of complexity regarding protein subcellular localization. In order to fully understand the in vivo functions of proteins, it is vital to ascertain where, within the cell, they are located. Several methods exist that can be applied to approach this challenge, including fluorescence microscopy imaging, organelle proteomics, proximity biotinylation methods, and protein correlation profiling methods; all of which have been applied in Saccharomyces cerevisiae in different contexts to determine the subcellular localization of yeast proteins. Whole-cell, high-throughput studies based on fluorescence microscopy have been performed using GFP- or other fluorescence protein-tagged protein libraries (Huh et al., 2003; Tkach et al., 2012; Breker et al., 2013; Denervaud et al., 2013; Chong et al., 2015; Weill et al., 2018; Yofe et al., 2016). Although powerful, these methods have documented problems in that the fluorescence signal of a particular protein of interest may not exceed the background level of cellular autofluorescence, particularly in yeast, and therefore it may not be possible to determine the location of such proteins. Additionally, some proteins may require their unmodified C- or N-terminus for correct localization (Weill et al., 2018; Yofe et al., 2016). As such, performing protein localization studies where proteins are tagged with a fluorescent protein either N- or C-terminally to the ORF in question may result in a given protein being reported with different localizations within the same biological system, depending on which strategy was used (Stadler et al., 2013).
Organelle enrichment strategies have also been employed to define organelle proteomes, thereby contributing to our knowledge on protein localization. These strategies often exploit the characteristic buoyant densities of organelles in their enrichment. The relative ease with which mitochondria can be highly enriched, for example, has been exploited in many studies that have attempted to define the mitochondrial proteome, with varying levels of specificity, in S. cerevisiae. This includes some studies that used more crude approaches to attempt to define the mitochondrial proteome (Sickmann et al., 2003; Reinders et al., 2006) or specific sub-mitochondrial proteomes (Zahedi et al., 2006; Vogtle et al., 2012) in some cases without control for other organelle proteins that were co-enriched along with the mitochondrial proteins. Improvements have been made in recent studies that mapped the entirety of the mitochondrial sub-organellar proteome using more complex fractionation and proteome quantitation methods (Morgenstern et al., 2017; Vogtle et al., 2017). For organelles of more heterogeneous density, however, the difficulty of such studies can be compounded by the presence of contaminating membranes from other organelles that co-enrich with the organelle of interest (Wiederhold et al., 2010). In focusing on a specific organelle, such methods fail to report on proteins that may be present at multiple locations in vivo. Further, there is no control for the proportion of an organelle that is lost upon enrichment.
Recently, new proximity labeling methods based on promiscuous biotinylation have shown promise in determining protein subcellular localization in organisms other than yeast. This includes the more detailed study of sub-organellar proteomes (Rhee et al., 2013; Hung et al., 2014), including cytoplasmic faces of organelles (Hung et al., 2017). The BirA-based BioID method (Roux et al., 2012) has been applied in S. cerevisiae to define the subcellular proteins that interact with a ribosomal scaffold protein (Opitz et al., 2017) and the APEX2 method (Lam et al., 2015) has been demonstrated to traverse the cell wall in proof-of-principle experiments in both S. cerevisiae and the fission yeast Schizosaccharomyces pombe (Hwang and Espenshade, 2016). Methods such as these focus only on the proximal interactors of a protein of interest, that are currently in the range of tens of nanometres (Rhee et al., 2013; Kim et al., 2014) and do not provide a whole-cell picture of the spatial organization of the proteome.
To overcome some of the inherent limitations of the methods described above, it is imperative to look at protein subcellular localization in the context of the whole cell. This has been commonly studied using whole-cell protein correlation profiling methods. Such methods are predicated on the observation that, when cell lysates are fractionated by some means, such as differential or density gradient centrifugation, proteins that localize to the same organelle will exhibit similar distributions when the protein distributions across the entire fractionation scheme are monitored (de Duve, 1971). This was originally monitored using enzyme assays that are representative of the organelles in question, but now employs quantitative mass spectrometry methods. Several such methods have been published, which offer differing levels of proteome coverage and subcellular resolution, employing different fractionation and mass spectrometry quantitation methods (Christoforou et al., 2016; Itzhak et al., 2016; Jean Beltran et al., 2016; Mulvey et al., 2017; Jadot et al., 2017; Geladaki et al., 2019). The hyperplexed Localization of Organelle Proteins by Isotope Tagging (hyperLOPIT) method uses density gradient centrifugation for subcellular fractionation, followed by multiplexed proteome quantitation using 10-plex TMT isobaric tags (Thompson et al., 2003) and SPS-MS3 quantitation on an Orbitrap Fusion Lumos mass spectrometer. The exquisite subcellular resolution offered by the density gradient coupled with multiplexed quantitation, followed by sophisticated machine learning analysis methods means that hyperLOPIT allows unbiased protein localization determination of thousands of proteins in parallel in a single experiment. Indeed the method has afforded the highest resolution spatial map of any whole-cell correlation profile-based spatial proteome study to date (Thul et al., 2017; Gatto et al., 2019).
We recently published a yeast spatial proteome mapping study in which we mapped the proteome in high throughput using hyperLOPIT, under nitrogen-sufficient conditions (Nightingale et al., 2019). In our study, we produced a dataset that provided localization data for 2,846 proteins. Only 936 of these proteins were found to map to a unique subcellular location, a number which corresponded to 32% of our observable spatial proteome. This suggests, in common with previous studies (Christoforou et al., 2016; Thul et al., 2017), that a large proportion of the proteome is dynamic or resides at multiple locations.
We provide here a protocol that describes how to perform nitrogen cavitation lysis (Hunter and Commerford, 1961; Simpson, 2010; Wang et al., 2014) subcellular fractionation, and a version of the hyperLOPIT protocol that has been modified for use with the yeast S. cerevisiae (see Figure 1 for an overview of the protocol). We further draw the reader’s attention to the fact that the subcellular fractionation and Western blotting portion of this protocol may be performed as a standalone experiment without TMT labeling. Further to this, in cases where a mass spectrometer capable of resolution of TMT 10- or 11-plex tags (such as the Thermo Fusion series) is not available, isobaric tags with lower multiplexing capability may instead be used. This may be achieved using iTRAQ (Ross et al., 2004) 4- or 8-plex, or TMT 6-plex, but, importantly, will result in reduced overall subcellular resolution relative to TMT 10- or 11-plex, as less of the subcellular fractionation gradient will be sampled in the experiment. HyperLOPIT and its predecessor, LOPIT, have been employed to great effect to map the subcellular proteomes of multiple different species and under numerous conditions (Sadowski et al., 2006; Nikolovski et al., 2012; Groen et al., 2014; Christoforou et al., 2016; Thul et al., 2017). We envisage that this protocol will therefore be applicable and modifiable to study the spatial proteomes of other yeasts, including the industrial yeast Komagataella phaffii (syn. Pichia pastoris) and pathogenic yeasts such as Candida albicans.
We recently published a protocol in Methods in Molecular Biology (Nightingale et al., 2018) that describes how to perform hyperLOPIT in S. cerevisiae. The lysis and subcellular fractionation portions of this protocol are substantially updated from a previous protocol by Wang and colleagues (2014). Here we provide a protocol with updated mass spectrometry data processing parameters using Proteome Discoverer 2.1 (Thermo Fisher Scientific, www.thermofisher.com), and with more focus on post-mass spectrometry informatics analysis. For a more in-depth and detailed informatics protocol than is presented here, we direct the reader to the publication of Breckels and colleagues (Breckels et al., 2016b). The subcellular fractionation portion of the hyperLOPIT protocol is technically challenging, requiring an unbroken stretch of experimental work lasting from cell harvest to gradient fraction collection. We strongly recommend that experimental work not be paused until the gradient fractions have been collected.
Figure 1. Overview of the hyperLOPIT method for Saccharomyces cerevisiae. Also included in parentheses are approximate timescales for each step, and potential stopping points.
Materials and Reagents
- 96-well plates (Thermo Fisher Scientific, catalog number: 167008)
- Falcon tubes, 15 and 50 ml capacity (Corning, catalog numbers: 430791 and 430291)
- Luer-lock syringes, 2 ml, 5 ml and 10 ml capacity (BD Plastipak, catalog numbers: 300185, 302187 and 300912, respectively)
- Microcentrifuge tubes, 1.5 ml capacity (Eppendorf, catalog number: 0030120086)
- Pipette tips (Rainin LTS 1,000 μl, LTS 250 μl and LTS 20 μl; catalog numbers: 17001864, 17001863 and 17001865; or equivalent tips compatible with pipettes used for this protocol)
- Polycarbonate thickwall ultracentrifuge tubes, 32 ml capacity (Beckman Coulter, catalog number: 355631)
- Polypropylene OptiSealTM ultracentrifuge tubes, 11.2 ml capacity (Beckman Coulter, catalog number: 362181) and associated tube adaptors (Beckman Coulter, catalog number: 362202)
- Semi-micro cuvettes (Sarstedt, catalog number: 67.742)
- Silicone tubing (1 mm inner diameter, 1 mm wall diameter) (Fisher Scientific, catalog number: 10430313)
- Stainless steel blunt-ended needle, 14 gauge (Sigma-Aldrich, catalog number: Z261408)
- Total recovery glass vials and caps with pre-slit septa (Waters, catalog number: 186000385C)
- Waste container
- X-ray film (Fujifilm, catalog number: 4741019289)
- Trans-Blot® TurboTM polyvinylidene fluoride (PVDF) membranes (Bio-Rad, catalog number: 1704157)
- Saccharomyces cerevisiae strain appropriate to experimental aims (e.g., BY4741 (Baker Brachmann et al., 1998), which we used for our recent hyperLOPIT study; Nightingale et al., 2019)
- Acetic acid (Sigma-Aldrich, catalog number: 320099)
- Acetone, analytical grade (Sigma-Aldrich, catalog number: 32201)
- Acetonitrile, HPLC gradient-grade (Fisher Scientific, catalog number: A/0627/17)
- Adenine hemisulfate (Sigma-Aldrich, catalog number: A3159)
- AmershamTM ECLTM Prime Enhanced Chemiluminescent Western Blotting Detection Reagent (GE Healthcare, catalog number RPN2232)
- Ammonium formate (Sigma-Aldrich, catalog number: 70221)
- Ammonium hydroxide (Sigma-Aldrich, catalog number: 30501)
- Arginine (Sigma-Aldrich, catalog number: A8094)
- DifcoTM BactoTM agar (Fisher Scientific, catalog number: 10455513)
- Difco Bacto peptone (Fisher Scientific, catalog number: DF0118-17-0)
- Difco Bacto yeast extract (Fisher Scientific, catalog number: DF0127-17-9)
- cOmpleteTM Mini protease inhibitor tablets (Roche, catalog number: 11836170001)
- cOmpleteTM protease inhibitor tablets (Roche, catalog number: 11873580001)
- D-(+)-Glucose (Sigma-Aldrich, catalog number: G7021)
- Dibasic potassium phosphate (Sigma-Aldrich, catalog number: P3786)
- DL-Dithiothreitol (DTT) (Melford Laboratories, catalog number: D11000)
- Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, catalog number: E5134)
- Ficoll® PM-400 (Sigma-Aldrich, catalog number: F4375)
- Formic acid (VWR, catalog number: 20318.297)
- HEPES free acid (Melford Laboratories, H75030)
- Histidine (Sigma-Aldrich, catalog number: H6034)
- Hydrochloric acid, 37% (v/v) (Fisher Scientific, catalog number: H/1200/PB15)
- Hydroxylamine 50% (w/v) solution (Thermo Fisher Scientific, catalog number: 90115)
- Iodoacetamide (Sigma-Aldrich, catalog number: I6125)
- Isoleucine (Sigma-Aldrich, catalog number: I5281)
- Leucine (Sigma-Aldrich, catalog number: L8912)
- Lysine (Sigma-Aldrich, catalog number: L5501)
- Magnesium acetate (Sigma-Aldrich, catalog number: M5661)
- Magnesium chloride (Sigma-Aldrich, catalog number: M8266)
- Methionine (Sigma-Aldrich, catalog number: M5308)
- Non-fat dry milk powder, such as MarvelTM
- OptiPrepTM (Sigma-Aldrich, catalog number: D1556)
- Phenylalanine (Sigma-Aldrich, catalog number: P5482)
- Potassium acetate (Sigma-Aldrich, catalog number: P1190)
- Potassium chloride (Sigma-Aldrich, catalog number: 31248)
- Protein concentration estimation assay, such as PierceTM BCA protein assay (Thermo Fisher Scientific, catalog number: 23225), DCTM protein assay (Bio-Rad, catalog number: 5000112) or Quick StartTM Bradford assay (Bio-Rad, catalog number: 5000202)
- Sequencing Grade Modified Trypsin (Promega, catalog number: V5111)
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: 436143)
- Sodium hydroxide (Sigma-Aldrich, catalog number: 06203)
- Sorbitol (Sigma-Aldrich, catalog number: W302902)
- Sucrose (Sigma-Aldrich, catalog number: S0389)
- Threonine (Sigma-Aldrich, catalog number: T8441)
- TMT 10-plex or 11-plex isobaric tagging reagents (Thermo Fisher Scientific, catalog number: 90111 or A37725, respectively)
- Trichloroacetic acid (TCA) (Fisher Scientific, catalog number: 421455000)
- Trifluoroacetic acid (TFA) (Thermo Fisher Scientific, catalog number: 85183)
- Tris base (Melford Laboratories, catalog number: T60040)
- Tris-(2-carboxyethyl) phosphine (TCEP) (Generon, catalog number: GEN-TCEP)
- Tryptophan (Sigma-Aldrich, catalog number: T8941)
- Tween® 20 (Sigma-Aldrich, catalog number: P1379)
- Tyrosine (Sigma-Aldrich, catalog number: T8566)
- Uracil (Sigma-Aldrich, catalog number: U1128)
- Valine (Sigma-Aldrich, catalog number: V4638)
- Water, HPLC gradient-grade (Fisher Scientific, catalog number: W/0106/17)
- Yeast nitrogen base without amino acids (Sigma-Aldrich, catalog number: Y0626)
- Zymolyase 100-T (Nacalai-Tesque, catalog number: 07665-55)
- Anti-Pgk1p antibody, for use as cytosol Western blotting marker (Abcam, catalog number: ab113687)
- Anti-Dpm1p antibody, for use as endoplasmic reticulum Western blotting marker (ThermoFisher Scientific, catalog number: A-6429)
- Anti-Pep1p antibody, for use as Golgi apparatus Western blotting marker (Abcam, catalog number: ab113690)
- Anti-Cox4p antibody, for use as mitochondrial Western blotting marker (ThermoFisher Scientific, catalog number: 459150)
- Anti-Hh3p antibody, for use as nuclear Western blotting marker (Abcam, catalog number: ab1791)
- Anti-Pma1p antibody, for use as plasma membrane Western blotting marker (Abcam, catalog number: ab4645)
- 5x SDS-PAGE sample buffer (Jena Biosciences, catalog number: BU-117)
- 4-20% Mini-PROTEAN® TGXTM Precast Protein Gels (Bio-Rad, catalog number: 4561096)
- TGS buffer 10x (Bio-Rad, catalog number: 1610732)
- 10x yeast nitrogen base (YNB) (see Recipes)
- 10x complete supplement mixture (see Recipes)
- 40% (w/v) glucose (see Recipes)
- Synthetic medium with 2% glucose (see Recipes)
- YPD agar medium (see Recipes)
- TCEP reduction buffer (see Recipes)
- Spheroplasting medium (see Recipes)
- Zymolyase 100-T solution (see Recipes)
- Spheroplast wash medium (see Recipes)
- (Optional) Ficoll lysis buffer (Kizer et al., 2006) (see Recipes)
- (Optional) Buffer NP (Kizer et al., 2006) (see Recipes)
- 1x LB (see Recipes)
- 6x LB (see Recipes)
- Iodixanol working solution (IWS) (see Recipes)
- 18% (w/v) OptiPrep solution (see Recipes)
- 16% (w/v) OptiPrep solution (see Recipes)
- Protein resolubilization buffer (see Recipes)
- Digestion buffer (see Recipes)
- 5% (v/v) hydroxylamine (see Recipes)
- TTBS (see Recipes)
- Blocking solution (see Recipes)
- Dithiothreitol (DTT) solution (see Recipes)
- Iodoacetamide (IAA) solution (see Recipes)
- Equilibration buffer (see Recipes)
- Desalting buffer 1 (see Recipes)
- Desalting buffer 2 (see Recipes)
- Elution buffer (see Recipes)
- RP mobile phase stock solution (see Recipes)
- RP mobile phase A (see Recipes)
- RP mobile phase B (see Recipes)
- RP resolubilization buffer (see Recipes)
Equipment
- 4 °C refrigerator (Polestar cooling, model: MR 100 E)
- -20 °C freezer (Hotpoint, model: FZS175)
- -80 °C freezer (New Brunswick Scientific, model: U725-G)
- Acquity Ultra Performance Liquid Chromatography (UPLC) system with photodiode array (PDA) detector (Waters)
- Acquity UPLC BEH C18 column (Waters, catalog number: 186002353)
- Analytical balance ( Explorer, model: EX124)
- Auto-Densiflow peristaltic pump with automatic meniscus detection (Labconco)
- Avanti® JXN-26 floor-top preparative centrifuge (Beckman Coulter)
- Bioruptor® Plus sonicator (Diagenode, catalog number: B01020001)
- Centrifuge bottle assemblies, 500 ml capacity (Beckman Coulter, catalog number: 355649)
- Cuvette spectrophotometer (Cecil Instruments, model: 2041)
- Dounce homogenizer, 7 ml capacity (Wheaton, catalog number: 357542)
- Erlenmeyer flasks, 250 ml, 500 ml and 2 L capacity (Fisher Scientific, catalog numbers: 15429103, 15439103 and 11383454)
- Handheld analog refractometer (Bellingham + Stanley, E-line, catalog number: 44-803)
- JLA-10.500 rotor (Beckman Coulter, catalog number: 369681)
- Nitrogen cavitation vessel (Parr Instrument Company, model: 4639)
- OptimaTM L80-XP floor-top ultracentrifuge (Beckman Coulter)
- Orbitrap FusionTM LumosTM TribridTM mass spectrometer (Thermo Fisher Scientific)
- Oxygen-free nitrogen cylinder (BOC, Nitrogen [Oxygen-Free] 230bar Cylinder, size: W)
- Pipettes (Rainin LTS L-1000 XLS, L-200 XLS, L-20 XLS and L-10 XLS, catalog numbers: 17014382, 17014391, 1704392 and 17014388; or equivalent pipettes capable of dispensing the same volumes)
- Plate-reader spectrophotometer, capable of measuring wavelengths between 250 nm and 850 nm (Molecular Devices, Spectramax M2e)
- Pre-column for mass spectrometer-coupled nanoLC (Thermo Fisher Scientific, catalog number: 160454)
- Proxeon EASY-Spray column (Thermo Fisher Scientific, catalog number: ES803)
- Refrigerated benchtop centrifuge, capable of centrifuging 15 ml and 50 ml Falcon tubes and running at 4,500 x g (Eppendorf, model: 5804R)
- Refrigerated benchtop microcentrifuge, capable of centrifuging microfuge tubes and running at 16,100 x g (Eppendorf, model: 5415R)
- Sep-Pak® tC18 cartridges, 100 mg sorbent per cartridge (Waters, catalog number: WAT036820)
- Shaking incubator for cell culture (Sartorius, Certomat, model: BS-1)
- SW32Ti rotor (Beckman Coulter, catalog number: 369650)
- Thermo ScientificTM SterilinTM Standard 90mm Petri Dishes (Fisher Scientific, catalog number: 15370366)
- Tube holder for microfuge and Falcon tubes (Starlab, catalog number: E2345-1000)
- Trans-Blot® TurboTM system (Bio-Rad, catalog number: 1704150)
- Type 70 Ti rotor (Beckman Coulter, catalog number: 337922)
- Ultimate RSLCnano chromatography system (Thermo Fisher Scientific)
- Vacuum centrifuge (Labconco, CentriVap concentrator)
- VTi65.1 rotor (Beckman Coulter, catalog number: 362759)
- Electrophoresis system and associated power pack (Bio-Rad, model: Mini-PROTEAN® II)
Software
- Proteome Discoverer version 2.1 (Thermo Fisher Scientific, www.thermofisher.com)
- R programming language (R Core Team, 2018) (www.r-project.org)
- Bioconductor (Gentleman et al., 2004) MSnbase package (Gatto and Lilley, 2012) (http://bioconductor.org/packages/release/bioc/html/MSnbase.html)
- Bioconductor (Gentleman et al., 2004) pRoloc package (Gatto et al., 2014) (http://bioconductor.org/packages/release/bioc/html/pRoloc.html)
- Bioconductor (Gentleman et al., 2004) pRolocGUI package (Breckels et al., 2018) (http://bioconductor.org/packages/release/bioc/html/pRolocGUI.html)
- RStudio R programming language editor (RStudio Team, 2016) (www.rstudio.com)
- Mascot Server (Matrix Science, www.matrixscience.com)
- Microsoft Excel
Procedure
- Cell culture
- At least 3 days before you plan to carry out the experiment, streak-plate an appropriate yeast strain from a cryostock onto a YPD agar plate. Incubate at 30 °C in a stationary incubator for approximately 2 days until colonies form and subsequently store at 4 °C for several weeks.
- The following day, transfer the starter culture to a flask filled with fresh medium. For a typical hyperLOPIT experiment we recommend culturing 720 OD units of yeast, which equates to 1.2 L of culture at OD600 of 0.6. For this volume of culture, we use 2 L Erlenmeyer flasks.
- Dilute such that the cells undergo a minimum of 2 doublings prior to harvest so that the desired cell density is reached at a convenient time.
Note: The amount of cells required for a new growth condition, which yields enough protein per collected density gradient fraction for downstream steps of the hyperLOPIT protocol (> 50 μg per fraction), should be empirically determined for each experiment in question. For experiments where the goal is not to perform hyperLOPIT, smaller cultures should be sufficient, but this should be determined by the investigator. - When the required OD600 has been reached, harvest cells by centrifugation at 3,000 x g for 5 min at room temperature and discard the supernatant. For this step, use 500 ml bottle assemblies and the JLA 10.500 rotor in the Avanti centrifuge.
- Cell pre-treatment for lysis
- Resuspend the cell pellet, still in the 500 ml bottle assemblies, in TCEP reduction buffer at 5 OD units per ml and incubate for 5 min at room temperature without shaking. Centrifuge at 3,000 x g for 5 min in the Avanti centrifuge to harvest the cells and discard the supernatant.
- Resuspend the cells in spheroplasting medium at 20 OD units per ml and transfer to 50 ml Falcon tubes. Add 1 μl of zymolyase 100-T solution per OD unit of yeast used in the experiment.
Note: Spheroplasting medium is composed of the same constituents as the medium that was used for cell culture, to maintain the same conditions as were used for culture. We do not, however, recommend the use of conditioned media for this step. The media should be made fresh and include 1.0-1.2 M sorbitol and 20 mM Tris-HCl, pH 7.5, to maintain osmotic support and the appropriate pH during spheroplasting. - Withdraw a 10 μl aliquot of the solution and dilute in 990 μl of water. Measure the OD600 after blanking the spectrophotometer with an equivalent dilution of spheroplasting medium in water.
Note: Spheroplast conversion efficiency is conveniently monitored by spectrophotometry. Absorbance of the cell suspension at 600 nm is proportional to the number of intact cells that are present in the suspension. Spheroplasts are very fragile and sensitive to changes in the tonicity of a solution. As such, transferring them to an osmotically unsupported solution will result in lysis of the spheroplasts. Only the remaining intact cells contribute to the absorbance at 600 nm, indicating the amount of cells that have undergone conversion to spheroplasts. - Incubate at 30 °C for 10 min with shaking at 200 rpm and measure OD600 of a 1:100 dilution. The OD600 should be around 10% of the pre-digestion OD600, indicating around 90% zymolyase digestion efficiency.
Note: This step should not be allowed to continue for more than 10 min, as incubation in the presence of zymolyase for too long can result in uncontrolled cell lysis. - Harvest cells, without transferring to new tubes, by centrifugation for 5 min at 1,500 x g, 4 °C and resuspend in spheroplast wash medium.
Note: The pellet can be sticky and difficult to resuspend, but do not vortex or pipette vigorously as this risks lysis of the fragile spheroplasts. Resuspend with a trimmed 1 ml pipette tip, use a glass rod or gently invert to resuspend. Due to the risk of lysis, do not aim for a homogeneous suspension of cells at this point. - Harvest the cells for 5 min at 1,500 x g, 4 °C and discard supernatant.
- Optional: Nuclear preparation
Note: If greater nuclear resolution is not of particular interest, skip to Section D.- Optionally perform a nuclear preparation according to Kizer et al. (2006) with modifications described subsequently. For this protocol, we use approximately 120 OD units of spheroplasts for a nuclear preparation and 600 OD units for the main subcellular fractionation.
Note: This step is optional and if other organelles are of special interest, enrichments for these organelles may be performed instead of a nuclear preparation, as was performed in two previous hyperLOPIT studies (Christoforou et al., 2016; Thul et al., 2017). - Resuspend the spheroplasts in Ficoll lysis buffer and transfer to a Dounce homogenizer. Lyse with 20 up-down strokes on ice.
Note: As in the original paper (Kizer et al., 2006), we use the “tight” Dounce pestle for the entirety of lysis, and do not advocate “pre-resuspension” with the “loose” pestle. - Pre-clear the lysate for 10 min at 3,220 x g, 4 °C and transfer the supernatant to a 32 ml round-bottomed ultracentrifuge tube. Balance the opposite position of a Type 70 Ti rotor (Beckman Coulter) with a tube of the same mass and density and centrifuge at 50,000 x g, 4 °C for 35 min. Use “MAX” acceleration and deceleration.
- Discard the supernatant and resuspend pellet in buffer NP with vigorous pipetting. Store resuspended nuclear-enriched pellet at -80 °C.
- Optionally perform a nuclear preparation according to Kizer et al. (2006) with modifications described subsequently. For this protocol, we use approximately 120 OD units of spheroplasts for a nuclear preparation and 600 OD units for the main subcellular fractionation.
- Lysis for density gradient centrifugation
- Resuspend the remaining 600 OD units of spheroplasts in 1x LB at 20 OD units per ml and transfer to the chamber of the nitrogen cavitation vessel.
- Lyse using a method modified from Wang et al. (2014). Charge the vessel with oxygen-free nitrogen to 500 psi and incubate for 3 min.
Note: The nitrogen within the cylinder is under pressure and if the inlet tap is opened too quickly, it can lead to the target pressure of 500 psi being overshot. Therefore, open the inlet tap slowly and closely monitor the pressure gauge to obviate the chance of this occurring. - Release the pressure by opening the gas inlet valve, so that the pressure drops to 300 psi.
- Allow a further 3 min of incubation and discharge the vessel by opening the outlet port at the base of the vessel. Collect the lysate at approximately 3 drops per second.
Note: As the nitrogen is released from solution during lysis, the pressure within the nitrogen cavitation vessel will drop and consequently the lysate will be released more slowly. Therefore monitor the rate at which the lysate is released from the outlet port and, if necessary, open the port more to maintain a steady rate of approximately 3 drops per second. - Clear the lysate of debris, aggregates, and unlysed cells and spheroplasts by centrifugation for 5 min at 1,000 x g, 4 °C. Subject the supernatant to another round of centrifugation for 10 min at 3,000 x g, 4 °C.
- Crude membrane preparation
- Prepare 18% (w/v) OptiPrep solution with the aid of a handheld refractometer and calibration curve of known OptiPrep concentrations and their refractive index measurements, measured in degrees Brix (°Bx) (see Note 1).
- Transfer the cleared lysate into four round-bottomed ultracentrifuge tubes (~7.5 ml lysate per tube). Underlay the contents of each tube with 5 ml 18% (w/v) OptiPrep cushion solution, prepared in 1x LB, using a syringe attached to a blunt-ended, wide-bore needle.
Notes:- Even though it is possible to fit the entirety of the lysate in a single round-bottomed centrifuge tube, we do not recommend this. If this is done, upon ultracentrifugation, the sheer amount of protein in the entirety of lysate will cause it to aggregate on top of the cushion solution. This may cause damage to organelle membranes and affect the results of the experiment as a whole. We find that if the lysate is split as described, this does not occur.
- When underlaying with 18% (w/v) OptiPrep cushion solution, take the utmost care to not to introduce any air bubbles into the cushion solution. If these are introduced it will result in mixing of the lysate with the OptiPrep solution, disrupting the crisp interface that should be present between the two solutions. Ultracentrifugation of a solution where this has occurred will prevent formation of a crude membrane interphase that would normally form between the two solutions, defeating the object of performing this step. Instead this will potentially cause the membranes to pellet and aggregate at the bottom of the tube. To prevent this happening, use the graduations on the syringe as a guide to the amount of OptiPrep solution withdrawn before underlaying. Before commencing the underlaying of the lysate, press down gently on the syringe plunger to allow a single drop of OptiPrep to be released from the wide-bore needle (to expel any air that may be present). Submerge the needle at the bottom of the ultracentrifuge tube and begin injection of the cushion solution, keeping a close eye on the level of the liquid within the syringe. As soon as the meniscus of the liquid drops to the level of the connection between the syringe and needle, stop injecting. Remove the needle and discard the remainder of the solution.
- Balance the four tubes to within 10 mg of each other and place the tubes in opposing sides of an SW32Ti swinging-bucket rotor.
- Centrifuge the tubes for 2 h at 28,000 rpm (96,300 x g), 4 °C. Select an acceleration profile of “MAX” and a deceleration profile of “9” (the slowest deceleration that uses a brake). This minimizes disruption of the membrane interphase that forms upon ultracentrifugation.
- Withdraw the cytosol-enriched supernatant from the tube, leaving between 1 and 2 cm of the supernatant above the membrane interphase, so as not to disturb the crude membrane interphase. Store the supernatant at -20 °C for further processing.
- Withdraw the membrane-containing interphase using a trimmed 1 ml pipette tip into a separate tube, taking care not to withdraw too much solution from either above or below the interphase.
- Isopycnic density gradient ultracentrifugation and fraction collection
- Adjust the concentration of OptiPrep in the collected membrane interphase solution to 16% (w/v), monitoring the concentration using a handheld refractometer (see Note 1).
Notes:- Using 600 OD units of culture typically yields around 2-3 ml of crude membranes at this step. The aim should be to adjust the refractive index of the crude membranes to the required value in as small a volume as possible and to make up the remainder of the 11.2 ml volume of the resolving gradient tube with pre-prepared 16% (w/v) OptiPrep. If this is not adhered to, it may result in the undesirable situation that the membranes, in the correct OptiPrep concentration, are split across multiple density gradients.
- We find that preparing a self-generating gradient using 16% (w/v) OptiPrep provides good separation of the major subcellular organelles in yeast (Figure 2). This step may be modified if a different gradient shape is desired or specific subcellular organelles are the main focus of the experiment.
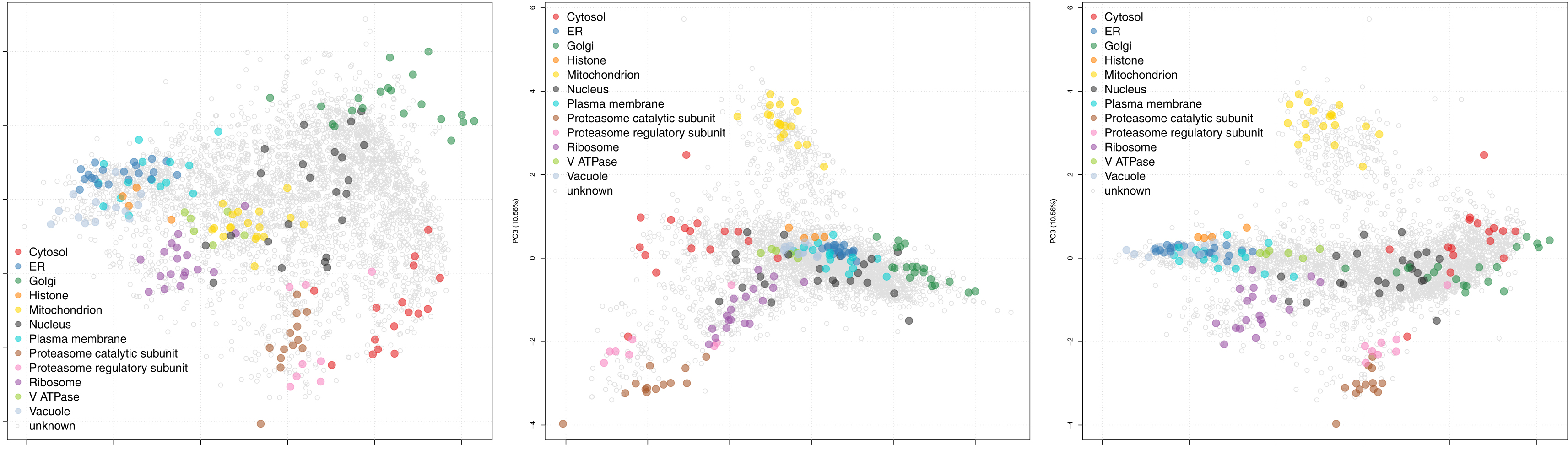
Figure 2. Representative data obtained using the protocol described here. After performing hyperLOPIT in S. cerevisiae with the optional nuclear preparation, two-dimensional Principal Components Analysis reveals the resolution of 12 separate organelles and protein complexes within the S. cerevisiae spatial proteome map, across three principal components. These encompass the main subcellular organelles of the organism (data reproduced from a single replicate performed in our previous study [Nightingale et al., 2019]). Within each plot each point represents a single protein group, points that are colored represent proteins determined by hyperLOPIT to localize to a specific organelle and light grey points represent protein that do not have a localization predicted by hyperLOPIT.
- Prepare a balance tube containing 16% (w/v) OptiPrep and use to fill a blank vertical rotor tube, which does not contain any membranes and acts as a balance tube.
- Balance the tubes to within 10 mg of each other, using 16% (w/v) OptiPrep solution. Place the two tubes in opposing sides of a VTi65.1 rotor and float aluminum spacers on top of the tubes.
- Screw the rotor lids on and centrifuge at 65,000 rpm (362,900 x g), for 4 h at 4 °C. Select an acceleration profile of “MAX” and a deceleration profile of “9” (the slowest deceleration that uses a brake). This acts to minimize disruption to the density gradient that has formed on ultracentrifugation.
- Remove the tubes from the VTi65.1 rotor. Remove and discard the plastic plugs.
- Prepare a Labconco Auto-Densiflow peristaltic pump with meniscus tracking probe for fraction collection by connecting a piece of collection tubing approximately 40 cm long to the probe. Clamp on to the peristaltic pump.
Note: If an Auto-Densiflow fraction collector is unavailable, fractions may be collected by puncturing the bottom of the tube and allowing the gradient fractions to drip out from the bottom and most dense, fraction to the top and least dense fraction. Alternative fractionators are available, from Brandel and Teledyne Isco, but these have not been tested for use with hyperLOPIT. We do NOT recommend collecting fractions by pipetting from the top of the gradient as this requires a high level of technical skill and is very susceptible to error, which will result in mixing of different parts of the gradient and ruin the entire experiment. - Flush the tubing with distilled water into a waste container and follow this by flushing through with air. Flush through with 1x LB and then air.
- Insert the gradient tube into the tube holder and set the probe direction to “down” until the probe finds the gradient meniscus, at which point the probe will stop moving.
- Set the probe to “deposit” and collect 23 fractions from the gradient into 1.5 ml microcentrifuge tubes, of which 22 should be 0.5 ml and one should be ~0.2 ml.
Note: This step is not automated, so volumes collected per fraction should be monitored closely by eye until the required volume has been collected for the fraction in question. Store fractions at -80 °C until further processing.
- Adjust the concentration of OptiPrep in the collected membrane interphase solution to 16% (w/v), monitoring the concentration using a handheld refractometer (see Note 1).
- Monitoring gradient shape
- Thaw gradient fractions and vortex well to ensure that the iodixanol content in the OptiPrep-containing fractions is uniformly suspended within the fraction.
- Determine refractive index of each fraction using a handheld, analog refractometer. Use approximately 20 μl of each fraction for this step.
Note: If the refractive index of any fraction exceeds the maximum refractive index of the refractometer, dilute that fraction with water and re-measure. Do not use 1x LB. Multiply the resultant refractive index by the dilution factor to obtain the undiluted refractive index measurement. - Monitor density gradient shape by plotting fraction number against the refractive index of that fraction. The result should be a slightly sigmoidal curve that is relatively flat in the middle of the gradient and becomes steeper in the last 2-3 fractions (Figure 3).
- If monitoring organelle distribution by enzyme assay, take an aliquot here with which to do this, and perform enzyme assays.
Note: We have not attempted such methods in S. cerevisiae but methods are available in the literature if this is desired.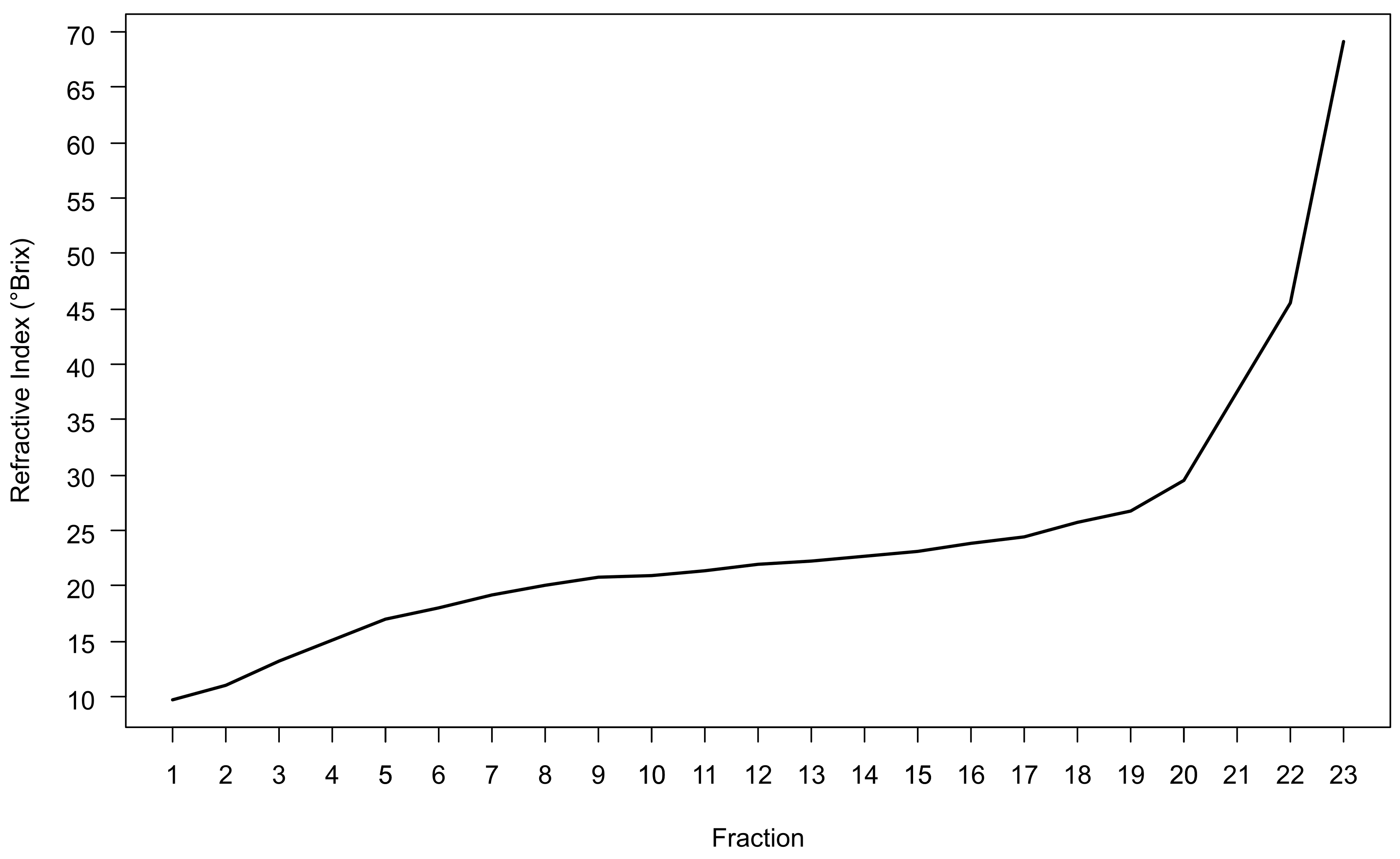
Figure 3. Typical gradient shape achieved using the concentration of OptiPrep and the density gradient ultracentrifugation parameters detailed within this protocol. Fractions were collected using an Auto-Densiflow fraction collector and are numbered in ascending order from the least to the most dense fraction of the gradient.
- Fraction processing
- Process all fractions (including separate organelle preparations and cytosol-enriched fraction) further by TCA precipitation, followed with acetone washing. For this add ¼ of the volume of 100% (w/v) TCA, prepared in water and vortex well.
- Incubate for 2 h at 4 °C to allow the protein content of each of the fractions to precipitate from solution.
- Harvest precipitates by centrifugation at maximum speed in a benchtop microcentrifuge at 4 °C for 10 min. Discard supernatants.
- Wash pellets in acetone that has been pre-chilled to -20 °C. Vortex extensively and subject to sonication cycles (30 s on and 30 s off) in a Diagenode BioRuptor Plus sonicator, until the pellets break down completely and become powdery.
Notes:- If the pellets do not break down completely, they may be difficult to resolubilize in downstream steps of this protocol.
- If a BioRuptor is not available, other sonicators may be used for this step.
- Repeat centrifugation, acetone washing, and sonication twice more; each time discarding the supernatant.
- Discard supernatants and allow pellets to dry briefly at room temperature until acetone evaporates (see Note 2).
- Sample resolubilization and protein concentration estimation
- Resolubilize each pellet in protein resolubilization buffer with extensive sonication.
Note: Resolubilize in the minimum volume in which the entire protein content is soluble. - For this step, begin with 50 μl of protein resolubilization buffer per pellet and monitor for the presence of any pellet in the fraction by centrifugation, as in the TCA precipitation step (Section H, step 3). Add further aliquots of protein resolubilization buffer, sonicate, and spin down to monitor the presence of any protein pellet. As soon as no pellet is visible after centrifugation, this indicates that the protein content has been resolubilized to completion.
- Estimate protein concentration in each fraction using a protein concentration estimation assay, preferably in 96-well plate format.
Notes:- If the recommended protein assay kits are not available, other protein estimation assays may be used, as long as they are compatible with 100 mM HEPES and 0.1% (w/v) SDS.
- We recommend the use of 96-well plate format assay as sample amounts in hyperLOPIT experiments can be extremely limited and this conserves the maximum amount of each sample for Western blotting and isobaric tag labeling.
- Determine overall protein yield. The ideal yield per fraction for isobaric tag labeling should be > 50 μg, allowing for an extra 1 μg per fraction used for downstream Western blotting. If fractionation is required without isobaric tag labeling, lower yields will be suitable.
- Resolubilize each pellet in protein resolubilization buffer with extensive sonication.
- Monitoring organelle resolution by Western blot
- Monitor organelle resolution using Western blotting against a panel of antibodies raised against marker proteins whose resolution is characteristic of the organelle which they represent (see Note 3) (Table 1).
Table 1. Suggested antibodies for use with hyperLOPIT in S. cerevisiae. The suggested primary and secondary antibody dilutions are based on Western blots that are carried out using 1 μg of total protein per lane of an SDS-PAGE gel.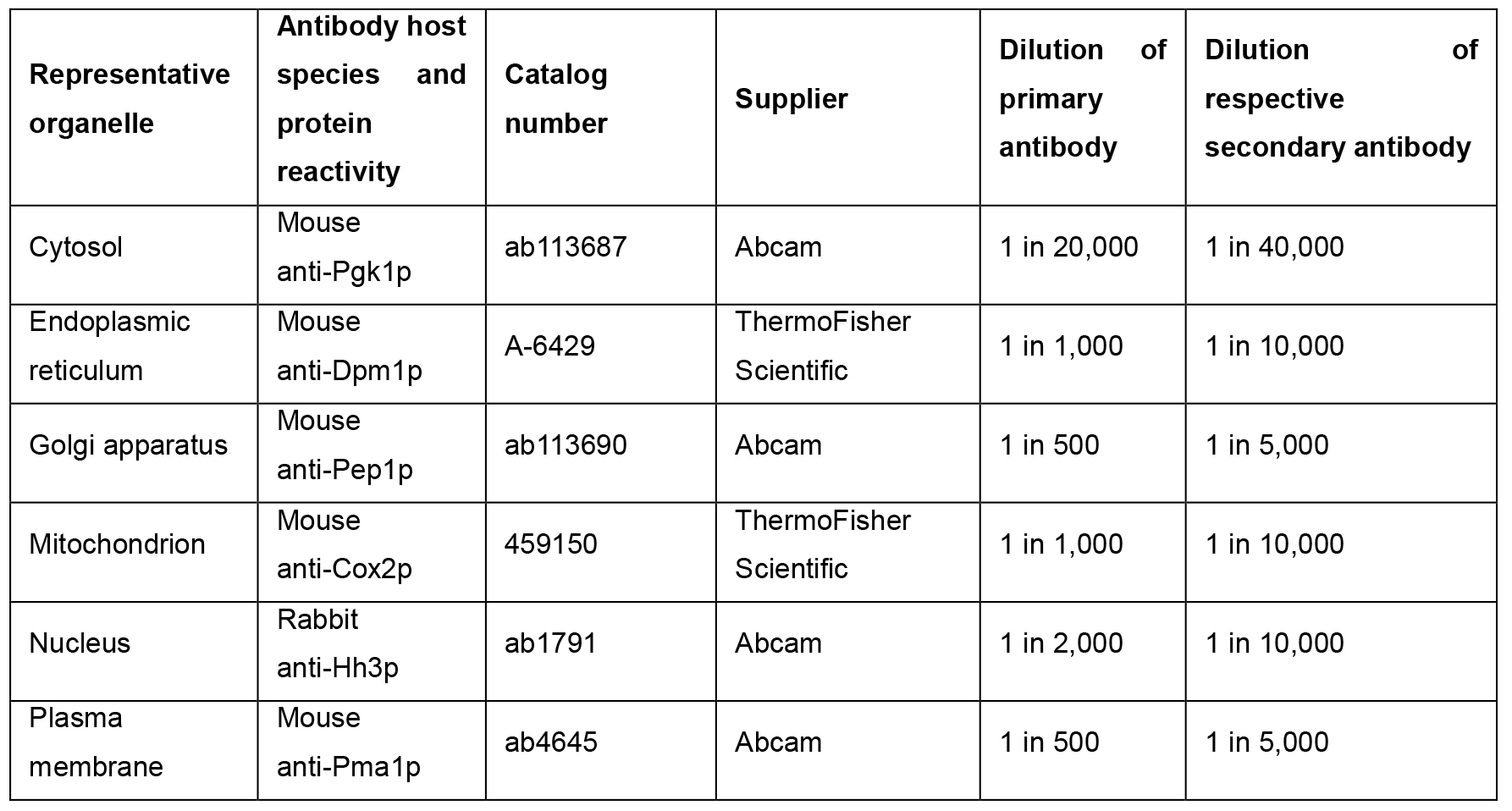
- Normalize the amounts of each fraction (including the nuclear preparation, cytosol-enriched fraction, and density gradient fractions) to 1 μg of total protein and heat to 95 °C in the presence of SDS-PAGE sample buffer for 5 min. Load each fraction on a single well of a pre-cast, gradient SDS-PAGE gel.
- Subject the samples to electrophoresis in SDS-PAGE minigels in TGS buffer and using the full length of the gels. Use a Mini-PROTEAN II electrophoresis system and power pack for this step and run the gel at 200 V.
- Transfer to Trans-Blot Turbo polyvinylidene fluoride (PVDF) membranes, using the mixed molecular weight program on a Bio-Rad Trans-Blot Turbo apparatus.
- Cut membranes into appropriate molecular weight ranges (see Note 3) and incubate each strip in blocking solution for 1 h at room temperature on a rotary mixer.
- Probe each strip with the appropriate primary antibody, diluted in blocking solution (see Note 4). Incubate overnight at 4 °C, with shaking on a rotary mixer (see Table 1 for suggested dilutions).
- The following day, place the membranes back at room temperature and wash three times for 5 min each in TTBS.
- Probe with the appropriate horseradish peroxidase-conjugated secondary antibody diluted in blocking solution (see Table 1 for suggested dilutions), for 1 h at room temperature.
- Discard secondary antibody and wash 3 times for ten minutes each in TTBS.
- Proceed to detection of signal using enhanced chemiluminescent (ECL) substrate.
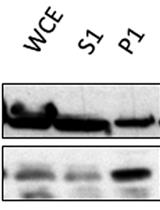
- Expose each membrane with X-ray film until the desired level of signal has been reached (see Figure 4 for an example Western blot).
- Assuming a suitable yield (see Note 5) has been obtained, proceed to the next section if it is desired to proceed with the hyperLOPIT protocol.
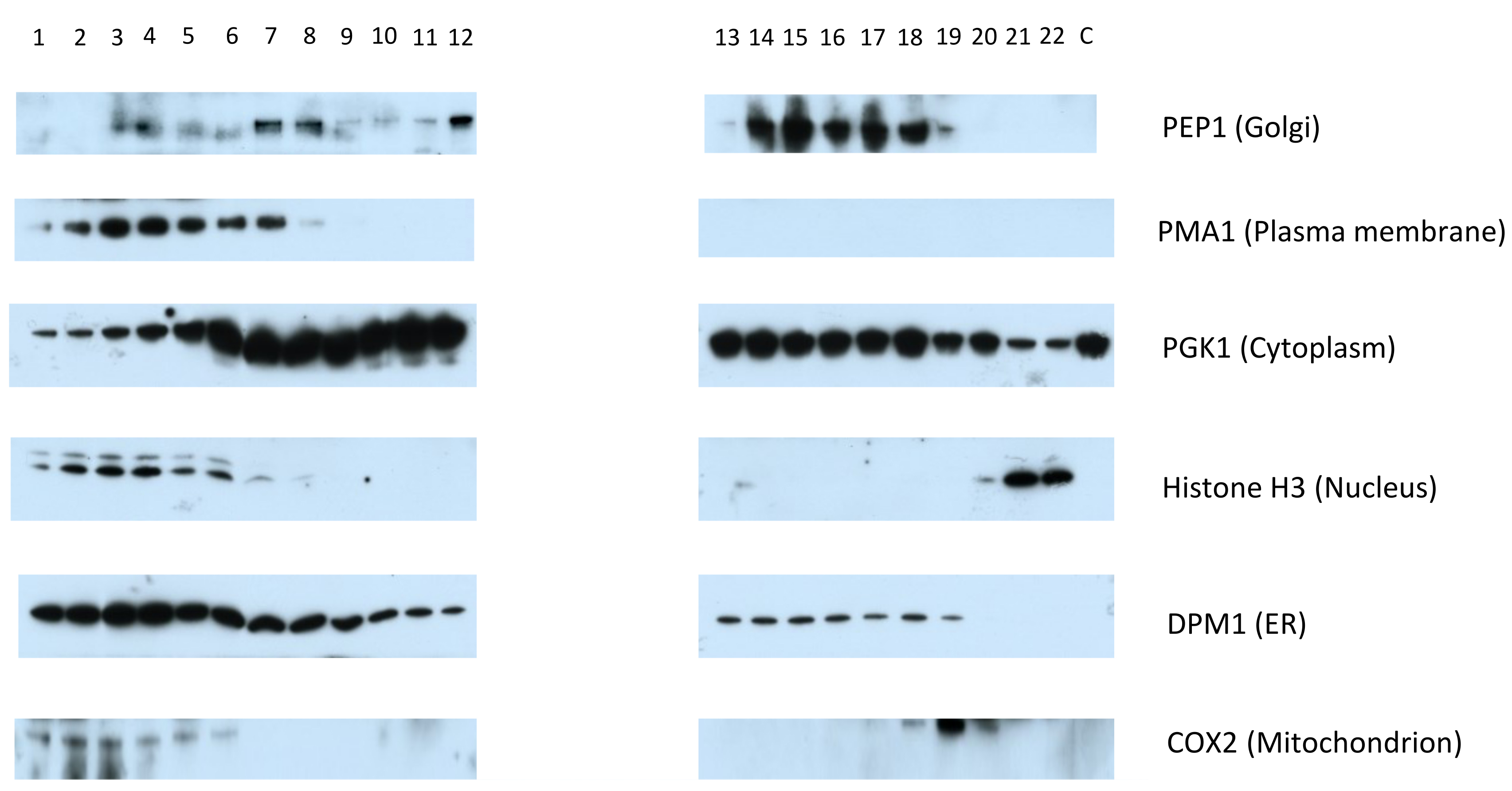
Figure 4. Representative Western blot typical of an experiment on S. cerevisiae, performed using the hyperLOPIT protocol described here, in the absence of a nuclear preparation and probing using the antibodies described in Table 1. The fractions are numbered in the order in which they were collected using the Labconco fraction collector, from the first and least dense fraction, to the twenty-third and most dense fraction. The separate cytosol-enriched fraction is denoted by “C”. Modified from (Nightingale et al., 2019).
- Monitor organelle resolution using Western blotting against a panel of antibodies raised against marker proteins whose resolution is characteristic of the organelle which they represent (see Note 3) (Table 1).
- Protein reduction, alkylation and digestion
- For experiments including nuclear preparations, select the nuclear preparation, cytosol-enriched fraction, and use the remainder of the isobaric tags to label fractions from the density gradient. In experiments that do not include nuclear preparations, select the cytosolic fraction and use the remainder of the tags to label gradient fractions (see Note 4). This recommendation is the same whether TMT 10- or 11-plex tags are being used, or isobaric tags of lower multiplexing capability.
Note: Fractions should be chosen for labeling on the basis of relative enrichment of organelles of interest, and relative depletion of other contaminating organelles, as determined by Western blotting. It should be noted that, even though proteins from only 7 organelles are probed in this Western blotting scheme, we are able to resolve 12 subcellular locations within our final dataset (Figure 2). Indeed, a well-designed fractionation scheme will result in the resolution of subcellular locations within the final spatial proteome map that were not included within the Western blotting experiment, as they will resolve discretely within the density gradient. - Normalize each fraction to the same amount of protein (50-100 μg) and make up to the same volume (100 μl) with protein resolubilization buffer.
Note: The amount of total protein may be reduced if yields are not high enough to use 100 μg of each fraction. We do not, however, recommend labeling < 50 μg of each of the selected fractions. - Add 11.1 μl of dithiothreitol (DTT) solution prepared in protein resolubilization buffer to each sample.
- Incubate for 1 h at 56 °C to allow reduction of disulfide bonds.
- Return to room temperature and add 12.3 μl of iodoacetamide (IAA) solution to each sample. Incubate for 1 h at room temperature in the dark.
- Add 1 ml of acetone to each fraction, vortex the fractions briefly and allow protein content to precipitate in acetone overnight at -20 °C.
- The following day, centrifuge all precipitated fractions at maximum speed in a benchtop microcentrifuge for ten minutes at 4 °C.
- Discard the supernatants and briefly air-dry the pellets (see Note 2).
- Resuspend the pellets in 87.5 μl of digestion buffer by vortexing.
- Digest with trypsin at 1:20 (w/w) protease:protein ratio, at 37 °C for 16 h. Add trypsin in two aliquots (1:40 (w/w) protease:protein ratio) spaced 1 h apart, such that the final volume of the digest is 100 μl.
- For experiments including nuclear preparations, select the nuclear preparation, cytosol-enriched fraction, and use the remainder of the isobaric tags to label fractions from the density gradient. In experiments that do not include nuclear preparations, select the cytosolic fraction and use the remainder of the tags to label gradient fractions (see Note 4). This recommendation is the same whether TMT 10- or 11-plex tags are being used, or isobaric tags of lower multiplexing capability.
- TMT labeling (modified from the TMT 10-plex kit user guide)
- This section describes labeling of digests with TMT tags. If using a different isobaric labeling chemistry, consult the relevant labeling protocol that comes with the kit.
- The following day, clear the digests of any insoluble material by centrifugation for 15 min in a microcentrifuge at maximum speed.
- Allow a TMT kit to equilibrate to room temperature. Before opening the vials, briefly pulse-centrifuge them.
- Add 41 μl of acetonitrile to each tag vial. Vortex well and allow to solubilize for 5 min.
- Transfer each resolubilized tag to a different digest and incubate for 2 h at room temperature on a shaking platform.
- Add 8 μl of 5% (v/v) hydroxylamine, prepared in digestion buffer to each sample and incubate for 30 min on a shaking platform at room temperature.
- Add 100 μl of HPLC-grade water to each sample and incubate for 1 h at 4 °C.
- Pool all labeled digests into a single tube and dry to completion in a vacuum centrifuge at 10 °C. Store at -80 °C until sample clean-up by solid-phase extraction (SPE).
- Sample clean-up by solid-phase extraction (SPE) (modified from a previous publication [Villen and Gygi, 2008])
- Equilibrate a Sep-Pak tC18 cartridge with 1.8 ml of 100% (v/v) HPLC-grade acetonitrile (ACN).
- Flush through with 600 μl Equilibration Buffer.
- Flush through with 1.8 ml of Desalting Buffer 1.
- Resolubilize sample in 1 ml 0.4% TFA and ensure that pH < 2. To achieve this, check pH using pH paper on a small aliquot (~10 μl) of sample. If pH > 2, keep adding aliquots of 10% TFA, checking pH after each addition until pH < 2.
Note: The buffering capacity of residual HEPES from the digestion and isobaric tag labeling steps can keep pH high, even in the presence of 0.4% TFA. Ensure that pH < 2 before loading on the SPE cartridge, or the labeled peptides will not bind to the tC18 material and will, instead, be lost. - Load resolubilized peptides onto the cartridge and allow to bind to the packing material.
- Desalt in 1.8 ml Desalting Buffer 1.
- Flush with 180 μl Desalting Buffer 2.
- Elute the desalted peptides from the cartridge using 1 ml Elution Buffer and remove a small aliquot corresponding to approximately 5-10 μg of labeled peptides.
- Dry the samples to completion in a vacuum centrifuge at 10 °C.
- Pre-fractionation by high pH reversed phase (RP) chromatography
- Fractionate the sample using high pH reversed phase chromatography prior to mass spectrometric analysis, on a Waters Acquity UPLC system.
- Resolubilize solid phase extracted sample in 100 μl RP resolubilization buffer by vortex mixing and spin down in a benchtop microcentrifuge for 10 min at maximum speed.
- Transfer supernatant to an Acquity autosampler vial and place in the autosampler of a Waters Acquity UPLC system, maintained at 10 °C.
Note: If desired another chromatography system may be used, that is capable of running in normal flow. - Inject the sample onto the system and run using the gradient parameters shown in Table 2. Monitor sample elution using a photodiode array (PDA) detector, scanning wavelengths from 210-400 nm. Collect fractions at 1-min intervals with the aid of a timed fraction collector and freeze to completion on dry ice.
Table 2. Parameters for use during high pH reversed phase pre-fractionation of hyperLOPIT samples on a Waters Acquity UPLC system. The entire LC gradient is 75 min long. Within the gradient, 0-10 min, 62-67.5 and 67.6-75 min should be isocratic. The gradient between each of the switching steps that is documented within this Table should be linear (represented by “6” with the Waters UPLC Inlet Method software).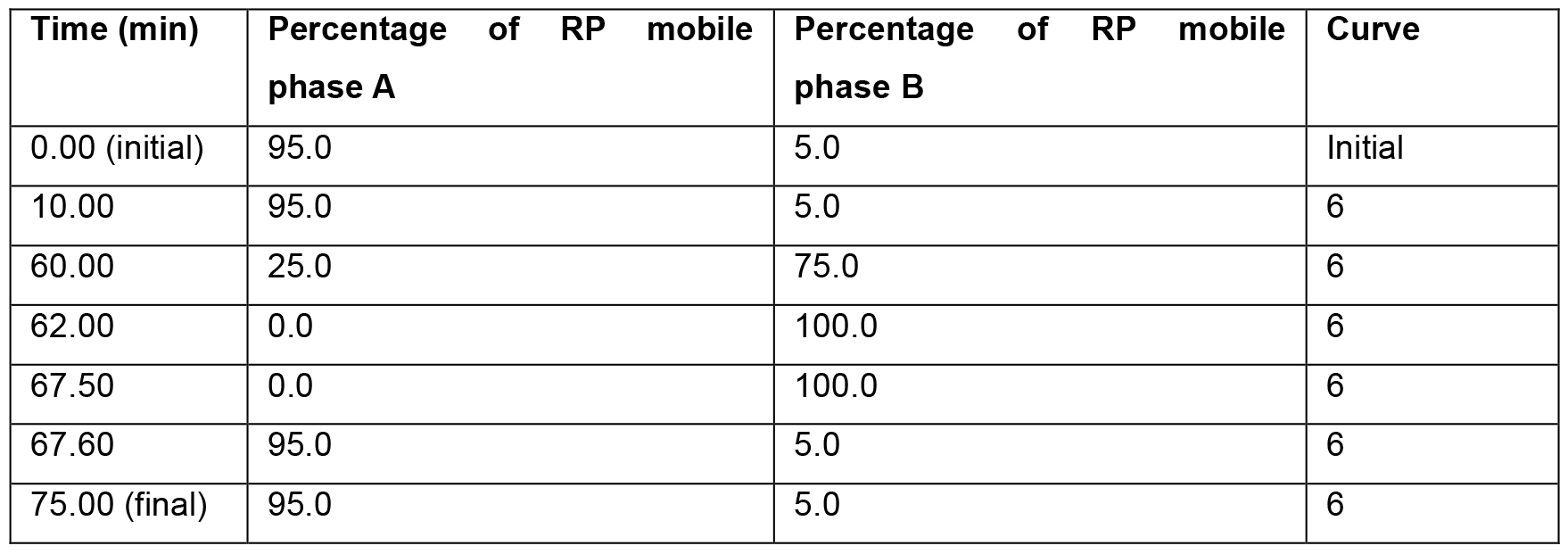
- Dry fractions to completion in a vacuum centrifuge at 10 °C. Store dried fractions at -80 °C.
- Use the collected chromatographic data to determine the collected fractions that correspond to the peptide-containing chromatographic space.
- LC-SPS-MS3 analysis of samples
- Resuspend the dried samples in 0.1% (v/v) formic acid and pool equal amounts in the following scheme: Pool the first with the middle fraction, the second with the second-from-middle, and so on until the end of the peptide-containing elution space.
Note: Pooling the samples in this fashion not only makes better use of the second dimension of peptide fractionation (low pH RP coupled in-line to the mass spectrometer) (Zhang et al., 2011), but also reduces mass spectrometry analysis time by half. - Load approximately 1 μg of each pooled fraction into the autosampler of the Dionex Ultimate 3000 RSLCnano chromatography system.
Note: Base this measurement either on the protein estimation that was performed prior to Western blotting and protein digestion or, perform a peptide estimation assay to determine the amount to load. - Analyze each pooled fraction by liquid chromatography-mass spectrometry using 120-min gradient on an Orbitrap Fusion Lumos Tribrid mass spectrometer coupled in line to the Dionex Ultimate 3000 RSLC nanoUPLC system. Acquire data using an LC-SPS-MS3 method (McAlister et al., 2014). For details about the methods used for LC and MS, see previous publications that have employed TMT 10-plex quantitation coupled with SPS-MS3 to perform hyperLOPIT studies (Christoforou et al., 2016; Thul et al., 2017; Mulvey et al., 2017). Alternatively, in the absence of a Fusion series mass spectrometer, use an appropriate LC-MS/MS method on the mass spectrometer that is being used for analysis.
Notes:- We recommend using a Thermo Fisher Scientific Fusion series mass spectrometer, as the SPS-MS3 capability (McAlister et al., 2014) offers greater quantitative accuracy and precision in the measurement of the abundance of each TMT 10- or 11-plex reporter ion in a specific scan, relative to the sole use of MS/MS. SPS-MS3 involves synchronous selection of multiple MS2 fragment ions, arising from the target precursor ion, for further fragmentation in an MS3 scan. This liberates TMT reporter ions that arise solely from the target precursor ion, in the absence of reporter ion signal arising from the fragmentation of contaminating ions that were co-selected in the MS1 scan. Other tandem mass spectrometers may be used for these experiments, but they will be more susceptible to issues (Ting et al., 2011; Wenger et al., 2011) with co-selection of contaminating precursor ions in addition to the ion targeted for fragmentation by MS/MS. This will result in a reduction in the overall quantitative accuracy and precision of quantitation in the experiment, a direct readout of which will be diminished organelle resolution in the final hyperLOPIT spatial map.
- Analysis of TMT labeled samples using mass spectrometers that are not of high enough resolution may preclude the use of 10- or 11-plex TMT tags due to an inability to resolve the isotopolog N and C tags, which will result in reporter ion coalescence (McAlister et al., 2012) and, consequently, in inaccurate quantitation. In such cases, isobaric tags with lower multiplexing capability should be used.
- Analyze the extra, non-fractionated, sample collected at the SPE step using the same mass spectrometry method as was used in Step O3 above. This will enable characterization of the isobaric tag labeling efficiency.
Note: It is recommended that the isobaric labeling efficiency be characterized for every hyperLOPIT experiment, independently of the analysis of the pre-fractionated samples by LC-SPS-MS3 or other LC-MS/MS method. The test ensures that the efficiency of labeling of both peptide N-termini and ε-amino groups of lysine residues with isobaric tag reagents is high enough (i.e., essentially complete). Indeed, we observe that labeling efficiencies are typically > 99% using this method. It should be noted that if either the pH or acetonitrile concentration is modified during isobaric tag labeling, it will result in diminished stability of the isobaric tag labeling reagents, hydrolysis of the reagents, or undesirable side-reactions with residues such as tyrosine, serine, and threonine. This will result in incomplete labeling of peptides, which will lead to inaccurate PSM-level and overall protein-level quantitation. This will result in non-representative and reduced overall organelle resolution in the final hyperLOPIT spatial map.
- Resuspend the dried samples in 0.1% (v/v) formic acid and pool equal amounts in the following scheme: Pool the first with the middle fraction, the second with the second-from-middle, and so on until the end of the peptide-containing elution space.
Data analysis
- Analysis of mass spectrometry data using Proteome Discoverer version 2.1
- Analyze the raw mass spectrometry data files using Proteome Discoverer 2.1 interfaced with an in-house Mascot server. Search the data against a canonical S. cerevisiae database, such as a UniProt or SwissProt database, either in the presence or absence of protein isoformal information. Alternatively download a protein sequence database from the Saccharomyces Genome Database (SGD–www.yeastgenome.org) (Cherry et al., 2012). Search the non-fractionated test sample file separately from the remainder of the fractionated samples. Perform the search for the fractionated sample files together, such that there is a single results report for the entire group of hyperLOPIT experimental mass spectrometry runs.
- Perform the labeling test search before carrying out the search of the fractionated hyperLOPIT raw data files. For the labeling test search, specify TMT (or other isobaric tags) labeling of both N-termini and lysine residues as dynamic modifications, but keep all other search parameters the same as described in remainder of this section. Export the Results files at peptide-spectral match (PSM) level and calculate the percentage of peptides that exhibit each isobaric tag modification, relative to all peptides that could theoretically exhibit that modification (i.e., all peptides with a lysine ε-amino group that can be labeled, and all peptide N-termini that are not otherwise modified). This gives an estimate of the labeling efficiency for that particular form of labeling, which should be > 99% using this method.
- For the fractionated hyperLOPIT data, load each set of raw data files into Proteome Discoverer as a set of Fractions, and not Files. Perform the Mascot search allowing a peptide tolerance of ±15 ppm and fragment match tolerance of ±0.6 Da if using LC-SPS-MS3. If using another LC-MS/MS method on a different mass spectrometry platform, use peptide and fragment match tolerances that are appropriate to the mass analyzers being used in the MS1 and MS2 scans. Include the Percolator algorithm (Brosch et al., 2009) node within the software, to allow filtering of peptide-spectral matches based on their respective false discovery rate.
- Set carbamidomethylation of cysteines, isobaric tag labeling of lysines and N-termini as static modifications. Set oxidation of methionine, and deamidation of asparagine and glutamine as dynamic modifications.
- For analyses that use LC-SPS-MS3, in the reporter ion quantitation node, use the sum of centroided peaks within a window of ±2 mmu around m/z of each monoisotopic reporter ion for quantitation and set MS order for activation to MS3. For experiments using other tandem mass spectrometric analysis methods and/or isobaric tags, set MS order for activation to MS2 and adjust the window accordingly for summing around the m/z of the reporter ion. Set protein group-level quantitation to be equal to the median of all quantified PSMs for any given protein group and report quantitation using the S/N measurement option that is available in Proteome Discoverer version 2.1.
- Export the protein group-level results information to an Excel workbook or tab-delimited text file. If exported to an Excel workbook, convert the workbook to CSV format before proceeding to the next section.
- Data analysis using the MSnbase and pRoloc suite of packages
- Analyze the results files from Proteome Discoverer using an R programming language editor such as RStudio (RStudio Team, 2016) that is running the Bioconductor (Gentleman et al., 2004) pRoloc (Gatto et al., 2014), pRolocGUI (Breckels et al., 2018) and MSnbase packages (Gatto and Lilley, 2012). For an exhaustive analysis protocol, we direct the reader to recent work published by Breckels and colleagues (Breckels et al., 2016b).
- Read the raw data into the R environment language, using the “readMSnSet2()” function, where “ecol” are specified as the columns that contain the quantitation data.
- Filter protein groups from the experiment that contain missing values for quantitation, using the “filterNA()” function. Alternatively, allow a certain number of missing values for quantitation and impute them in some way.
Note: The missing values may be set to zero, as has been performed previously (Christoforou et al., 2016) or they may be imputed using one of the “impute()” methods that are described in the MSnbase package (Gatto, and Lilley, 2012). - Normalize the quantitation data for each row (i.e., protein group) such that the summed ratios are equal to 1. Use the “normalize ()” function and the “sum” method.
Note: Other normalization methods are available within the MSnbase package, which may be used to normalize the Proteome Discoverer quantitation data but we find the “sum” method useful for this particular Data analysis. - Observe the data using dimensionality reduction such as Principal Components Analysis (PCA) or t-Distributed Stochastic Neighbor Embedding (t-SNE) (Van Der Maaten and Hinton, 2008) plots in the absence of any annotation. Note any cluster resolution that is evident from the data.
- Curate a list of marker proteins that are known to localize to subcellular organelles in S. cerevisiae. Alternatively, use the markers that are provided within the pRoloc package (using pRolocmarkers(“scer”)).
Note: Ensure that marker annotation is included for all organelles that are expected to resolve discretely within the experiment. When performing classification of proteins to organelle classes using supervised machine learning approaches, proteins can only be classified to one of the organelle classes that was defined prior to starting the analysis. Therefore, if annotation is omitted for a specific organelle or set of organelles, it will result in proteins from that organelle or set of organelles being subsumed into another organelle that resolved similarly to the organelle that has been omitted within the experiment. The net result will be not only lack of the organelle in question in the final dataset, but also failure to classify any proteins to that organelle. - Append the marker annotation to the MSnSet and observe the resolution within the data at the level of subcellular organelles.
- Optionally, perform automated protein marker annotation using the “addGOAnnotation()” function within pRoloc, in which the user can add Gene Ontology (GO) (Ashburner et al., 2000; The Gene Ontology Consortium, 2017) annotation from any GO name space to search for clustering of proteins based on common molecular function, biological process, or cellular component. The resolution of GO annotated clusters that resolve discretely after this analysis can be observed using the pRolocGUI package. This can further be used to augment the marker protein data used for classification of proteins of unknown localization to subcellular organelles.
- Perform protein classification using a binary classifier to classify proteins of unknown localization to one of the pre-defined organelle classes. This may encompass use of a supervised machine learning method, such as a Support Vector Machine (SVM) (Vapnik, 1995) as has been widely used in multiple previous hyperLOPIT studies (Christoforou et al., 2016; Thul et al., 2017). Newer methods may instead be employed which include a semi-supervised machine learning method (“phenoDisco”) (Breckels et al., 2013), which can predict presence of novel protein clusters without a priori knowledge of their existence. A variant method is available that uses additional, lower quality, auxiliary data in addition to the highly curated marker protein list to improve classification performance (Breckels et al., 2016a). Alternatively, a new probabilistic classification method may be used, which is based on Bayesian statistics (Crook et al., 2018) and is now available for use within the pRoloc package.
- Observe resolution of all proteins after classification to organelles.
- Export all classification data on an organelle-by-organelle basis and sort SVM scores in descending order for all proteins classified as belonging to a given organelle.
- Consult the original literature to look for corroboration between protein localizations predicted by hyperLOPIT and low throughput studies. Allow a certain permitted percentage of false discoveries for protein localization to a given organelle, based on the literature (e.g., 5% or 1%).
Note: Due to the nature of such machine-learning methods, all proteins in the dataset will be classified to a single location that was pre-defined in the organelle marker list training data. This does not faithfully recapitulate what happens in vivo, where proteins can localize to multiple subcellular organelles or traffic between multiple organelles. - Set the SVM score cut-off for each organelle at this threshold and use the resultant data as the final spatial proteome map. See Figure 2 for representative data from a single biological replicate performed in reference (Nightingale et al., 2019), without any protein classification. Further, see the Bioconductor (Gentleman et al., 2004) pRolocdata (Gatto et al., 2014) package (version 1.19.4 onwards) and https://proteome.shinyapps.io/yeast2018/ (Nightingale et al., 2019) for a fully processed dataset comprising data from four biological replicate experiments, performed with and without optional nuclear preparations, that can be interactively explored.
- Finally, optionally validate the localizations of a subset of proteins that have been predicted by the hyperLOPIT analysis in a low throughput manner and using an orthogonal method. This may be performed using fluorescence microscopy, affinity purification-mass spectrometry, subtractive proteomics, proximity labeling, or density gradient protein co-fractionation experiments.
Notes
- Prepare this solution by mixing iodixanol working solution (IWS) with 1x LB. To increase the OptiPrep concentration and refractive index, add IWS. To decrease, add 1x LB.
- Do not over-dry pellets as they will become very difficult to resolubilize in subsequent steps. Drying for 5 min at room temperature should enable most of the acetone to evaporate. If the protein pellet begins to change from white to colorless, it is a sign that it is starting to over-dry.
- To minimize the amount of protein used for Western blotting, run as few gels as possible. Cut membranes, post-blotting, into strips of appropriate molecular weight range based on their reported molecular weight and probe each strip with a single antibody. We typically find that we can probe for all of the markers in Table 1 running only two SDS-PAGE gels.
- Primary antibodies diluted in blocking solution may be stored at -20 °C after use and reused at least 5 times, with no appreciable loss of signal. Do not re-use horseradish peroxidase-conjugated secondary antibodies.
- If it is necessary to obtain high enough protein yield, the protein contents of adjacent fractions can be pooled prior to labeling. If pooling is performed multiple times within the same gradient, however, it will diminish the resolving ability of the density gradient, meaning that overall organelle resolution within the experiment will be impacted.
Recipes
- Cell culture
- 10x yeast nitrogen base (YNB)
Dissolve 6.7 g yeast nitrogen base without amino acids in 1 L water and filter-sterilize - 10x complete supplement mixture
0.03% (w/v) isoleucine
0.15% (w/v) valine
0.04% (w/v) adenine hemisulfate
0.02% (w/v) arginine
0.02% (w/v) histidine
0.1% (w/v) leucine
0.03% (w/v) lysine
0.02% (w/v) methionine
0.05% (w/v) phenylalanine
0.2% (w/v) threonine
0.04% (w/v) tryptophan
0.03% (w/v) tyrosine
0.02% (w/v) uracil
Dissolve all components in water and filter sterilize - 40% (w/v) glucose
Dissolve 40 g glucose in 100 ml water and sterilize by autoclaving - Synthetic medium with 2% glucose (1.2 L per experiment)
Mix 120 ml 10x YNB, 60 ml 40% (w/v) glucose and 120 ml 10x complete supplement mixture
Make up to 1.2 L with sterile water - YPD agar medium
- Suspend 4 g Bacto agar, 4 g Bacto peptone and 2 g Bacto yeast extract in 190 ml water
- Sterilize by autoclaving
- Whilst still molten, add 10 ml 40% (w/v) glucose and pour agar plates (approximately 20 ml per plate)
- 10x yeast nitrogen base (YNB)
- Cell pre-treatment and spheroplast generation
- TCEP reduction buffer (150 ml per experiment)
20 mM Tris-HCl, pH 7.5
10 mM Tris-(2-carboxyethyl) phosphine (TCEP) - Spheroplasting medium (40 ml per experiment)
Composed of the same constituents as synthetic medium, but with the addition of 20 mM Tris-HCl, pH 7.5 and 1.2 M sorbitol
Freshly supplement with 1 Complete EDTA-free protease inhibitor tablet per 50 ml buffer - Zymolyase 100-T solution (700 μl per experiment)
5 μg/ml zymolyase 100-T prepared in spheroplasting medium
Prepare 1 μl for every OD unit of yeast used in the experiment - Spheroplast wash medium (150 ml per experiment)
Composed of the same constituents as spheroplasting medium, with the omission of the Tris-HCl buffer
Freshly supplement with 1 Complete EDTA-free protease inhibitor tablet per 50 ml buffer
- TCEP reduction buffer (150 ml per experiment)
- (Optional) Nuclear preparation
- Ficoll lysis buffer (Kizer et al., 2006) (15 ml per experiment)
18% (w/v) Ficoll PM-400
20 mM dibasic potassium phosphate, pH 6.8
1 mM magnesium chloride
0.5 mM EDTA
Freshly supplement with 1 Complete Mini EDTA-free protease inhibitor tablet per 10 ml buffer - Buffer NP (Kizer et al., 2006) (300 μl per experiment)
340 mM sucrose
20 mM Tris-HCl, pH 7.4
50 mM potassium chloride
5 mM magnesium chloride
Freshly supplement with 1 Complete Mini EDTA-free protease inhibitor tablet per 10 ml buffer
- Ficoll lysis buffer (Kizer et al., 2006) (15 ml per experiment)
- Cell lysis
- 1x LB (75 ml per experiment)
250 mM sucrose
50 mM potassium acetate
2 mM magnesium acetate
1 mM EDTA, pH 8.0
Freshly supplement with 1 Complete EDTA-free protease inhibitor tablet per 50 ml buffer - 6x LB (4 ml per experiment)
300 mM potassium acetate
12 mM magnesium acetate
6 mM EDTA, pH 8.0
- 1x LB (75 ml per experiment)
- Density gradient centrifugation
- Iodixanol working solution (IWS) (18 ml per experiment)
Mix 3 ml 6x LB with 15 ml OptiPrep - 18% (w/v) OptiPrep solution (25 ml per experiment)
- Mix 9 ml IWS with 16 ml 1x LB
- Double-check refractive index against a standard curve of OptiPrep concentrations of known refractive index
- Adjust if needed, using IWS to increase refractive index and 1 x LB to reduce refractive index
- 16% OptiPrep solution (20 ml per experiment)
- Mix 6.4 ml IWS with 13.6 ml 1x LB
- Double check refractive index against a standard curve of OptiPrep concentrations of known refractive index
- Adjust if needed, using IWS to increase refractive index and 1x LB to reduce refractive index
- Iodixanol working solution (IWS) (18 ml per experiment)
- Fraction processing, digestion and TMT labeling
- Protein resolubilization buffer (20 ml per experiment)
100 mM HEPES-NaOH, pH 8.5
0.1% (w/v) SDS - Digestion buffer (2 ml per experiment)
100 mM HEPES-NaOH, pH 8.5 - 5% (v/v) hydroxylamine (120 μl per experiment)
Mix 108 μl of digestion buffer with 12 μl 50% (w/v) hydroxylamine solution
Prepare just before use - TTBS (1 L per experiment)
25 mM Tris-HCl, pH 7.6
150 mM NaCl
0.1% (v/v) Tween-20 - Blocking solution (100 ml per antibody used)
5% (w/v) non-fat milk powder, prepared in TTBS - Dithiothreitol (DTT) solution
100 mM DL-dithiothreitol, prepared in protein resolubilization buffer - Iodoacetamide (IAA) solution
250 mM iodoacetamide, prepared in protein resolubilization buffer
- Protein resolubilization buffer (20 ml per experiment)
- Sample clean-up by solid phase extraction (SPE)
- Equilibration buffer (600 μl per experiment)
50% (v/v) acetonitrile
0.5% (v/v) acetic acid - Desalting buffer 1 (1.8 ml per experiment)
0.1% (v/v) trifluoroacetic acid - Desalting buffer 2 (180 μl per experiment)
0.5% (v/v) acetic acid - Elution buffer (1 ml per experiment)
80% (v/v) acetonitrile
0.5% (v/v) acetic acid
- Equilibration buffer (600 μl per experiment)
- High pH reversed-phase chromatography
- RP mobile phase stock solution (100 ml per experiment)
200 mM ammonium formate, pH 10.0. Adjust pH with ammonium hydroxide. - RP mobile phase A (500 ml per experiment)
20 mM ammonium formate, pH 10.0
Prepare by mixing 50 ml RP mobile phase stock solution with 450 ml HPLC-grade water
Re-adjust to pH 10.0 with ammonium hydroxide, if necessary - RP mobile phase B (500 ml per experiment)
20 mM ammonium formate, pH 10.0
80% (v/v) acetonitrile
Prepare by mixing 50 ml RP mobile phase stock solution with 400 ml HPLC-grade ACN and 50 ml HPLC-grade water
Do not adjust pH - RP resolubilization buffer (100 μl per experiment)
95% (v/v) RP mobile phase A
5% (v/v) RP mobile phase B
- RP mobile phase stock solution (100 ml per experiment)
Acknowledgments
We acknowledge funding from the BBSRC (Strategic Longer and Larger grant awarded to KSL (award BB/L002817/1) and CASE studentship awarded to KSL and SGO (award BB/I016147/1)). We thank Owen Vennard for critically reading this protocol.
Competing interests
There are no conflicts of interest or competing interest.
References
- Alepuz, P. M., de Nadal, E., Zapater, M., Ammerer, G. and Posas, F. (2003). Osmostress-induced transcription by Hot1 depends on a Hog1-mediated recruitment of the RNA Pol II. EMBO J 22(10): 2433-2442.
- Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., Harris, M. A., Hill, D. P., Issel-Tarver, L., Kasarskis, A., Lewis, S., Matese, J. C., Richardson, J. E., Ringwald, M., Rubin, G. M. and Sherlock, G. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25(1): 25-29.
- Brachmann, C. B., Davies, A., Cost, G. J., Caputo, E., Li, J., Hieter, P. and Boeke, J. D. (1998). Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14(2): 115-132.
- Breckels, L. M., Holden, S. B., Wojnar, D., Mulvey, C. M., Christoforou, A., Groen, A., Trotter, M. W., Kohlbacher, O., Lilley, K. S. and Gatto, L. (2016a). Learning from heterogeneous data sources: an application in spatial proteomics. PLoS Comput Biol 12(5): e1004920.
- Breckels, L. M., Mulvey, C. M., Lilley, K. S. and Gatto, L. (2016b). A Bioconductor workflow for processing and analysing spatial proteomics data. F1000Res 5: 2926.
- Breckels, L. M., Naake, T. and Gatto L. (2018). Interactive visualisation of spatial proteomics data. R package version 1.14.0.
- Breckels, L. M., Gatto, L., Christoforou, A., Groen, A. J., Lilley, K. S. and Trotter, M. W. (2013). The effect of organelle discovery upon sub-cellular protein localisation. J Proteomics 88: 129-140.
- Breker, M., Gymrek, M. and Schuldiner, M. (2013). A novel single-cell screening platform reveals proteome plasticity during yeast stress responses. J Cell Biol 200(6): 839-850.
- Brosch, M., Yu, L., Hubbard, T. and Choudhary, J. (2009). Accurate and sensitive peptide identification with Mascot Percolator. J Proteome Res 8(6): 3176-3181.
- Cherry, J. M., Hong, E. L., Amundsen, C., Balakrishnan, R., Binkley, G., Chan, E. T., Christie, K. R., Costanzo, M. C., Dwight, S. S., Engel, S. R., Fisk, D. G., Hirschman, J. E., Hitz, B. C., Karra, K., Krieger, C. J., Miyasato, S. R., Nash, R. S., Park, J., Skrzypek, M. S., Simison, M., Weng, S. and Wong, E. D. (2012). Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res 40(Database issue): D700-705.
- Chen, X. J., Wang, X., Kaufman, B. A. and Butow, R. A. (2005). Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science 307(5710): 714-717.
- Chong, Y. T., Koh, J. L., Friesen, H., Duffy, S. K., Cox, M. J., Moses, A., Moffat, J., Boone, C. and Andrews, B. J. (2015). Yeast proteome dynamics from single cell imaging and automated analysis. Cell 161(6): 1413-1424.
- Christoforou, A., Mulvey, C. M., Breckels, L. M., Geladaki, A., Hurrell, T., Hayward, P. C., Naake, T., Gatto, L., Viner, R., Martinez Arias, A. and Lilley, K. S. (2016). A draft map of the mouse pluripotent stem cell spatial proteome. Nat Commun 7: 8992.
- Crook, O. M., Mulvey, C. M., Kirk, P. D. W., Lilley, K. S. and Gatto, L. (2018). A Bayesian mixture modelling approach for spatial proteomics. PLoS Comput Biol 14(11): e1006516.
- Denervaud, N., Becker, J., Delgado-Gonzalo, R., Damay, P., Rajkumar, A. S., Unser, M., Shore, D., Naef, F. and Maerkl, S. J. (2013). A chemostat array enables the spatio-temporal analysis of the yeast proteome. Proc Natl Acad Sci U S A 110(39): 15842-15847.
- de Duve, C. (1971). Tissue fraction-past and present. J Cell Biol 50(1): 20.
- Gancedo, C., Flores, C. L. and Gancedo, J. M. (2016). The expanding landscape of moonlighting proteins in yeasts. Microbiol Mol Biol Rev 80(3): 765-777.
- Gatto, L. and Lilley, K. S. (2012). MSnbase-an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 28(2): 288-289.
- Gatto, L., Breckels, L. M., Wieczorek, S., Burger, T. and Lilley, K. S. (2014). Mass-spectrometry-based spatial proteomics data analysis using pRoloc and pRolocdata. Bioinformatics 30(9): 1322-1324.
- Gatto, L., Breckels, L. M. and Lilley, K. S. (2019). Assessing sub-cellular resolution in spatial proteomics experiments. Curr Opin Chem Biol 48: 123-149.
- Groen, A. J., Sancho-Andres, G., Breckels, L. M., Gatto, L., Aniento, F. and Lilley, K. S. (2014). Identification of trans-golgi network proteins in Arabidopsis thaliana root tissue. J Proteome Res 13(2): 763-776.
- Gentleman, R. C., Carey, V. J., Bates, D. M., Bolstad, B., Dettling, M., Dudoit, S., Ellis, B., Gautier, L., Ge, Y., Gentry, J., Hornik, K., Hothorn, T., Huber, W., Iacus, S., Irizarry, R., Leisch, F., Li, C., Maechler, M., Rossini, A. J., Sawitzki, G., Smith, C., Smyth, G., Tierney, L., Yang, J. Y. and Zhang, J. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5(10): R80.
- Geladaki, A., Kocevar Britovsek, N., Breckels, L. M., Smith, T. S., Vennard, O. L., Mulvey, C. M., Crook, O. M., Gatto, L. and Lilley, K. S. (2019). Combining LOPIT with differential ultracentrifugation for high-resolution spatial proteomics. Nat Commun 10(1): 331.
- Hope, I. A. and Struhl, K. (1986). Functional dissection of a eukaryotic transcriptional activator protein, GCN4 of yeast. Cell 46(6): 885-894.
- Huh, W. K., Falvo, J. V., Gerke, L. C., Carroll, A. S., Howson, R. W., Weissman, J. S. and O'Shea, E. K. (2003). Global analysis of protein localization in budding yeast. Nature 425(6959): 686-691.
- Hung, V., Lam, S. S., Udeshi, N. D., Svinkina, T., Guzman, G., Mootha, V. K., Carr, S. A. and Ting, A. Y. (2017). Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 6: e24463.
- Hung, V., Zou, P., Rhee, H. W., Udeshi, N. D., Cracan, V., Svinkina, T., Carr, S. A., Mootha, V. K. and Ting, A. Y. (2014). Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol Cell 55(2): 332-341.
- Hunter, M. J. and Commerford, S. L. (1961). Pressure homogenization of mammalian tissues. Biochim Biophys Acta 47: 580-586.
- Hwang, J. and Espenshade, P. J. (2016). Proximity-dependent biotin labelling in yeast using the engineered ascorbate peroxidase APEX2. Biochem J 473(16): 2463-2469.
- Itzhak, D. N., Tyanova, S., Cox, J. and Borner, G. H. (2016). Global, quantitative and dynamic mapping of protein subcellular localization. Elife 5: e16950.
- Jadot, M., Boonen, M., Thirion, J., Wang, N., Xing, J., Zhao, C., Tannous, A., Qian, M., Zheng, H., Everett, J. K., Moore, D. F., Sleat, D. E. and Lobel, P. (2017). Accounting for protein subcellular localization: A compartmental map of the rat liver proteome. Mol Cell Proteomics 16(2): 194-212.
- Jean Beltran, P. M., Mathias, R. A. and Cristea, I. M. (2016). A portrait of the human organelle proteome in space and time during cytomegalovirus infection. Cell Syst 3(4): 361-373 e366.
- Kim, D. I., Birendra, K. C., Zhu, W., Motamedchaboki, K., Doye, V. and Roux, K. J. (2014). Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc Natl Acad Sci U S A 111(24): E2453-2461.
- Kizer, K. O., Xiao, T. and Strahl, B. D. (2006). Accelerated nuclei preparation and methods for analysis of histone modifications in yeast. Methods 40(4): 296-302.
- Lam, S. S., Martell, J. D., Kamer, K. J., Deerinck, T. J., Ellisman, M. H., Mootha, V. K. and Ting, A. Y. (2015). Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 12(1): 51-54.
- McAlister, G. C., Huttlin, E. L., Haas, W., Ting, L., Jedrychowski, M. P., Rogers, J. C., Kuhn, K., Pike, I., Grothe, R. A., Blethrow, J. D. and Gygi, S. P. (2012). Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal Chem 84(17): 7469-7478.
- McAlister, G. C., Nusinow, D. P., Jedrychowski, M. P., Wuhr, M., Huttlin, E. L., Erickson, B. K., Rad, R., Haas, W. and Gygi, S. P. (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 86(14): 7150-7158.
- Morgenstern, M., Stiller, S. B., Lubbert, P., Peikert, C. D., Dannenmaier, S., Drepper, F., Weill, U., Hoss, P., Feuerstein, R., Gebert, M., Bohnert, M., van der Laan, M., Schuldiner, M., Schutze, C., Oeljeklaus, S., Pfanner, N., Wiedemann, N. and Warscheid, B. (2017). Definition of a high-confidence mitochondrial proteome at quantitative scale. Cell Rep 19(13): 2836-2852.
- Mulvey, C. M., Breckels, L. M., Geladaki, A., Britovsek, N. K., Nightingale, D. J. H., Christoforou, A., Elzek, M., Deery, M. J., Gatto, L. and Lilley, K. S. (2017). Using hyperLOPIT to perform high-resolution mapping of the spatial proteome. Nat Protoc 12(6): 1110-1135.
- Nikolaev, Y., Deillon, C., Hoffmann, S. R., Bigler, L., Friess, S., Zenobi, R., Pervushin, K., Hunziker, P. and Gutte, B. (2010). The leucine zipper domains of the transcription factors GCN4 and c-Jun have ribonuclease activity. PLoS One 5(5): e10765.
- Nightingale, D. J., Geladaki, A., Breckels, L. M., Oliver, S. G. and Lilley, K. S. (2019). The subcellular organisation of Saccharomyces cerevisiae. Curr Opin Chem Biol 48: 86-95.
- Nikolovski, N., Rubtsov, D., Segura, M. P., Miles, G. P., Stevens, T. J., Dunkley, T. P., Munro, S., Lilley, K. S. and Dupree, P. (2012). Putative glycosyltransferases and other plant Golgi apparatus proteins are revealed by LOPIT proteomics. Plant Physiol 160(2): 1037-1051.
- Nightingale, D. J., Oliver, S. G. and Lilley, K. S. (2018). Mapping the Saccharomyces cerevisiae spatial proteome with high resolution using hyperLOPIT. In: Methods in Molecular Biology. In: Oliver. S. G. (Ed.). Humana Press.
- Opitz, N., Schmitt, K., Hofer-Pretz, V., Neumann, B., Krebber, H., Braus, G. H. and Valerius, O. (2017). Capturing the Asc1p/Receptor for Activated C Kinase 1 (RACK1) Microenvironment at the head region of the 40S ribosome with quantitative BioID in yeast. Mol Cell Proteomics 16(12): 2199-2218.
- Stadler, C., Rexhepaj, E., Singan, V. R., Murphy, R. F., Pepperkok, R., Uhlen, M., Simpson, J. C. and Lundberg, E. (2013). Immunofluorescence and fluorescent-protein tagging show high correlation for protein localization in mammalian cells. Nat Methods 10(4): 315-323.
- Sickmann, A., Reinders, J., Wagner, Y., Joppich, C., Zahedi, R., Meyer, H. E., Schonfisch, B., Perschil, I., Chacinska, A., Guiard, B., Rehling, P., Pfanner, N. and Meisinger, C. (2003). The proteome of Saccharomyces cerevisiae mitochondria. Proc Natl Acad Sci U S A 100(23): 13207-13212.
- Reinders, J., Zahedi, R. P., Pfanner, N., Meisinger, C. and Sickmann, A. (2006). Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J Proteome Res 5(7): 1543-1554.
- Rhee, H. W., Zou, P., Udeshi, N. D., Martell, J. D., Mootha, V. K., Carr, S. A. and Ting, A. Y. (2013). Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339(6125): 1328-1331.
- Roux, K. J., Kim, D. I., Raida, M. and Burke, B. (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196(6): 801-810.
- Ross, P. L., Huang, Y. N., Marchese, J. N., Williamson, B., Parker, K., Hattan, S., Khainovski, N., Pillai, S., Dey, S., Daniels, S., Purkayastha, S., Juhasz, P., Martin, S., Bartlet-Jones, M., He, F., Jacobson, A. and Pappin, D. J. (2004). Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 3(12): 1154-1169.
- R Core Team. (2018). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
- RStudio Team. (2016). RStudio: Integrated Development Environment for R.
- Sadowski, P. G., Dunkley, T. P., Shadforth, I. P., Dupree, P., Bessant, C., Griffin, J. L. and Lilley, K. S. (2006). Quantitative proteomic approach to study subcellular localization of membrane proteins. Nat Protoc 1(4): 1778-1789.
- Simpson, R. J. (2010). Disruption of cultured cells by nitrogen cavitation. Cold Spring Harb Protoc 2010(11): pdb prot5513.
- Thompson, A., Schafer, J., Kuhn, K., Kienle, S., Schwarz, J., Schmidt, G., Neumann, T., Johnstone, R., Mohammed, A. K. and Hamon, C. (2003). Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 75(8): 1895-1904.
- Thul, P. J., Akesson, L., Wiking, M., Mahdessian, D., Geladaki, A., Ait Blal, H., Alm, T., Asplund, A., Bjork, L., Breckels, L. M., Backstrom, A., Danielsson, F., Fagerberg, L., Fall, J., Gatto, L., Gnann, C., Hober, S., Hjelmare, M., Johansson, F., Lee, S., Lindskog, C., Mulder, J., Mulvey, C. M., Nilsson, P., Oksvold, P., Rockberg, J., Schutten, R., Schwenk, J. M., Sivertsson, A., Sjostedt, E., Skogs, M., Stadler, C., Sullivan, D. P., Tegel, H., Winsnes, C., Zhang, C., Zwahlen, M., Mardinoglu, A., Ponten, F., von Feilitzen, K., Lilley, K. S., Uhlen, M. and Lundberg, E. (2017). A subcellular map of the human proteome. Science 356(6340).
- The Gene Ontology Consortium. (2017). Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res 45(D1): D331-D338.
- Ting, L., Rad, R., Gygi, S. P. and Haas, W. (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods 8(11): 937-940.
- Tkach, J. M., Yimit, A., Lee, A. Y., Riffle, M., Costanzo, M., Jaschob, D., Hendry, J. A., Ou, J., Moffat, J., Boone, C., Davis, T. N., Nislow, C. and Brown, G. W. (2012). Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat Cell Biol 14(9): 966-976.
- Van Der Maaten, L. and Hinton, G. (2008). Visualizing Data using t-SNE. J Mach Learn Res 9: 2579-2605.
- Vapnik, V. (1995). The nature of statistical learning theory. Springer.
- Villen, J. and Gygi, S. P. (2008). The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat Protoc 3(10): 1630-1638.
- Vogtle, F. N., Burkhart, J. M., Rao, S., Gerbeth, C., Hinrichs, J., Martinou, J. C., Chacinska, A., Sickmann, A., Zahedi, R. P. and Meisinger, C. (2012). Intermembrane space proteome of yeast mitochondria. Mol Cell Proteomics 11(12): 1840-1852.
- Vogtle, F. N., Burkhart, J. M., Gonczarowska-Jorge, H., Kucukkose, C., Taskin, A. A., Kopczynski, D., Ahrends, R., Mossmann, D., Sickmann, A., Zahedi, R. P. and Meisinger, C. (2017). Landscape of submitochondrial protein distribution. Nat Commun 8(1): 290.
- Wang, Y., Lilley, K. S. and Oliver, S. G. (2014). A protocol for the subcellular fractionation of Saccharomyces cerevisiae using nitrogen cavitation and density gradient centrifugation. Yeast 31(4): 127-135.
- Weill, U., Yofe, I., Sass, E., Stynen, B., Davidi, D., Natarajan, J., Ben-Menachem, R., Avihou, Z., Goldman, O., Harpaz, N., Chuartzman, S., Kniazev, K., Knoblach, B., Laborenz, J., Boos, F., Kowarzyk, J., Ben-Dor, S., Zalckvar, E., Herrmann, J. M., Rachubinski, R. A., Pines, O., Rapaport, D., Michnick, S. W., Levy, E. D. and Schuldiner, M. (2018). Genome-wide SWAp-Tag yeast libraries for proteome exploration. Nat Methods 15(8): 617-622.
- Wiederhold, E., Veenhoff, L. M., Poolman, B. and Slotboom, D. J. (2010). Proteomics of Saccharomyces cerevisiae organelles. Mol Cell Proteomics 9(3): 431-445.
- Wenger, C. D., Lee, M. V., Hebert, A. S., McAlister, G. C., Phanstiel, D. H., Westphall, M. S. and Coon, J. J. (2011). Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat Methods 8(11): 933-935.
- Yofe, I., Weill, U., Meurer, M., Chuartzman, S., Zalckvar, E., Goldman, O., Ben-Dor, S., Schutze, C., Wiedemann, N., Knop, M., Khmelinskii, A. and Schuldiner, M. (2016). One library to make them all: streamlining the creation of yeast libraries via a SWAp-Tag strategy. Nat Methods 13(4): 371-378.
- Zahedi, R. P., Sickmann, A., Boehm, A. M., Winkler, C., Zufall, N., Schonfisch, B., Guiard, B., Pfanner, N. and Meisinger, C. (2006). Proteomic analysis of the yeast mitochondrial outer membrane reveals accumulation of a subclass of preproteins. Mol Biol Cell 17(3): 1436-1450.
- Zhang, R., Sioma, C. S., Wang, S. and Regnier, F. E. (2001). Fractionation of isotopically labeled peptides in quantitative proteomics. Anal Chem 73(21): 5142-5149.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Nightingale, D. J. H., Lilley, K. S. and Oliver, S. G. (2019). A Protocol to Map the Spatial Proteome Using HyperLOPIT in Saccharomyces cerevisiae. Bio-protocol 9(14): e3303. DOI: 10.21769/BioProtoc.3303.
Category
Microbiology > Microbial proteomics > Whole organism
Systems Biology > Proteomics > Spatial Proteomics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.