- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Dynamic and Sequential Protein Reconstitution on Negatively Curved Membranes by Giant Vesicles Fusion

Published: Vol 9, Iss 13, Jul 5, 2019 DOI: 10.21769/BioProtoc.3294 Views: 5221
Reviewed by: Samantha E. R. DundonAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
In vitro investigation of the interaction between proteins and positively curved membranes can be performed using a classic nanotube pulling method. However, characterizing protein interaction with negatively curved membranes still represents a formidable challenge. Here, we describe our recently developed approach based on laser-triggered Giant Unilamellar Vesicles (GUVs) fusion. Our protocol allows sequential addition of proteins to a negatively curved membrane, while at the same time controlling the buffer composition, lipid composition and membrane tension. Moreover, this method does not require a step of protein detachment, greatly simplifying the process of protein encapsulation over existing methods.
Keywords: GUVBackground
Cellular membrane remodeling processes including vesicle trafficking can be topologically split into either outside-in budding such as endocytosis and inside-out budding such as intraluminal vesicle formation at early endosomes, enveloped virus budding and cytokinesis. A budding event induces changes in membrane shape, resulting in two distinct topologies: a convex surface or a concave surface, conventionally defined as positive and negative curvature, respectively (Figure 1).
In vitro methods for studying outside-in budding processes based on membrane nanotube pulling such as the interaction of proteins with positively curved membranes during endocytosis are well-established (Sorre et al., 2012; Prévost et al., 2017). In contrast, investigating protein interaction with negatively curved membranes is much more challenging because it is in general not straightforward to obtain such a topology–i.e., to incorporate proteins inside a membrane nanotube pulled from a GUV in a classic tube-pulling experiment. Indeed, the membrane has to be deformed away from the compartment where proteins are present (Figure 1). Encapsulating the protein of interest inside the GUV can in principle be achieved during GUV swelling, and by pulling out a nanotube afterwards (Prévost et al., 2015). This methodology, however, requires the protein to be detached from the Outer leaflet of the GUV, which can only be performed if the binding is reversible, and the procedure has to be adjusted for each individual protein. Moreover, when a protein is encapsulated, it also interacts with the membrane during swelling, potentially hampering the process of swelling itself. Another limitation of this protocol is that sequential addition of proteins (or lipids) is not possible.
An alternative method consists in pulling a nanotube inside the GUV, and adding the protein from the outside by micropipette injection (Dasgupta and Dimova, 2014). But this method requires very flaccid GUVs, a combination of flow and optical tweezers to form the tube, and is not suitable when the tube diameter has to be controlled down to 15-20 nm.
Here we describe a protocol to investigate the interaction of proteins with negatively curved membranes and membrane necks (De Franceschi et al., 2018). We apply this method to study the endosomal sorting complex required for transport (ESCRT) machinery, which catalyzes inside-out membrane budding processes by polymerizing on negatively curved membranes that carry a negative charge (Henne et al., 2013; McCullough et al., 2018; Caillat et al., 2019). Characterizing the ESCRT-III complex is particularly challenging, because these proteins exhibit strong binding to the membrane, so that their removal from the Outer membrane of a GUV after the encapsulation process is impossible (De Franceschi et al., 2018), making other methods (Prévost et al., 2015) unsuitable. Our method relies on laser-triggered GUV fusion (Rørvig-Lund et al., 2015), and presents a number of advantages over existing methods. First, protein encapsulation is performed in conditions that inhibit protein binding to the membrane, avoiding inhibition of GUV swelling and protein/lipids aggregation. Thus, the protein can be encapsulated using a buffer that keeps it stable and avoids unwanted in-bulk polymerization. However, upon fusion with a protein-free GUV, optimal binding conditions, including lipid species and salt concentration, can be achieved. Importantly, GUV fusion is triggered after a membrane nanotube has been pulled, therefore allowing visualizing dynamic interaction of proteins with a negatively-curved membrane topology. Moreover, by performing multiple fusion events, additional proteins can be added sequentially. Similarly, the lipid composition can be modulated in a controlled fashion by sequential fusion events.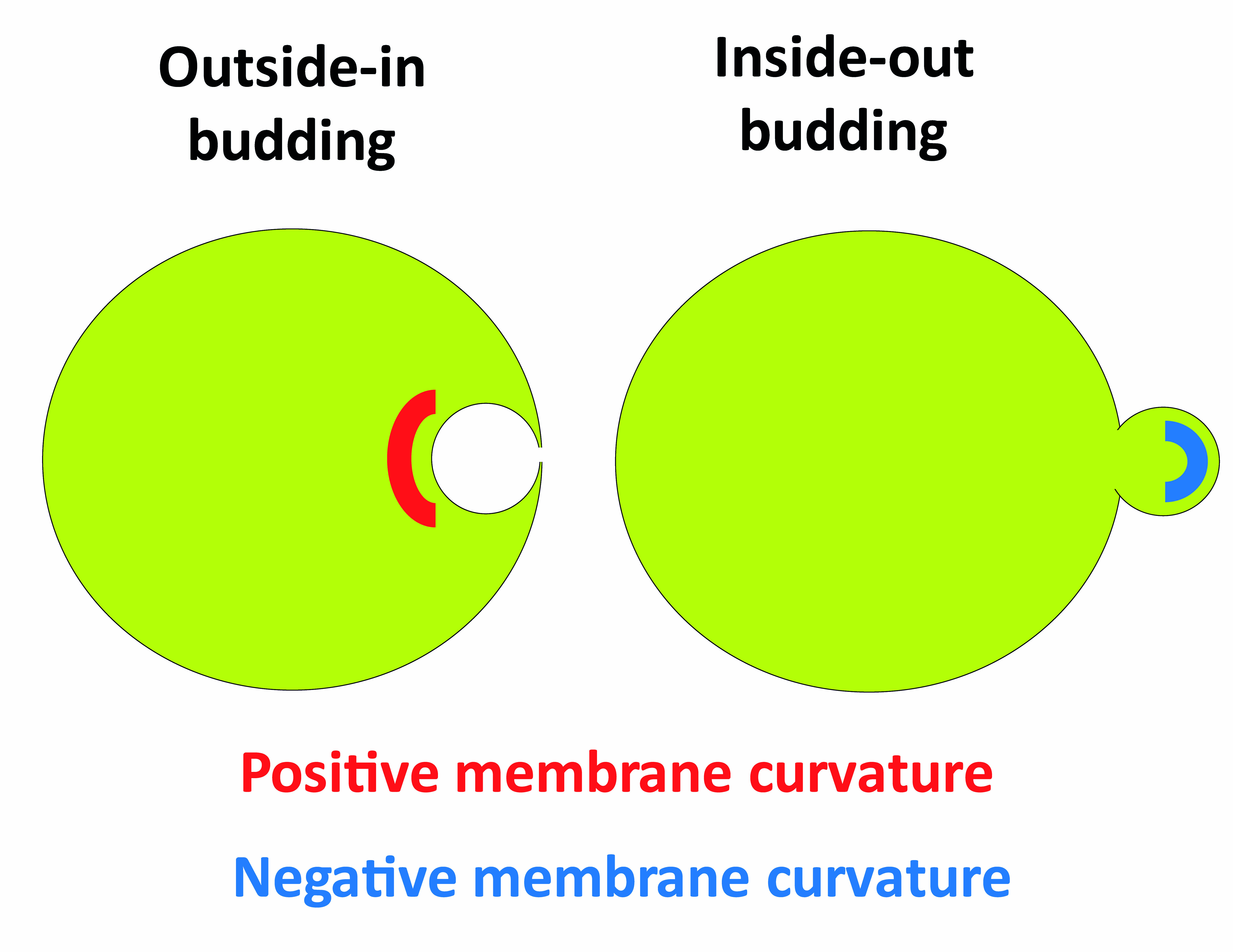
Figure 1. Schematic representing outside-in and inside-out budding events, and the membrane curvatures created during these processes
Method outline: The principle in this methodology is to have proteins inside one neutral GUV (green in Figure 2) and a second protein-free GUV made of negatively charged lipids (magenta in Figure 2) until GUV fusion is triggered. A tube is pulled beforehand from the negatively charged GUV (magenta), so that upon GUV fusion negative membrane curvature becomes accessible to the proteins. Therefore, the lipid mix and buffer in the green GUV are designed to maintain proteins' stability and allow their encapsulation avoiding premature binding and aggregation. At the same time, the final lipid and buffer composition resulting from GUV fusion must create the appropriate conditions for protein-membrane interaction.
We exemplify this concept by explaining the design of buffers and lipid mixes used for investigating binding between the ESCRT-III protein CHMP2B and PI(4,5)P2 (De Franceschi et al., 2018).
CHMP2B is stable at relatively high salt concentration. Lowering NaCl concentration to 50 mM in the presence of negatively charged lipids such as PI(4,5)P2 triggers polymerization and membrane binding. Therefore, CHMP2B is encapsulated in Buffer E containing 70 mM NaCl using Lipid Mix E, which does not contain any negatively charged lipids. In these conditions, CHMP2B does not interact with the membrane. Therefore, the proteins present outside the GUVs can be easily washed away by dilution (Figure 2).
The second population of GUVs contains PI(4,5)P2 in a lipid mix that mimics the composition of the inner leaflet of the plasma membrane (Prévost et al., 2015), Here, the buffer P has a low NaCl concentration, so that, upon GUV fusion, it will mix with buffer E resulting in optimal NaCl concentration to promote CHMP2B:PI(4,5)P2 interaction.
A further constrain in the buffers' composition is that the inner buffers (E and P) must be denser than the Outer buffer, to allow the GUVs to sediment both during centrifugation and in the observation chamber (Mathivet et al., 1996). Moreover, all buffers must have the same osmolarity. By using a combination of sucrose, glucose and NaCl, these conditions can be achieved (see Recipes section).
The proteins present outside the GUVs are removed by sequential dilution, and at the same time streptavidin-coated nanorods that interact with biotinylated lipids, are added (Figure 2). Once the GUVs are properly positioned, irradiation of the nanorods at the GUVs' interface will induce a strong local heating, in turn causing membrane instability and thus fusion (Rørvig-Lund et al., 2015). We improved the efficiency of this method by directly tethering the nanorods to the membrane via streptavidin:biotin interaction.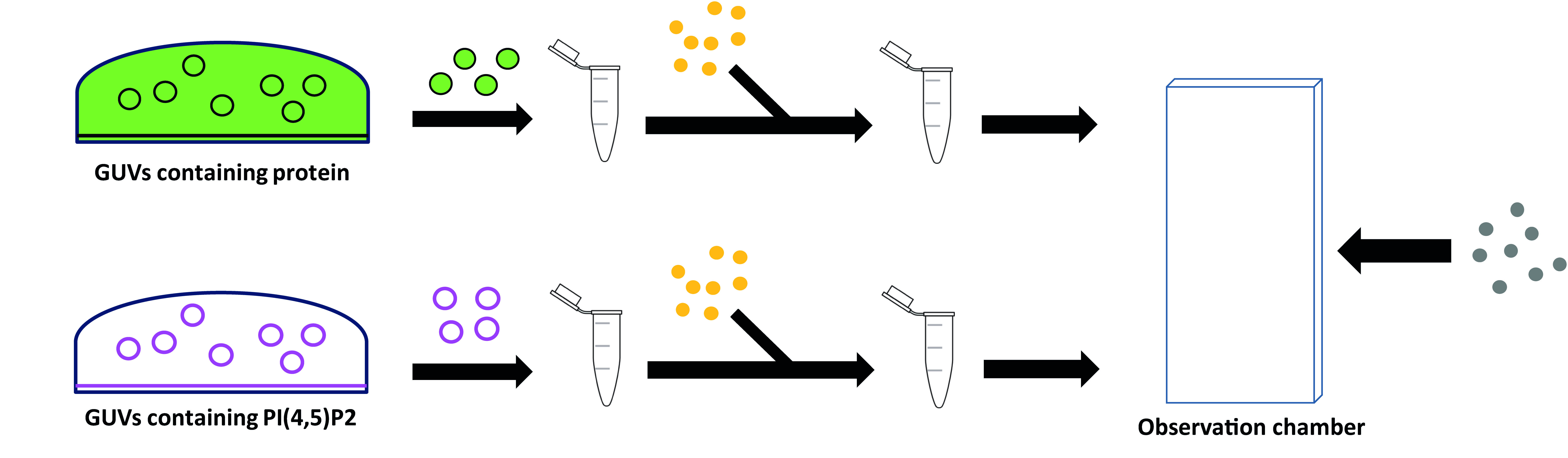
Figure 2. Stepwise procedure for protein encapsulation and washing. Two populations of GUVs are grown separately, then diluted in Outer buffer and centrifuged. GUVs collected from the bottom of the Eppendorf are added to Streptavidin-coated Gold nanorods (in yellow) and further diluted in Outer buffer before being introduced to the observation chamber together with the Streptavidin-coated microbeads (in grey).
Materials and Reagents
- Consumables
- Pipette tips
- 18 x 18 mm glass coverslips (Menzel, catalog number: BB018018A1)
- Hamilton syringe (Dominique Dutscher, Type 701/N)
- 3.5 mm culture dish (Sigma-Aldrich, catalog number: CLS430165)
- 1.5 ml centrifuge tube (Sigma-Aldrich, catalog number: T9661)
- 0.5 ml tube (Sigma-Aldrich, catalog number: T9036)
- 0.22 µm membrane filter (MF-MilliporeTM, catalog number: GSWP04700)
- 1 ml syringe (Sigma-Aldrich, catalog number: Z230723).
- Kimtech precision tissue task wipe (Kimtech, catalog number: 05511)
- Lipids
Note: All lipids were purchased in powder from Avanti Polar Lipids, dissolved in chloroform (unless otherwise indicated) and stored at -20 °C.- Egg L-α-phosphatidylcholine [EPC] (Avanti Polar Lipids, catalog number: 840051)
- 1,2-dioleoylsn-glycero-3-phospho-L-serine [DOPS] (Avanti Polar Lipids, catalog number: 840035)
- 1,2-dioleoyl-sn-glycero-3-phosphatidylethanol-amine [DOPE] (Avanti Polar Lipids, catalog number: 850725)
- Cholest-5-en-3β-ol [cholesterol] (Avanti Polar Lipids, catalog number: 700000)
- L-α-phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2] (Avanti Polar Lipids, catalog number: 840046)
Note: Dissolved in chloroform:methanol solution (70:30). - L-α-phosphatidylethanolamine-N- (lissamine Rhodamine B sulfonyl) [egg PE–Rhodamine] (Avanti Polar Lipids, catalog number: 810146)
- 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[biotinyl(polyethyleneglycol)-2000] [DSPE-PEG(2000)-biotin] (Avanti Polar Lipids, catalog number: 880129)
- Other reagents
- β-casein from bovine milk (> 99%) (Sigma-Aldrich, catalog number: C6905)
- Sucrose (Sigma-Aldrich, catalog number: S7903)
- D+ glucose (Sigma-Aldrich, catalog number: G7021)
- NaCl (Sigma-Aldrich, catalog number: S7653)
- Tris (Sigma-Aldrich, catalog number: 10708976001)
- Streptavidin-functionalized gold nanorods (NanopartzTM, catalog number: C12-10-850-TS-DIH-50)
- Polyvinyl alcohol (PVA; Merck, catalog number: 8.14894.0101)
- Polystyrene streptavidin coated beads (diameter 3.2 μm) (Spherotech, catalog number: SVP-30-5)
- Chloroform (Sigma-Aldrich, catalog number: 288306)
- Mineral oil (Sigma-Aldrich, catalog number: M5310)
- Ethanol (96%, Sigma-Aldrich, catalog number: 16368)
- Lipid mix P (see Recipes)
- Lipid mix E (see Recipes)
- Buffer P (see Recipes)
- Buffer E (see Recipes)
- Outer buffer (see Recipes)
- PVA solution (see Recipes)
- β-casein solution (see Recipes)
Equipment
The equipment has been accurately described in Prévost et al. (2017).
- Micropipettes: borosilicate capillaries with internal and external radii of 0.78 mm and 1 mm, respectively (Harvard apparatus, catalog number: 30-0036)
- Phase-contrast microscope: Nikon TE2000 inverted microscope, eC1 confocal system (Nikon), with two laser lines (λ = 488 nm and 543 nm); optical tweezers induced by a 5 W ytterbium fiber continuous wave laser (λ > 1070 nm; IPG GmBH Germany)
- Plasma cleaner (SPI Supplies, Plasma Prep III)
- Oven: Series B Classic.Line (Binder, Mode B 28)
- Vacuum pump: Pfeiffer Balzers Turbo Molecular Pumps DRAG 020 and Pfeiffer Balzers TCP 015 Turbo Molecular Pump Controller
- Glass syringe: Hamilton 701N (10 µl) (VWR, catalog number: 549-1135)
Procedure
- Preparation of GUVs by PVA-assisted swelling, washing and nanorods incubation
- Clean 18 x 18 mm glass slides by rinsing twice with ethanol and water. Dry them with Kimtech tissue. A separate slide is needed for each GUV population.
Optional: Plasma-clean the glass slides for 5 min using air as plasma source. This facilitates PVA spreading and attachment. - Warm up the PVA solution to ~55 °C and spread 120 µl of it on the glass slides. Drain the excess of PVA by leaning the side of the slide on Kimtech tissue. PVA can also be spin-coated on the slide at 90 x g.
- Incubate the slides at 60 °C for 50 min in oven. Cool down to room temperature before use.
- Prepare lipid mixes P and E (Recipes 1 and 2) in chloroform to a total final concentration of 1 mg/ml. For multiple fusion events with different proteins or lipids, prepare additional mixes.
- Clean the Hamilton syringe with chloroform thoroughly and spread 15 μl of each lipid mix on each corresponding slide using the side of the needle until the chloroform has completely evaporated and the slide appears dry.
- Position each slide into a separate 3.5 mm culture dish and remove the residual organic solvent using a vacuum pump.
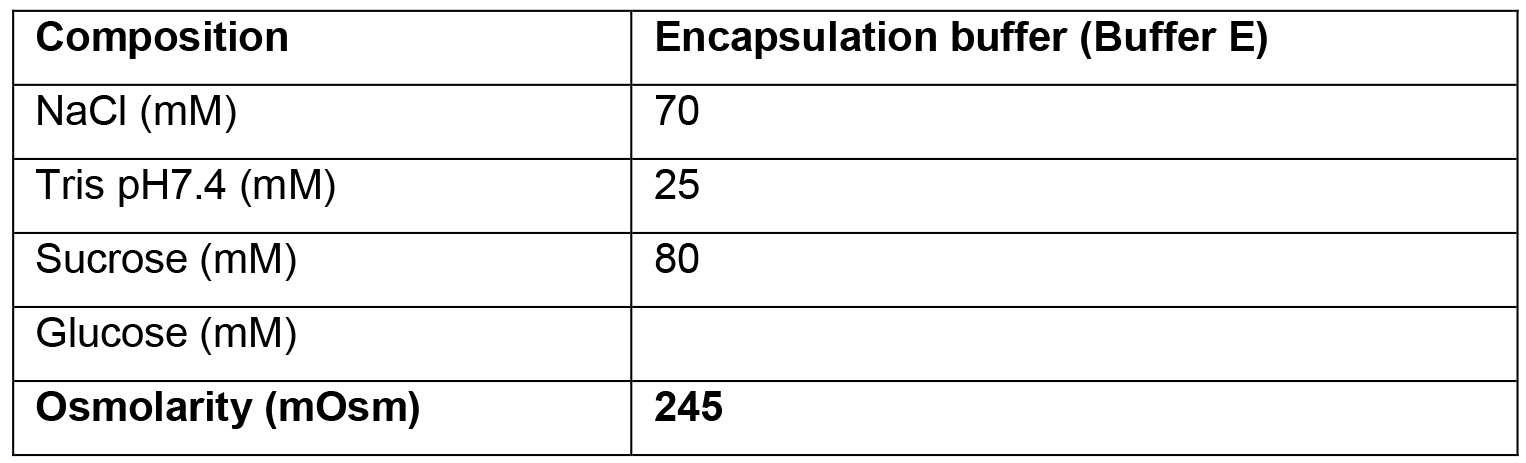
Note: For sensitive lipids such as PI(4,5)P2, 20 min in vacuum are sufficient to remove the residual organic detergent without compromising the integrity of the lipids. - Dilute the protein(s) of interest in Buffer E (Recipe 4).
Note: Take into account the potential effect of the storage buffer on the final osmolarity and salt concentration of Buffer E. - Deposit 360 μl of Buffers P and E (Recipes 3 and 4) on the corresponding slides. Avoid overflowing of the buffer out of the slide.
- Incubate the slides for 30-60 min at the desired temperature to allow swelling. GUV growth can be easily visualized with a phase-contrast microscope.
- Collect 200 μl of GUVs form each slide, by tilting the dish and gently rinsing the slide by gentle pipetting. Transfer each GUVs preparation to a separate 1.5 ml centrifuge tube.
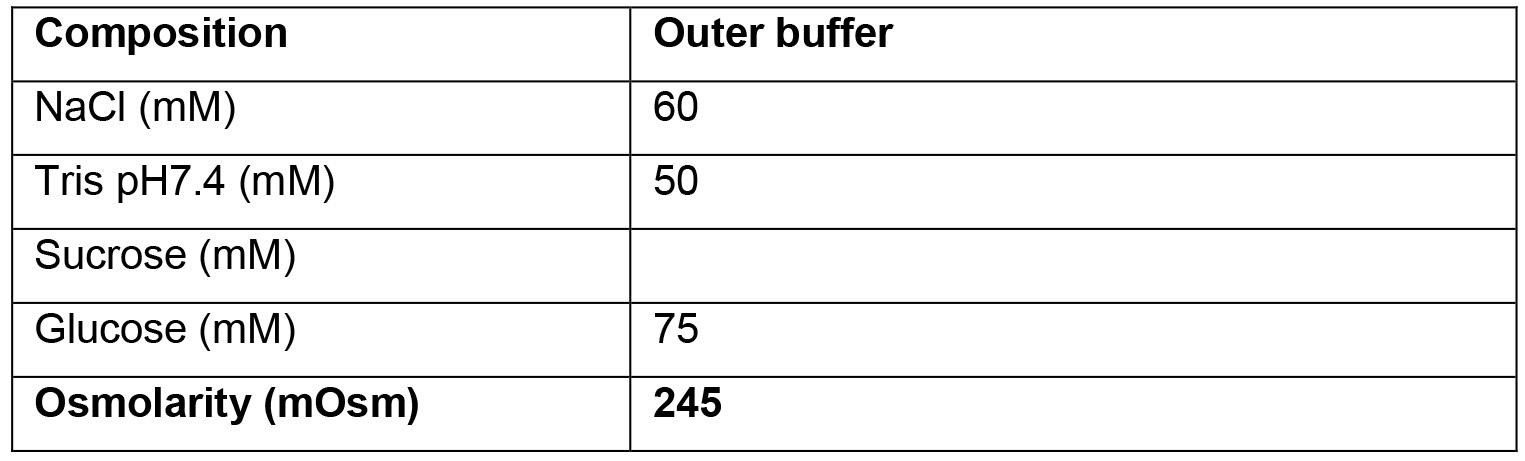
Note: GUVs are very fragile. Pipetting should be as gentle as possible. - Add 1.2 ml of Outer buffer (Recipe 5) to each tube; gently mix by inverting the tube several times.
- Centrifuge for 10 min at 100 x g. The GUVs will sink to the bottom. Transfer 20 μl of GUV from the bottom of the tube to a new 0.5 ml tube.
- Add 80 μl of Outer buffer and 0.8 μl of streptavidin-coated gold nanorods to the GUVs from the previous step. Mix gently by pipetting until the nanorods are uniformly suspended. Incubate for 20 min at room temperature.
Note: The dilutions in Steps A11 and A13 lower the concentration of the protein present in Buffer E outside the GUVs and helps in removing lipid aggregates and small vesicles.
- Clean 18 x 18 mm glass slides by rinsing twice with ethanol and water. Dry them with Kimtech tissue. A separate slide is needed for each GUV population.
- Preparation of the experimental chamber
- Prepare the experimental chamber and micropipettes as previously described, also step-by-step by a movie (Prévost et al., 2017). At least three micropipettes are necessary to perform the experiment (Figure 3). While a precise pressure control is needed for R- and L-pipettes since each aspirates a GUV by connecting them to water reservoirs of controlled height (Kwok and Evans, 1981), the pressure of the T-pipette used to hold the microbead can be adjusted manually using a syringe. The tip diameter of the R- and L-pipette should be in the range 5-10 μm, while the T-pipette should have an opening < 3 μm. Position the tip of all three micropipettes at the center of the stage, within the field of view, ~100 μm above the bottom of the chamber.
- Passivate the chamber and pipettes by incubating with β-casein solution (Recipe 7) for 20 min. The volume of the chamber is ~200 μl. Remove the β-casein from the chamber, and rinse with Outer buffer. Fill the chamber with 170 μl of the buffer.
Note: About 1 ml of β-casein solution is needed to fill both the chamber and the micropipettes. - Carefully aspirate15 μl of each population of GUVs from the bottom of the Eppendorf tube and add them slowly to the chamber. Place the two populations on separate regions of the chamber (Figure 3).
- Add 2 μl of streptavidin-coated polystyrene beads (diameter ~3 μm) to the chamber to a final concentration of 0.1 x 10-3% (w/v). Place the beads in a separate region of the chamber, to avoid mixing with the GUVs (Figure 3).
- Keep the GUVs in the chamber for 20-30 min. During this time, the GUVs and the beads settle to the bottom of the chamber. Moreover, evaporation from buffer creates an osmotic gradient, which renders the GUVs “floppy” and allows their aspiration by the micropipettes.
- In order to prevent further evaporation during the experiment, seal the chamber with mineral oil.
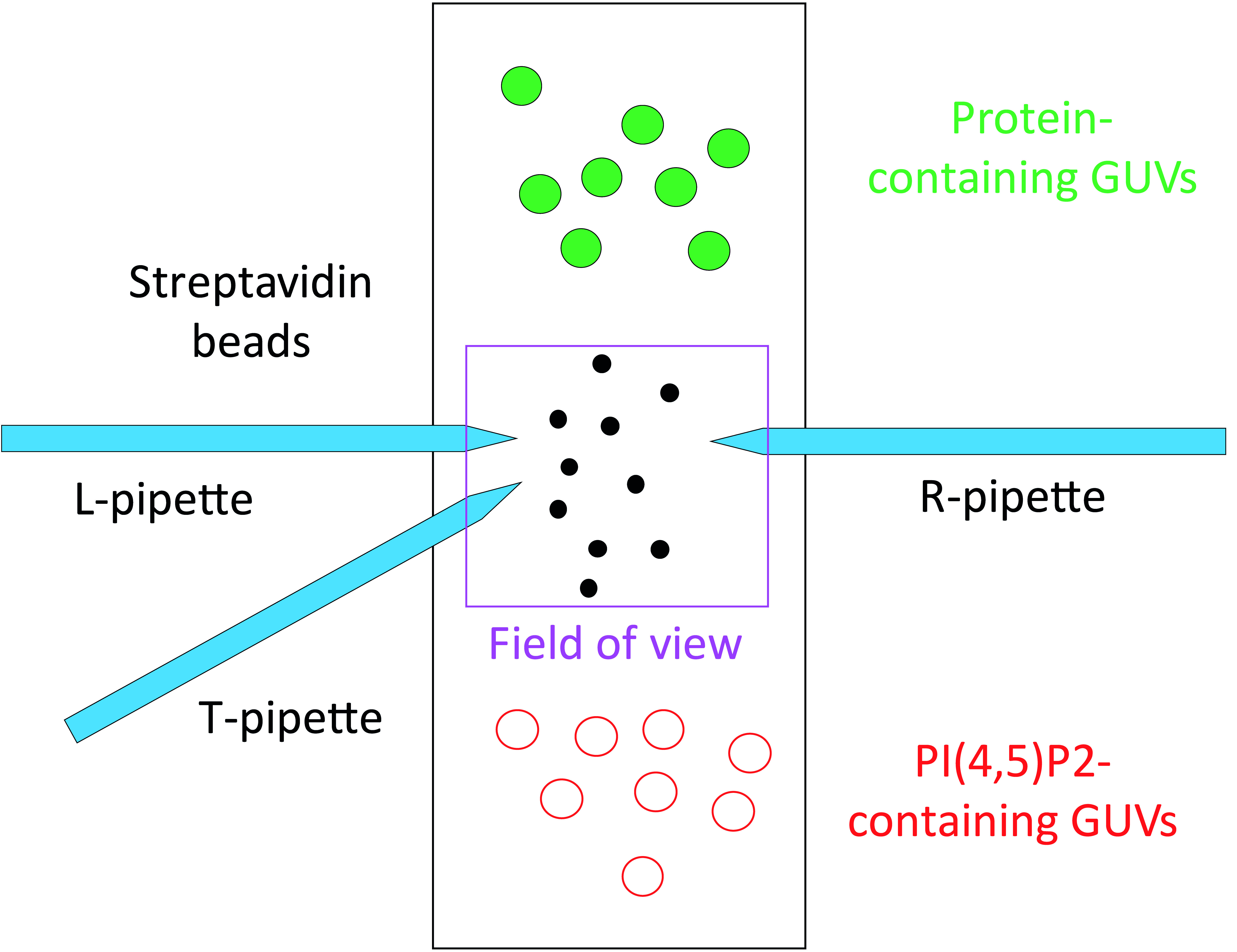
Figure 3. Schematic representing the arrangement of the experimental chamber including pipettes, GUVs and microbeads. L (left) and R (right) micropipettes hold both GUVs before fusion, whereas the T-pipette is used to firmly hold a micro-bead and pull the nanotube.
- GUVs fusion experiment (for movies depicting the fusion event, please see Supplementary data from De Franceschi et al., 2018):
- Lower the tip of the T-pipette to the bottom of the chamber. By moving the microscope stage, find a microbead and aspirate it strongly with the T-pipette. Lock the pressure by closing the valve on the tubing between pipette and syringe. Raise the tip of the T-pipette 100 μm from the bottom of the chamber.
- Set the R- and L-pipette aspiration pressure to zero. By moving the microscope stage, find a PI(4,5)P2-containing GUV and aspirate it with the R-pipette. Raise the GUV 100 μm above the bottom of the chamber.
- By moving the microscope stage, find a protein-containing GUV. Lower the L-pipette and aspirate the GUV. Raise this pipette 100 μm from the bottom of the chamber.
- Adjust the focus on the microbead held by the T-pipette. Release the pressure of the R-pipette to almost zero and bring it in contact with the bead. By moving the R-pipette with the micromanipulator away from the bead, pull a nanotube from the PI(4,5)P2-containing GUV. Adjust the height of the R-pipette so that both the microbeads and the nanotube are in focus.
- By using the micromanipulators, position laterally and vertically the R- and L-pipettes so that the infra-red laser is focused at the interface between both GUVs. Release the aspiration pressure of both R- and L-pipettes to nearly zero.
Important: This is the most critical step, since fusion will occur only if the GUVs are placed correctly and if the aspiration pressure of both the R- and L-pipettes is nearly zero. Observing the GUVs by DIC rather than fluorescence, both during positioning and during fusion, greatly simplify this procedure (Figure 4).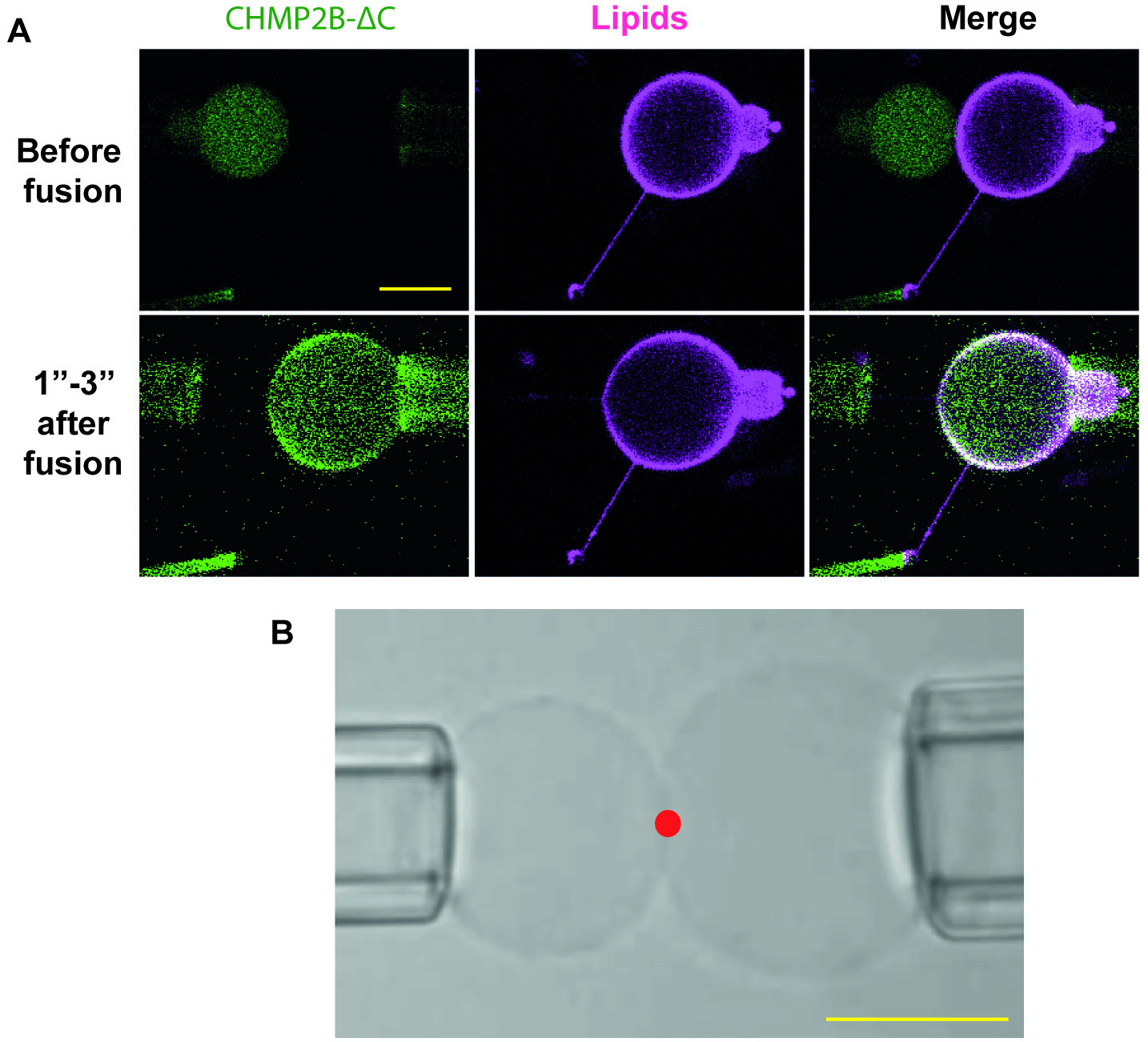
Figure 4. Positioning and fusion of GUVs. A. Fluorescent image of GUVs before and immediately after fusion. Lipid mixing is still not completed. The contrast in the image after fusion has been enhanced to show that fusion occurs without protein leaking outside the GUVs. Scale bar = 10 µm. B. DIC imaging of GUVs optimal position before fusion. The red dot indicates the position of the optical tweezers. Scale bar = 10 µm. - Trigger fusion by turning on the infra-red laser. If the GUVs are aligned correctly, fusion is instantaneous. If fusion does not occur, switch off the laser and fine-tune the position of the GUVs by using the micromanipulators. Then repeat the process.
Note: It is also possible to leave the laser on while adjusting the position of the pipettes. However, this occasionally induces strong adhesion between the two GUVs, which can inhibit fusion. Moreover, prolonged irradiation can cause membrane heating (due to nanorods absorption) and thus degradation of sensitive lipids such as PI(4,5)P2. - After fusion, content mixing is almost instantaneous, while lipid mixing might require tens of seconds to be completed (Figure 4). This may cause fluctuations in membrane tension independently from the aspiration pressure of the pipettes. Wait 2-3 min until the tension stabilizes before performing any measurement. The focus may have to be readjusted in order to visualize the nanotube. There is no release of GUV content during or after fusion.
- Once membrane tension is stable, tube radius can be changed to study recruitment of proteins as a function of tube radius, as usually done with nanotube assays (Prévost et al., 2017).
Data analysis
The fusion protocol described here is used to reconstitute proteins inside GUVs. Once fusion has been accomplished, measurements and data analysis are performed similarly to a classical tube-pulling experiment (Prévost et al., 2017). In our setup, the optical trap (hence the infra-red laser) was used to induce fusion, and therefore force measurement was not possible. However, in a setup having multiple optical traps, force measurements can be performed.
Notes
- This protocol is independent of the method used to grow GUVs, but we recommend using the PVA-swelling method, given a good protein encapsulation efficiency and a high yield of GUVs. GUVs are very fragile and a significant amount of the GUVs produced are lost during the washing procedure, making other methods, such as electroformation on platinum wires (Prévost et al., 2017) less suitable, since less GUVs are produced. Moreover, PVA-assisted GUV swelling can work over a wide range of temperatures (4-60 °C) and thus can be adapted to the experimental conditions needed for essentially all proteins and lipids.
- It is generally easier to fuse GUVs of similar size, because they are easier to align.
- In doing a tube pulling experiment, it is important to make sure that the vesicles are unilamellar. The PVA-swelling method generally produces unilamellar vesicles. However, confirming the unilamellarity of vesicles is not necessary when using our protocol, since occasional fusion of multilamellar vesicles is very easy to recognize: only the outer membrane will undergo fusion, thus revealing the presence of inner membranes (Figure 5).
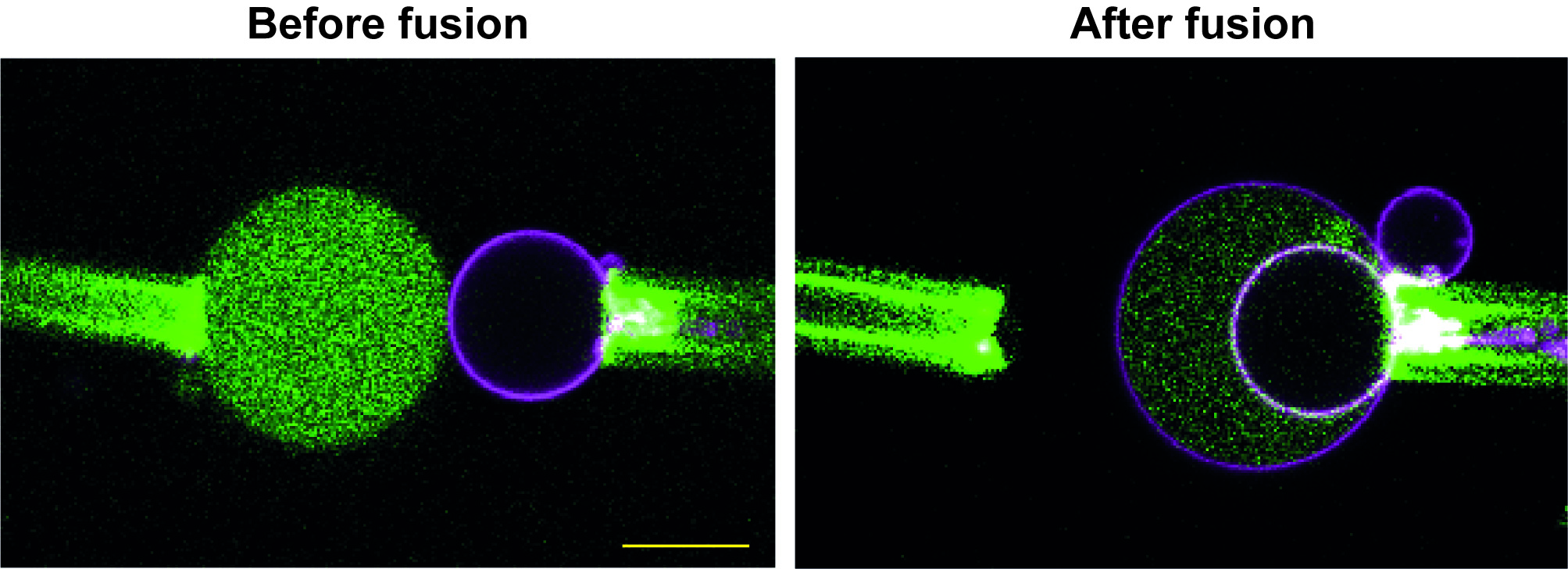
Figure 5. Fusion event with a multilamellar GUV. The GUV in magenta is multilamellar, but this becomes evident only after fusion, since only the outer membrane can fuse with the other GUV. This creates an additional compartment, filled with proteins, between the membranes. Scale bar = 10 µm. - There is a significant batch-to-batch variation in the degree of streptavidin-functionalization of gold nanoparticles. This can severely affect the efficiency of GUV fusion, and the amount of nanoparticles added needs to be adjusted for each new batch. However, the amount of nanoparticles that are generally supplied in each batch is sufficient for hundreds of experiments.
Recipes
Notes:
- The lipid mixes P and E and the P, E and Outer buffers described in this protocol are needed for a fusion experiment between the ESCRT-III protein CHMP2B and GUVs containing PI(4,5)P2. Different proteins, lipids or binding condition might require adjustment of the lipid mixes and buffers.
- For each GUV production, 360 μl of buffer is needed. About 3 ml of Outer buffer are needed for each experiment. Buffer composition can vary widely depending on the requirements and aims of the experiments. Always make sure that all buffers have a similar osmolarity, and that buffers P and E contain enough sucrose compared to the glucose content of the Outer buffer, in order to make the GUV sink.
- In this example, 23 μl of CHMP2B stock solution at 30 μM is added to 337 μl of Buffer E. The protein stock buffer contains 100 mM NaCl and 25 mM HEPES, thus it does not significantly affect the osmolarity.
- Lipid mix P
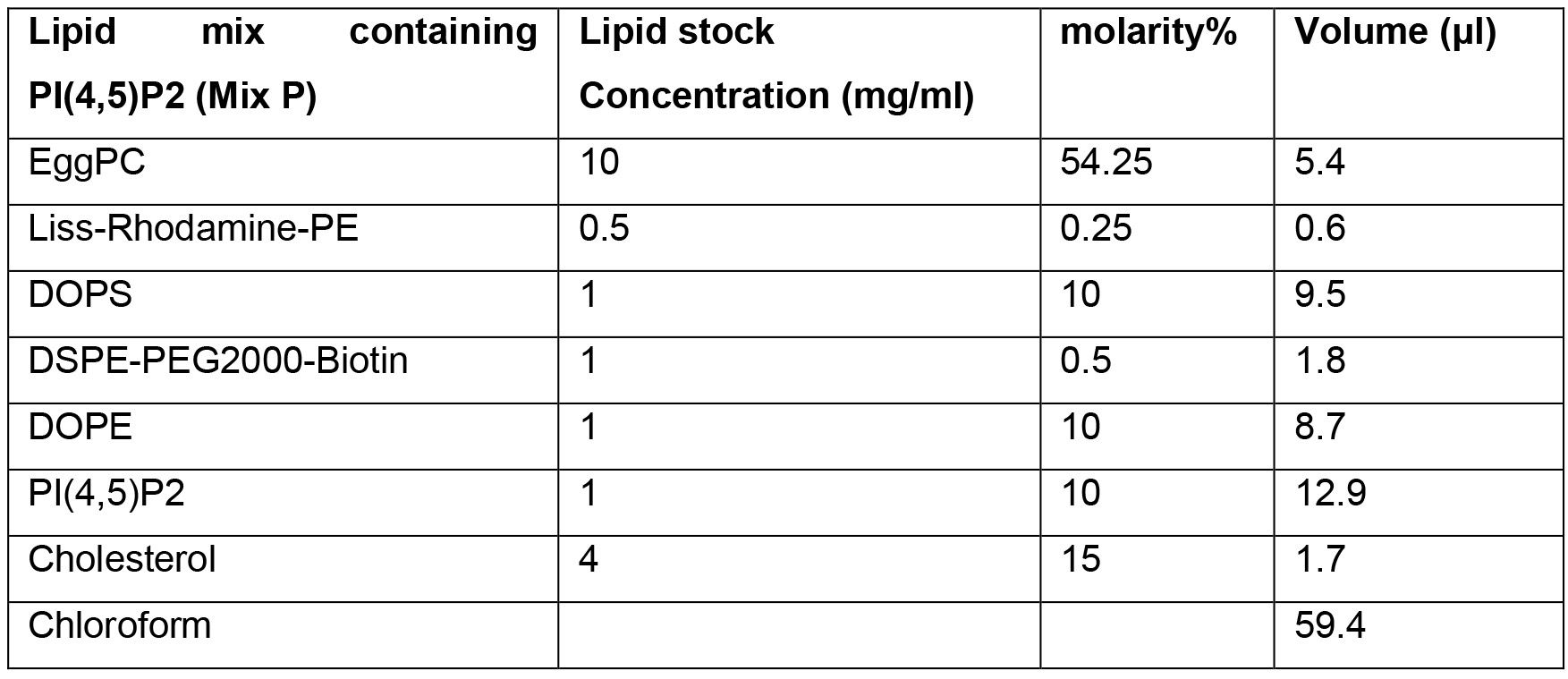
- Lipid mix E
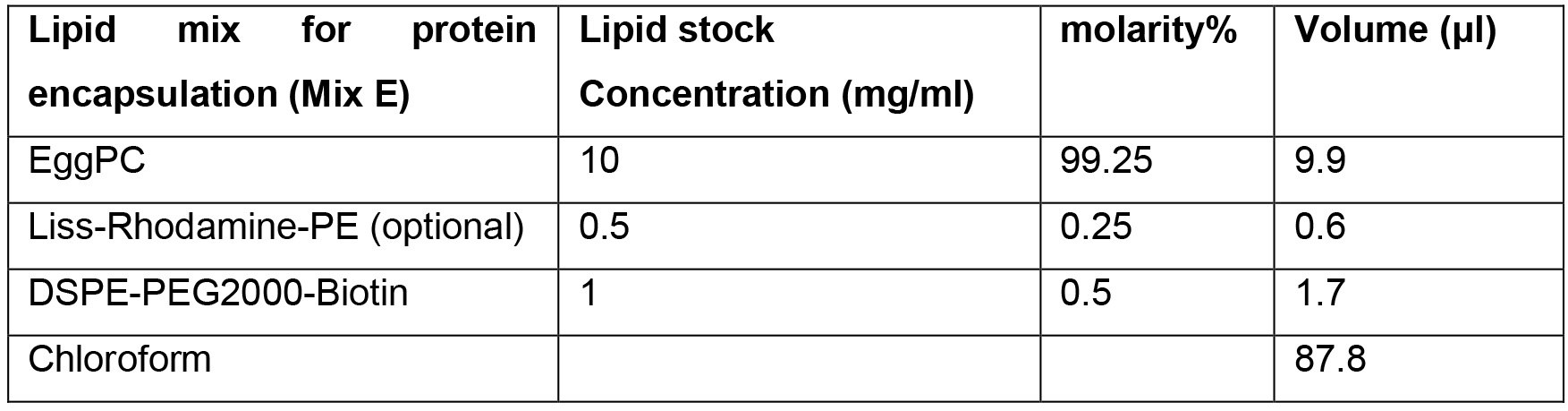
- Buffer for PI(4,5)P2-containing GUVs (Buffer P)

- Encapsulation buffer (Buffer E)
- Outer buffer
- PVA solution
- Dissolve 5% w/v of PVA in the following buffer: 100 mM Sucrose, 50 mM NaCl and 50 mM Tris pH 7.4
- Dissolve PVA by gentle stirring at ~80 °C
- β-casein solution
- Dissolve β-casein at 5 mg/ml in the following buffer: 100 mM NaCl and 50 mM Tris pH 7.4
- Filter the solution with a 0.22 µm membrane filter
Acknowledgments
This work was supported by the Agence Nationale de la Recherche (ANR-14-CE09-0003-01). Post-doctoral fellowships from the Institut Curie, the Fondation pour la Recherche Médicale (SPF20160936338), European Molecular Biology Organization (EMBO) non-stipendiary long term fellowship (ALTF 818-2016), MSCA Grant 751715 (ESCRT model), are acknowledged for funding the development of this method and its dissemination.
This method, first employed in De Franceschi et al. (2018), combines and optimizes three techniques: classic tube pulling assay (Prévost et al., 2017), laser-triggered GUV fusion (Rørvig-Lund et al., 2015) and protein encapsulation by PVA-assisted gel swelling (Weinberger et al., 2013).
Competing interests
The authors declare no conflict of interest.
References
- Caillat, C., Maity, S., Miguet, N., Roos, W. H. and Weissenhorn, W. (2019). The role of VPS4 in ESCRT-III polymer remodeling. Biochem Soc Trans 47(1): 441-448.
- Dasgupta, R. and Dimova, R. (2014). Inward and outward membrane tubes pulled from giant vesicles. J Phys D Appl Phys 47(28): 282001.
- De Franceschi, N., Alqabandi, M., Miguet, N., Caillat, C., Mangenot, S., Weissenhorn, W. and Bassereau, P. (2018). The ESCRT protein CHMP2B acts as a diffusion barrier on reconstituted membrane necks. J Cell Sci 132(4).
- Henne, W. M., Stenmark, H. and Emr, S. D. (2013). Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb Perspect Biol 5(9).
- Kwok, R. and Evans, E. (1981). Thermoelasticity of large lecithin bilayer vesicles. Biophys J 35(3): 637-652.
- Mathivet, L., Cribier, S. and Devaux, P. F. (1996). Shape change and physical properties of giant phospholipid vesicles prepared in the presence of an AC electric field. Biophys J 70(3): 1112-1121.
- McCullough, J., Frost, A. and Sundquist, W. I. (2018). Structures, functions, and dynamics of ESCRT-III/Vps4 membrane remodeling and fission complexes. Annu Rev Cell Dev Biol 34: 85-109.
- Prévost, C., Tsai, F. C., Bassereau, P. and Simunovic, M. (2017). Pulling membrane nanotubes from giant unilamellar vesicles. J Vis Exp (130).
- Prévost, C., Zhao, H., Manzi, J., Lemichez, E., Lappalainen, P., Callan-Jones, A. and Bassereau, P. (2015). IRSp53 senses negative membrane curvature and phase separates along membrane tubules. Nat Commun 6: 8529.
- Rørvig-Lund, A., Bahadori, A., Semsey, S., Bendix, P. M. and Oddershede, L. B. (2015). Vesicle fusion triggered by optically heated gold nanoparticles. Nano Lett 15(6): 4183-4188.
- Sorre, B., Callan-Jones, A., Manzi, J., Goud, B., Prost, J., Bassereau, P. and Roux, A. (2012). Nature of curvature coupling of amphiphysin with membranes depends on its bound density. Proc Natl Acad Sci U S A 109(1): 173-178.
- Weinberger, A., Tsai, F. C., Koenderink, G. H., Schmidt, T. F., Itri, R., Meier, W., Schmatko, T., Schroder, A. and Marques, C. (2013). Gel-assisted formation of giant unilamellar vesicles. Biophys J 105(1): 154-164.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
de Franceschi, N., Alqabandi, M., Weissenhorn, W. and Bassereau, P. (2019). Dynamic and Sequential Protein Reconstitution on Negatively Curved Membranes by Giant Vesicles Fusion. Bio-protocol 9(13): e3294. DOI: 10.21769/BioProtoc.3294.
Category
Biophysics > Bioengineering > Protein encapsulation
Molecular Biology > Protein > Protein-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.