- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Detection of Heteroplasmic Variants in the Mitochondrial Genome through Massive Parallel Sequencing

(*contributed equally to this work) Published: Vol 9, Iss 13, Jul 5, 2019 DOI: 10.21769/BioProtoc.3283 Views: 5663
Reviewed by: Chiara AmbrogioEnrico PatruccoMauro Sbroggio'
Original research article
The authors used this protocol in:

Jul 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Detecting heteroplasmies in the mitochondrial DNA (mtDNA) has been a challenge for many years. In the past, Sanger sequencing was the main option to perform this analysis, however, this method could not detect low frequency heteroplasmies. Massive Parallel Sequencing (MPS) provides the opportunity to study the mtDNA in depth, but a controlled pipeline is necessary to reliably retrieve and quantify the low frequency variants. It has been shown that differences in methods can significantly affect the number and frequency of the retrieved variants.
In this protocol, we present a method involving both wet lab and bioinformatics that allows identifying and quantifying single nucleotide variants in the full mtDNA sequence, down to a heteroplasmic load of 1.5%. For this, we set up a PCR-based amplification of the mtDNA, followed by MPS using Illumina chemistry, and variant calling with two different algorithms, mtDNA server and Mutect.
The PCR amplification is used to enrich the mitochondrial fraction, while the bioinformatic processing with two algorithms is used to discriminate the true heteroplasmies from background noise. The protocol described here allows for deep sequencing of the mitochondrial DNA in bulk DNA samples as well as single cells (both large cells such as human oocytes, and small-sized single cells such as human embryonic stem cells) with minor modifications to the protocol.
Background
In the past, the methods used for studying the mtDNA were amongst others PCR-RFLP (PCR-restriction fragment length polymorphism), Sanger sequencing and mitochondrial DNA re-sequencing using Affymetrix’s MitoChip. However, these methods are not able to accurately quantify the heteroplasmic load under 10%. Massive Parallel Sequencing (MPS) represents, in all likelihood, the best solution to investigate variants in the mitochondrial genome. However, when analyzing mtDNA there are two key factors that make mtDNA analysis less straightforward compared to the nuclear genome. First, the mtDNA contains regions with significant homology to regions dispersed within the nuclear genome called nuclear mitochondrial DNA sequences (NuMTS). Second, multiple mtDNA copies are present within a cell, and variants can be present at frequencies ranging between 0 and 100%. For this reason, when performing MPS analysis of the mtDNA different strategies might be applied to accurately identify and quantify single nucleotide variants (SNVs). The best approach to overcome the first problem is to enrich the sample for its mitochondrial genome only. This can be achieved by selectively amplifying the mtDNA by long-range PCR, by isolating the mtDNA using mtDNA enrichment kits, or by isolating the mitochondria themselves prior to DNA extraction (Just et al., 2015). The second issue is more challenging. Whilst MPS provides the ideal type of data to simultaneously identify SNVs and/or rearrangements and calculating their loads, the manner in which the data are generated and processed will not only determine the type of variants that can be detected, but also their lower threshold. If an SNV is present at a very low frequency, its signal will be undistinguishable from the systems sequencing errors (Bai and Wong, 2004; Rohlin et al., 2009; Zhang et al., 2012; Ye et al., 2014). Recently, many pipelines have been released to identify and quantify variants. However, bioinformatic processing does not represent the only critical step in these analyses. In our hands, we found that the initial amplification of the template is an extremely important step for the correct evaluation of SNV frequencies, such that a suboptimal PCR amplification leads to a gross overestimation of the retrieved frequencies (Zambelli et al., 2017). This is especially the case for PCR protocols with higher cycle numbers and primer sets that do not result in a linear amplification. The method we present here can be used to accurately detect and quantify single nucleotide variants at a low heteroplasmic load (as low as 1.5%) in both bulk DNA samples and single cells (Zambelli et al., 2017 and 2018). We here describe amplification conditions and bioinformatic processing for both bulk DNA and single cells with detailed information and screenshots about the bioinformatic steps.
Materials and Reagents
- 96-well plate
- 200 μl Eppendorf tubes
- LongAmp Buffer (LongAmp Taq DNA Polymerase kit) (New England Biolabs, catalog number: M0323L), stored at -20 °C
- Taq LongAmp (LongAmp Taq DNA Polymerase kit) (New England Biolabs, catalog number: M0323L), stored and kept at all times at -20 °C
- dNTP’s (dNTP sets, IllustraTM) (VWR, catalog number: 28-4065-57), diluted to 2 mM and stored at -20 °C
- Tricine (Sigma-Aldrich, catalog number: T9784), diluted to 200 mM and stored at 4 °C
- DTT (DL-Dithiothreitol) (Sigma, catalog number: D-0632), stored at 4 °C
- NaOH (Stock solution: 1 M)
- TBE buffer (1x) (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15581044)
- Agarose (Agarose DNA Pure Grade, VWR, catalog number: 443666A)
- PCR Purification Kit (AMPure XP for PCR purification, Beckman Coulter, catalog number: A63882)
- Primer set 1 (5042f-1424r) (Integrated DNA Technologies, diluted to 10 μmol, aliquoted and stored at -20 °C)
Forward primer: 5′-AGC AGT TCT ACC GTA CAA CC-3′
Reverse primer: 5′-ATC CAC CTT CGA CCC TTA AG-3′ - Primer set 2 (528f-5789r) (Integrated DNA Technologies, diluted to 10 μmol, aliquoted and stored at -20 °C)
Forward primer: 5′-TGC TAA CCC CAT ACC CCG AAC C-3′
Reverse primer: 5′-AAG AAG CAG CTT CAA ACC TGC C-3′
Note: These two primer sets (Items 12 and 13) were selected because they were able to amplify large fragments of the mtDNA and tested negative when amplifying Rho Zero cells, indicating that they did not amplify NuMTS in the nuclear genome. These primer sets were also evaluated for their performance in low frequency heteroplasmy calling by performing spike-in experiments and were shown to give the better estimation of the low frequency variants (Zambelli et al., 2017).
- QubitTM dsDNA Broad Range Assay Kit (InvitrogenTM, catalog number: Q32853)
- Ethanol (80%)
- Tris-HCl solution (10 mM, pH 8.0)
- Library Preparation kit (KAPA HyperPlus kit, Roche, catalog number: 07962436001)
- Fragmentation Kit (HS NGS Fragment Kit, Agilent, catalog number: DNF-474)
- Tween 20 (Sigma-Aldrich, catalog number: P9416)
- MPS Reagent Kit (NovaSeq6000 S2 Reagent Kit [200 cycles]) (Illumina, catalog number: 20012861)
- Gel electrophoresis (see Recipes)
- Alkaline Lysis Buffer (see Recipes)
- EBT (Elution Buffer with Tris) buffer (see Recipes)
Equipment
- Agarose Gel electrophoresis apparatus
- Power Supply (Electrophoresis Power Supply–EPS 301) (GE Health Care, catalog number: 18113001)
- Thermal Cycler (VeritiTM 96-Well, Applied Biosystems, catalog number: 4375786)
- Magnet
- Qubit Fluorometric Quantification (InvitrogenTM, catalog number: Q33238)
- Fragment Analyzer (Agilent formerly Advanced Analytical Technologies, Agilent, https://www.aati-us.com/instruments/fragment-analyzer/)
- Sequencing system (e.g., NovaSeq6000, Illumina, catalog number: 20012850)
Software
- bcl2fastq conversion software v2.19 script (Illumina, http://emea.support.illumina.com/downloads/bcl2fastq-conversion-software-v2-19.html)
- Seqtk (https://bioconda.github.io/recipes/seqtk/README.html)
- BWA-MEM
- GATK v3.3 (Broad Institute, https://software.broadinstitute.org/gatk/)
- GATK v3.6 Mutect2 (Broad Institute, https://software.broadinstitute.org/gatk/documentation/tooldocs/3.7-0/org_broadinstitute_gatk_tools_walkers_cancer_m2_MuTect2.php)
- Mpileup (SAMtools, http://samtools.sourceforge.net/mpileup.shtml)
- mtDNA server (https://mtdna-server.uibk.ac.at/index.html)
- MitoWheel (http://mitowheel.org/mitowheel.html)
- MitImpact (http://mitimpact.css-mendel.it/)
- MutPred2 (http://mutpred.mutdb.org/)
Procedure
- Extraction of bulk DNA: extract DNA from the sample of interest.
- The sample can be a bulk of tissue or cell lines. No special kit is required for bulk analysis, for instance, we routinely use either the DNeasy kit from Qiagen or a phenol/chlorophorm extraction followed by ethanol precipitation (Zambelli et al., 2018).
- For single cells, we manually collect them with the aid of a stereomicroscope under a horizontal flow in 2.5 μl of alkaline lysis buffer (200 mM NaOH and 50 mM DTT) in 200 μl Eppendorf tubes (Spits et al., 2006) and store them at -20 °C. Before amplification, the single cells are incubated for 10 min at 65 °C for lysis, then prepared for amplification as under Step 3.
- Dilute the DNA sample to a working solution of 10 ng/μl (only for bulk DNA).
- Prepare the master mix as following:
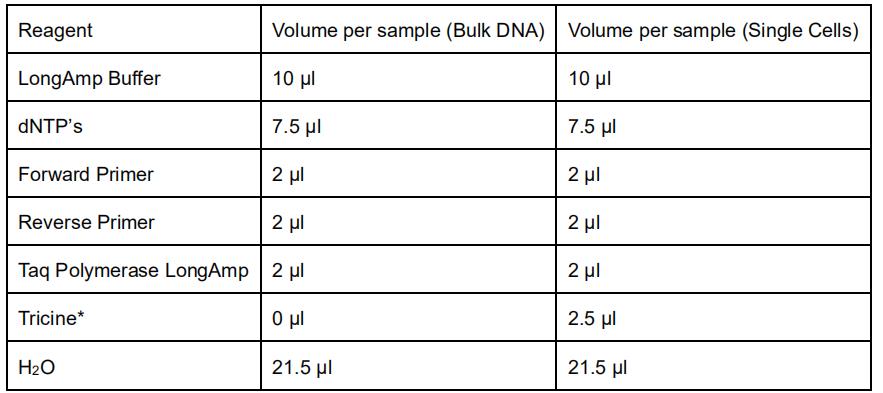
*Tricine is used in the PCR reaction to buffer the alkaline lysis buffer used in the single cell collection. - Aliquot 45 μl of the master mix to PCR tubes and add 5 μl of 10 ng/μl DNA for bulk.
Note: For single cells, add 47.5 μl of the master mix to the Eppendorf tubes with the cells collected in the alkaline lysis buffer. - Use the following PCR program:
- step 1 (Initiation): 30 s at 94 °C
- step 2 (8 cycles): 15 s at 94 °C
30 s at 64 °C minus 0.4 °C per cycle (this is the touchdown step)
11 min at 65 °C (for primer set 1) or 5 min at 65 °C (for primer set 2) - step 3 (22 cycles for bulk; 27 cycles for large single cells (human oocytes), 37 cycles for other single cells):
15 s at 94 °C
30 s at 61 °C
11 min at 65 °C (for primer set 1) or 5 min at 65 °C (for primer set 2) - step 4 (Final): 11 min at 65 °C (for primer set 1) or 5 min at 65 °C (for primer set 2)
- step 5: cooling step at 4 °C until storage.
- Keep the amplicons stored at 4 °C (for short-term storage) or -20 °C (long-term storage) until further processing.
- Confirm successful amplification by running a gel-electrophoresis (1% for bulk, 1.5% for single cells) (see Recipes).
- Load 9 μl per sample on the gel and run the gel at 80 V for 1 h. Successful amplification should be as shown in Figure 1.
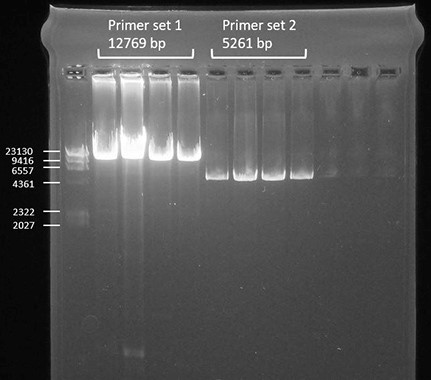
Figure 1. Successful amplification of the two primer sets compared to a DNA ladder shown by gel-electrophoresis - Clean up the samples with AMPure beads as follows.
Note: This is usually done in batches of 96 samples in a 96-well plate.- Add 54 μl of AMPure beads to 30 μl of each amplicon (the ratio is 1.8x).
- Mix the samples by pipetting up and down and incubate the samples for 5 min at room temperature.
- Place the samples on the magnet for 5 min or until the solution is clear.
- Remove the supernatant without disturbing the beads.
- Add 200 μl ethanol (80%).
- Incubate the samples for 30 s on the magnet.
- Remove the supernatant without disturbing the beads.
- Repeat Steps 9e-9g.
- Let the beads dry for 5 min at room temperature.
- Remove the samples from the magnet.
- Add 30 μl of Tris-HCl (10 mM, pH 8.0) and incubate the samples for 2 min at room temperature.
- Place the samples on the magnet until the solution is clear.
- Transfer the supernatant into a new 96-well plate.
- Quantify the DNA concentration of the samples using the Qubit according to the manufacturer’s instructions.
- Pool the amplicons of primer sets 1 and 2 together per sample, and to maintain a uniform coverage, the amplicons need to be mixed in a 0.35/0.65 ratio (35% of the shorter amplicon generated by primer set 2 and 65% of the longer amplicon generated by primer set 1). Use a total of 500 ng DNA in 17.5 μl Tris-HCl (10 mM, pH 8.0).
Note: The procedure is also possible for each amplicon separately, again with a total of 500 ng. - Prepare the library with the KAPA HyperPlus Kit using half of the reagent volumes as specified by the supplier. This is the protocol in summary:
- Prepare the fragmentation mix (on ice):
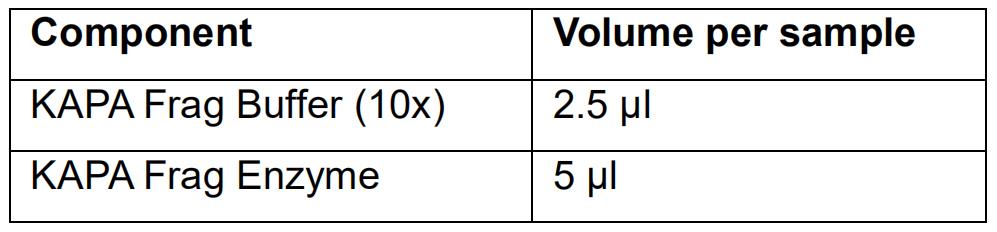
- Vortex the fragmentation mix and spin down.
- Add 7.5 μl of the fragmentation mix to 17.5 μl of the purified DNA sample and mix by pipetting (work on ice).
- Incubate the samples in a thermocycler (lid heated at 50 °C).
10 min at 4 °C
15 min at 37 °C
Keep the samples at 4 °C - Proceed immediately to the End Repair and A-tailing.
- Prepare the End Repair and A-tailing mix as follows:
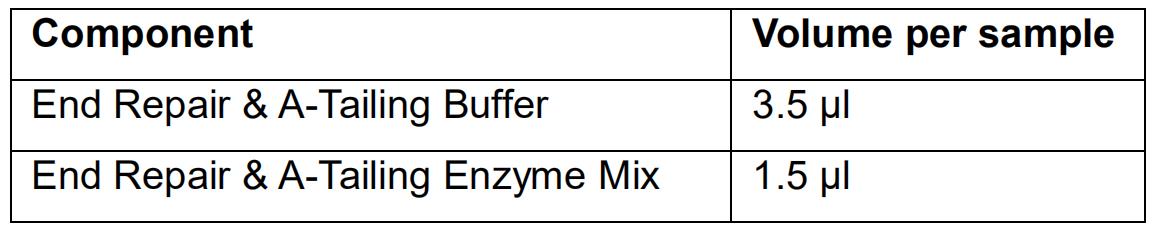
- Mix the End Repair and A-tailing mix by vortexing and spin down.
- Add 5 μl of the End Repair and A-tailing mix to the fragmented DNA.
- Incubate the samples in the thermocycler (put the lid on 85 °C).
30 min at 65 °C
Keep the samples at 4 °C. - Proceed immediately to the Ligation step.
- Prepare the Ligation mix as follows:
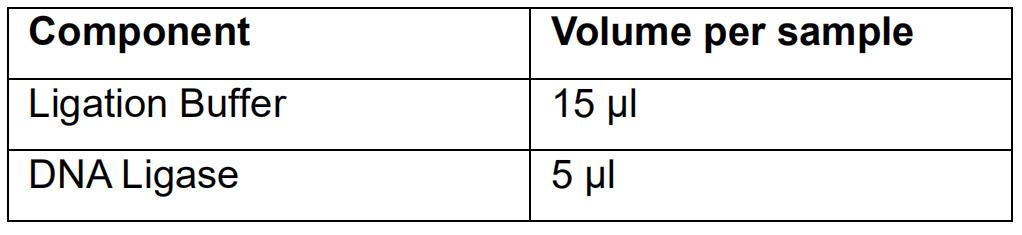
- Mix the Ligation mix by pipetting up and down.
- Add 20 μl of the Ligation mix to 30 μl of the End Repaired and A-tailed DNA.
- Add a unique adapter per sample: 5 μl of 7.5 μM Illumina TruSeq Unique Dual (UD) indexed adapter.
Note: These adapters may be custom produced by an oligo supplier (e.g., Integrated DNA Technologies) or may be purchased as a kit (e.g., IDT for Illumina–TruSeq DNA UD Indexes). Be aware that most suppliers deliver ready-made adapters at 15 μM while in this protocol we use 7.5 μM. - Mix thoroughly by pipetting.
- Incubate the samples in a thermocycler (no heated lid) for 15 min at 20 °C.
- Proceed immediately with the bead sample clean up.
- Prepare the fragmentation mix (on ice):
- Clean up the libraries with AMPure beads as follows:
- Add 44 μl of AMPure beads to 35 μl of the library (the ratio is 0.8x).
- Mix the samples by pipetting up and down and incubate the samples for 5 min at room temperature.
- Place the samples on a magnet for 5 min or until the solution is clear.
- Remove the supernatant without disturbing the beads.
- Add 200 μl of ethanol (80%).
- Incubate the samples for 30 s on the magnet.
- Remove the supernatant without disturbing the beads.
- Repeat Steps 13e-13g.
- Let the beads dry for 5 min at room temperature.
- Remove the samples from the magnet.
- Add 27 μl of Tris-HCl (10 mM, pH 8.0).
- Incubate the samples for 2 min at room temperature.
- Place the samples on the magnet until the solution is clear.
- Transfer 25 μl of the eluate to a new 96-well plate.
- Size-select the libraries with AMPure beads as follows:
- Add 75 μl of nuclease-free water to the purified libraries.
- Add 50 μl of AMPure beads to 100 μl of the library (the ratio is 0.5x).
- Mix the samples by pipetting up and down and incubate the samples for 5 min at room temperature.
- Place the samples on the magnet for 5 min or until the solution is clear.
- Transfer 140 μl of the supernatant to a new 96-well plate.
- Add 20 μl of AMPure beads to 140 μl of supernatant (the ratio is 0.7x).
- Mix the samples by pipetting up and down and incubate the samples for 5 min at room temperature.
- Place the samples on the magnet for 5 min or until the solution is clear.
- Remove the supernatant without disturbing the beads.
- Add 200 μl of ethanol (80%).
- Incubate the samples for 30 s on the magnet.
- Remove supernatant without disturbing the beads.
- Repeat Steps 14j-14I.
- Let the beads dry for 5 min at room temperature.
- Remove the samples from the magnet.
- Add 15 μl of Tris-HCl (10 mM; pH 8.0).
- Incubate the samples for 2 min at room temperature.
- Place the samples on the magnet until the solution is clear.
- Transfer 13 μl of the eluate to a new 96-well plate.
- Check the quality of the final library (1/10 dilution of the prepared library) by electrophoresis on the AATI Fragment Analyzer using the HS NGS Fragment Kit. An example of successful libraries is shown in Figure 2.
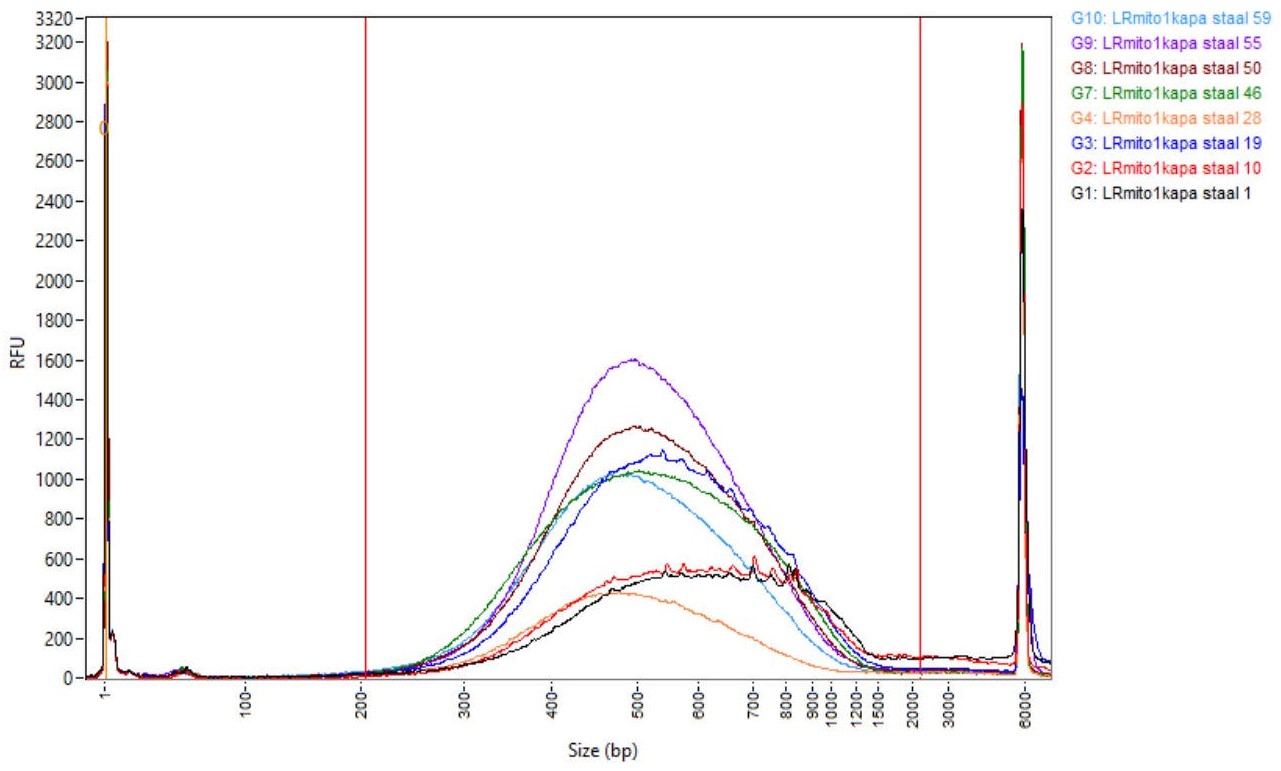
Figure 2. Successfully prepared libraries shown by electrophoresis on an AATI Fragment Analyzer - Quantify all samples using the Qubit according to the supplier’s instructions.
- Use the average size (smear analysis on the AATI Fragment Analyzer between 200 and 1500 base pairs) and the Qubit concentration, to calculate the molarity of the obtained library. Use the formula below:
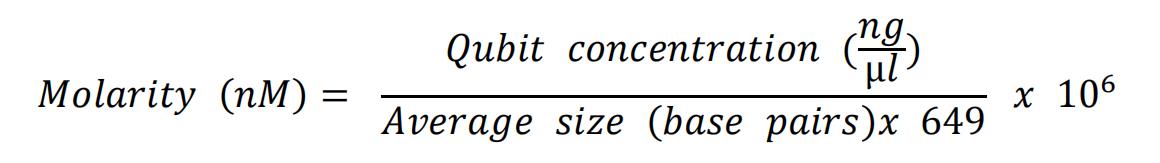
- Dilute all libraries to 2 nM in EBT buffer.
- Pool all the samples by combining equal volumes of each 2 nM library.
- Load the samples on the Illumina platform of choice. For the Illumina NovaSeq6000, perform the library denaturation as mentioned in the NovaSeq6000 Sequencing System Guide (Document 1000000019358v11). For 96 samples, use 6% capacity (matching the denaturation of 9 μl of 2 nM library pool) of the NovaSeq6000 S2 Reagent Kit (200 cycles).
Data analysis
- Demultiplex the base call (.bcl) files to .fastq files with Illumina bcl2fastq v2.19 script.
- Extract ad random 1.5 million reads from the .fastq files with the ‘seqtk’ tool.
Note: This is to reduce the number of reads because a high number can cause computational problems in Step 6. - Align the .fastq file to the reference genome (NC_012920.1) and generate a .bam file using BWA-MEM.
- Realign for insertions and deletions and base recalibrations (from .bam to .bam) with GATK v3.3.
- For variant calling, use the tool GATK v3.6Mutect2 (from .bam to .vcf).
Note: The code for the previous steps 1-5 can be found in the Supplementary File 1. - Upload the .bam file to mtDNA server (https://mtdna-server.uibk.ac.at/index.html).
- The mtDNA server report shows:
- Heteroplasmic variants (an example is shown in Figure 3).
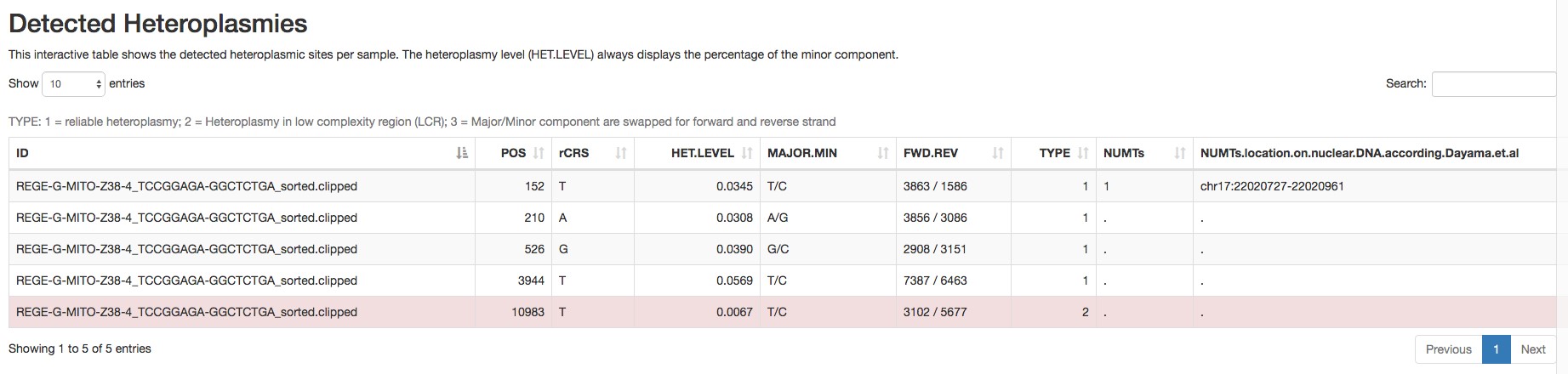
Figure 3. Report of the detected heteroplasmic variants by mtDNA server - Homoplasmic variants (an example is shown in Figure 4).
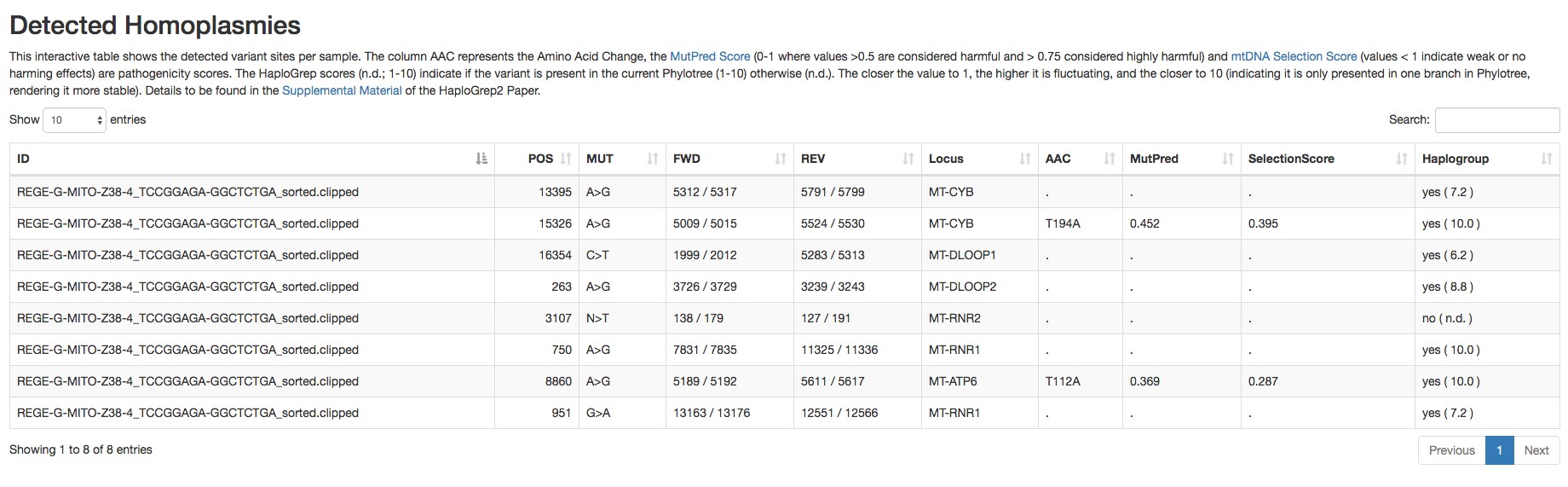
Figure 4. Report of the detected homoplasmic variants by mtDNA server - Haplogroup and possible contamination with other haplogroups (an example is shown in Figure 5).
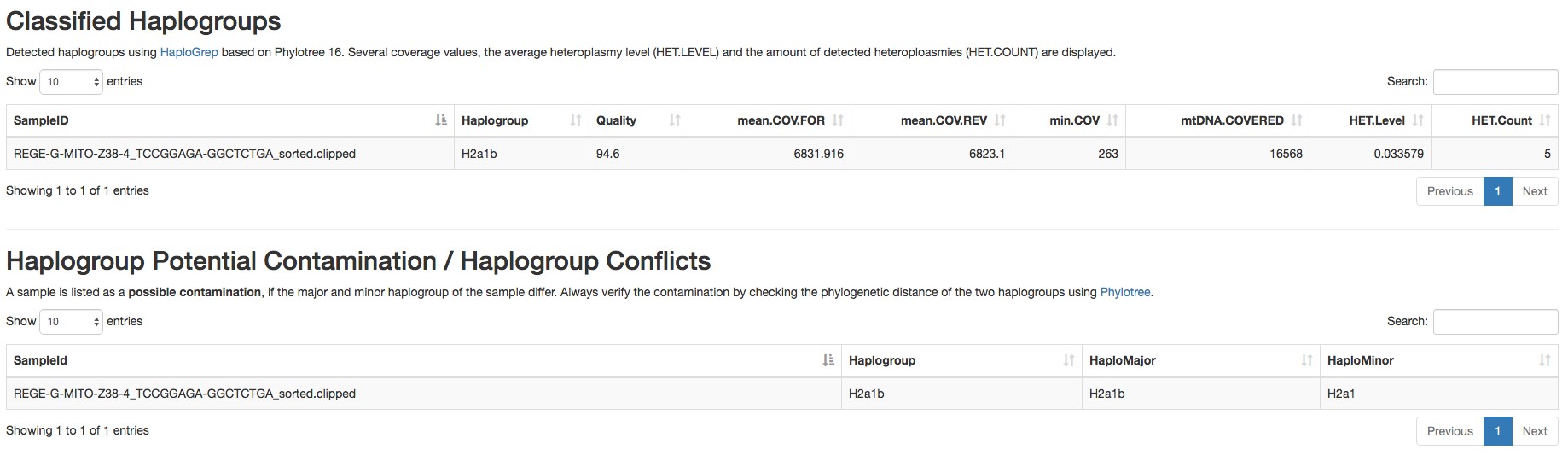
Figure 5. Report of the haplogroup detected by mtDNA server - Coverage plot (an example is shown in Figure 6).
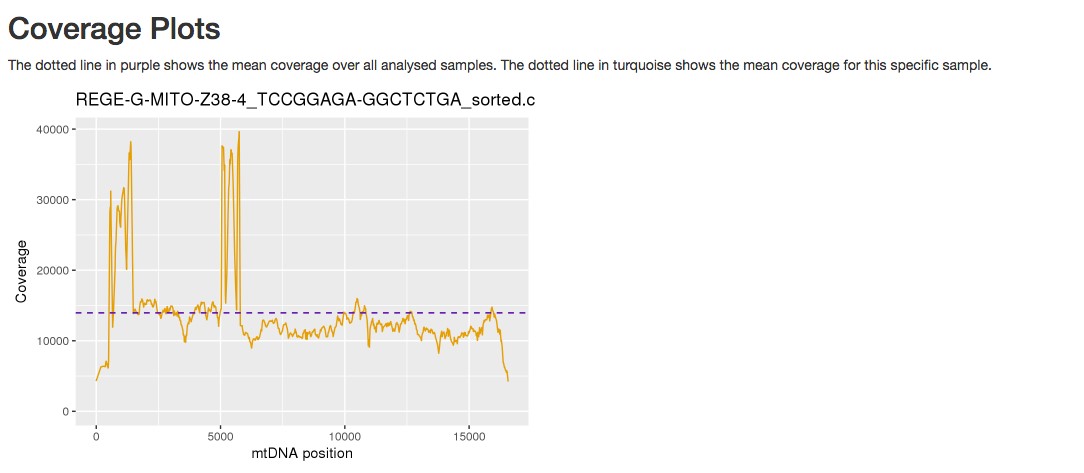
Figure 6. Example of a coverage plot provided by mtDNA server
- Heteroplasmic variants (an example is shown in Figure 3).
- Open the Mutect file and sort the variants per frequency.
- Open the Mutect file and follow the process shown in Figure 7:
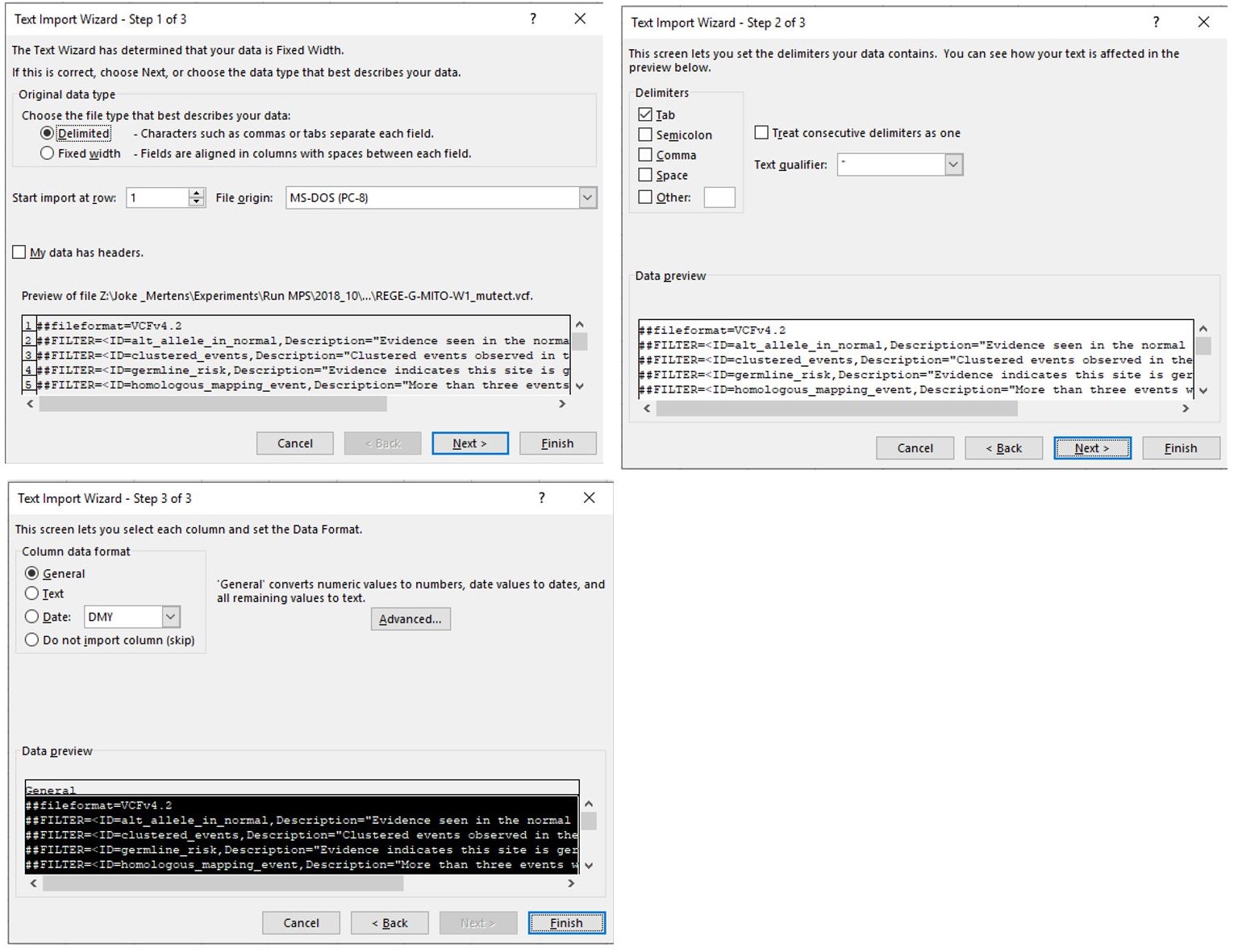
Figure 7. Sequential steps to open a Mutect file in a Microsoft Office Excel. Make sure that “Delimited” is indicated in step 1 and follow the standard settings. - When the file is opened, select the complete column below cell J39 as shown in Figure 8.
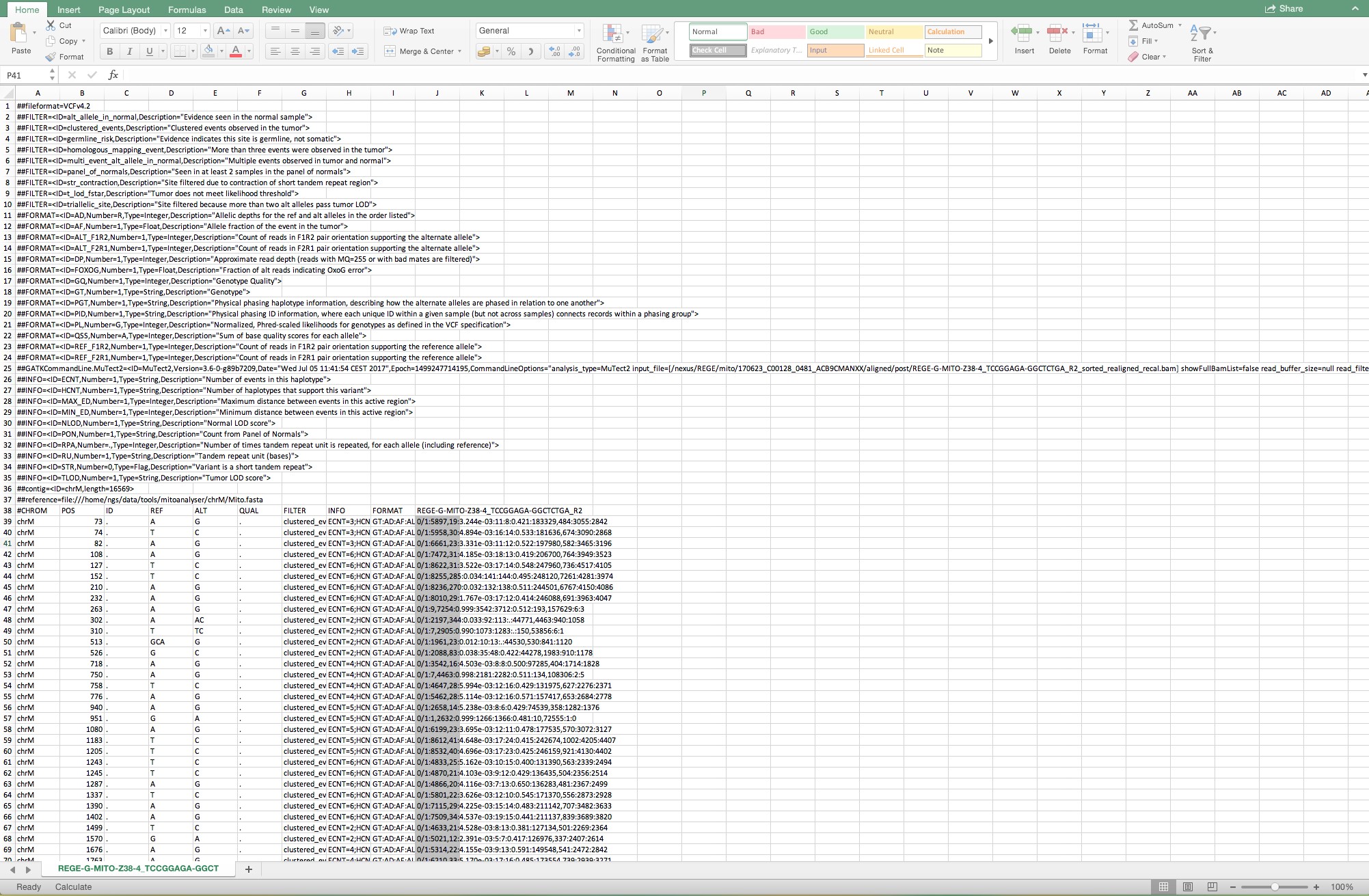
Figure 8. Lay-out when a Mutect file is opened and the complete column below cell J39 is selected - Convert the selected area to columns by selecting the tool “Convert text to columns” as shown in Figure 9.
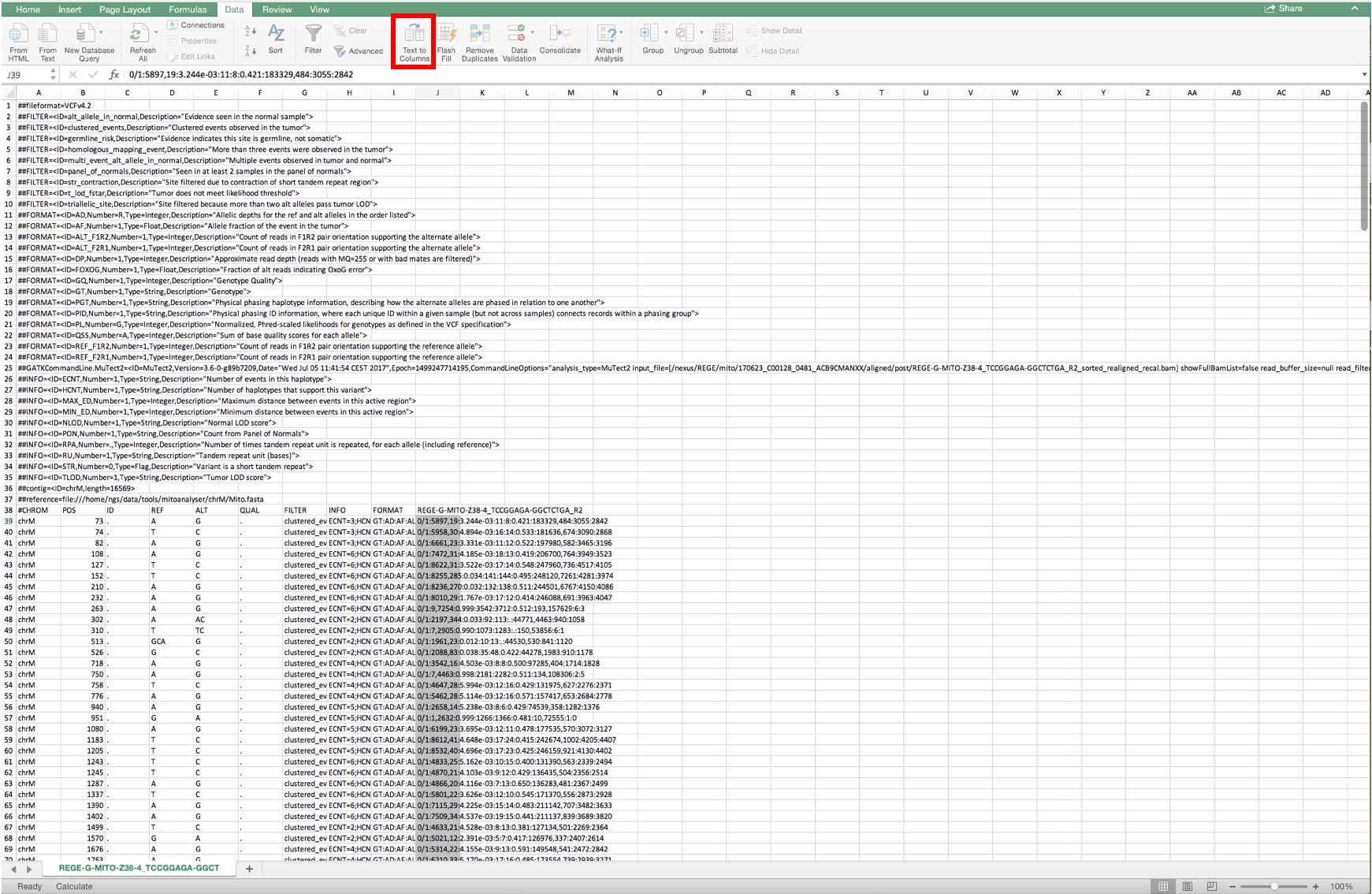
Figure 9. Conversion of text to columns in the complete column below cell J39
- Follow these setting to complete the conversion as shown in Figure 10.
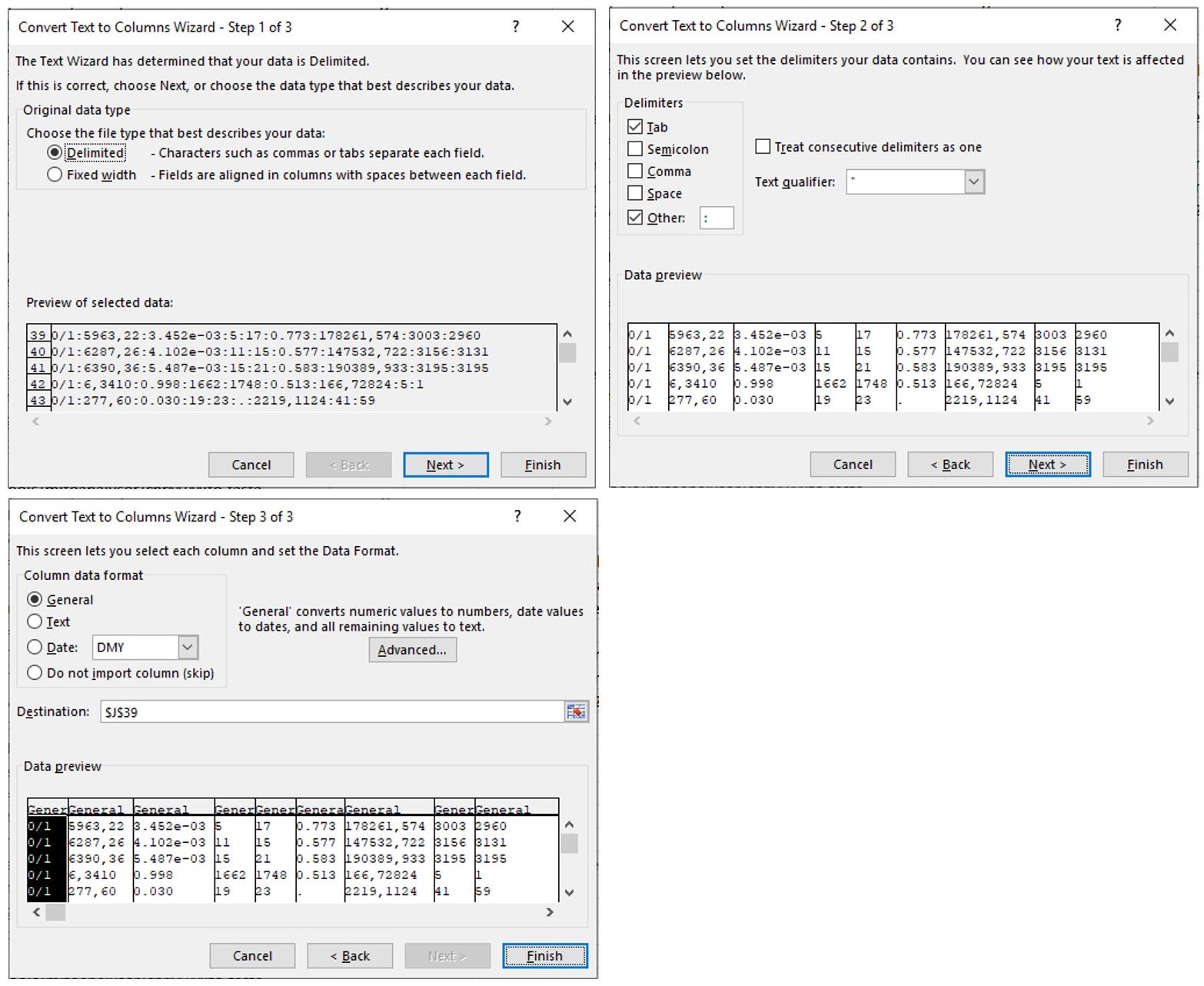
Figure 10. Sequential steps to complete the conversion from text to columns. Make sure that “Delimited” is indicated in step 1 and that the delimiter “other” is indicated with a semicolon as the delimiter. Follow the standard settings in step 3. - Select all rows from Row 39 and sort in “Column L” for “Largest to smallest” as shown in Figure 11.
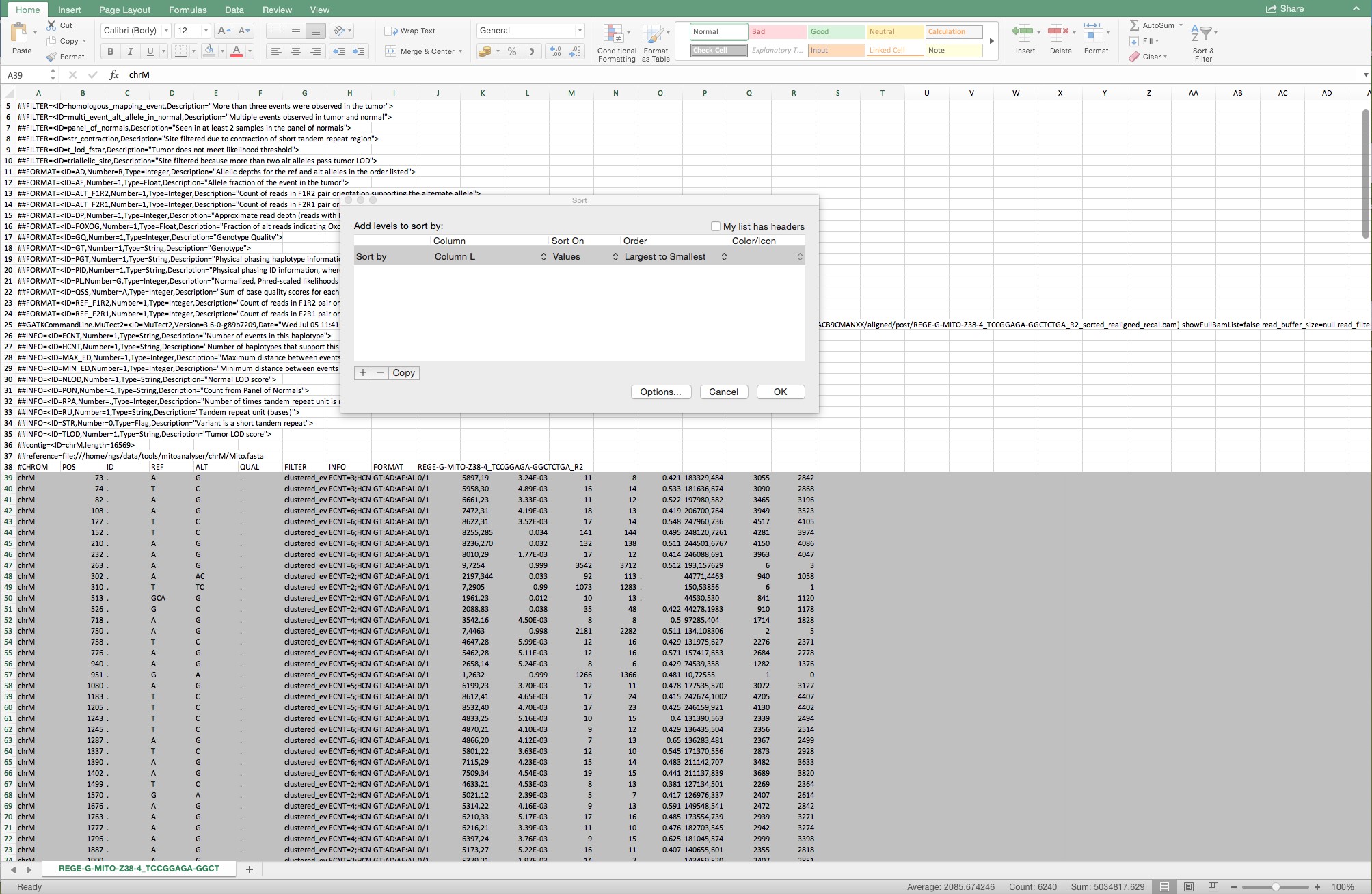
Figure 11. Example of the selected rows from Row 39 and of the sorting from largest to smallest from all rows below Row 39
- The variants > 0.015 in column L are selected for further analysis. Column K represents the forward and reverse reads of the position divided by a comma. Only variants with total reads (forward + reverse) above 1000 are considered (Figure 12).
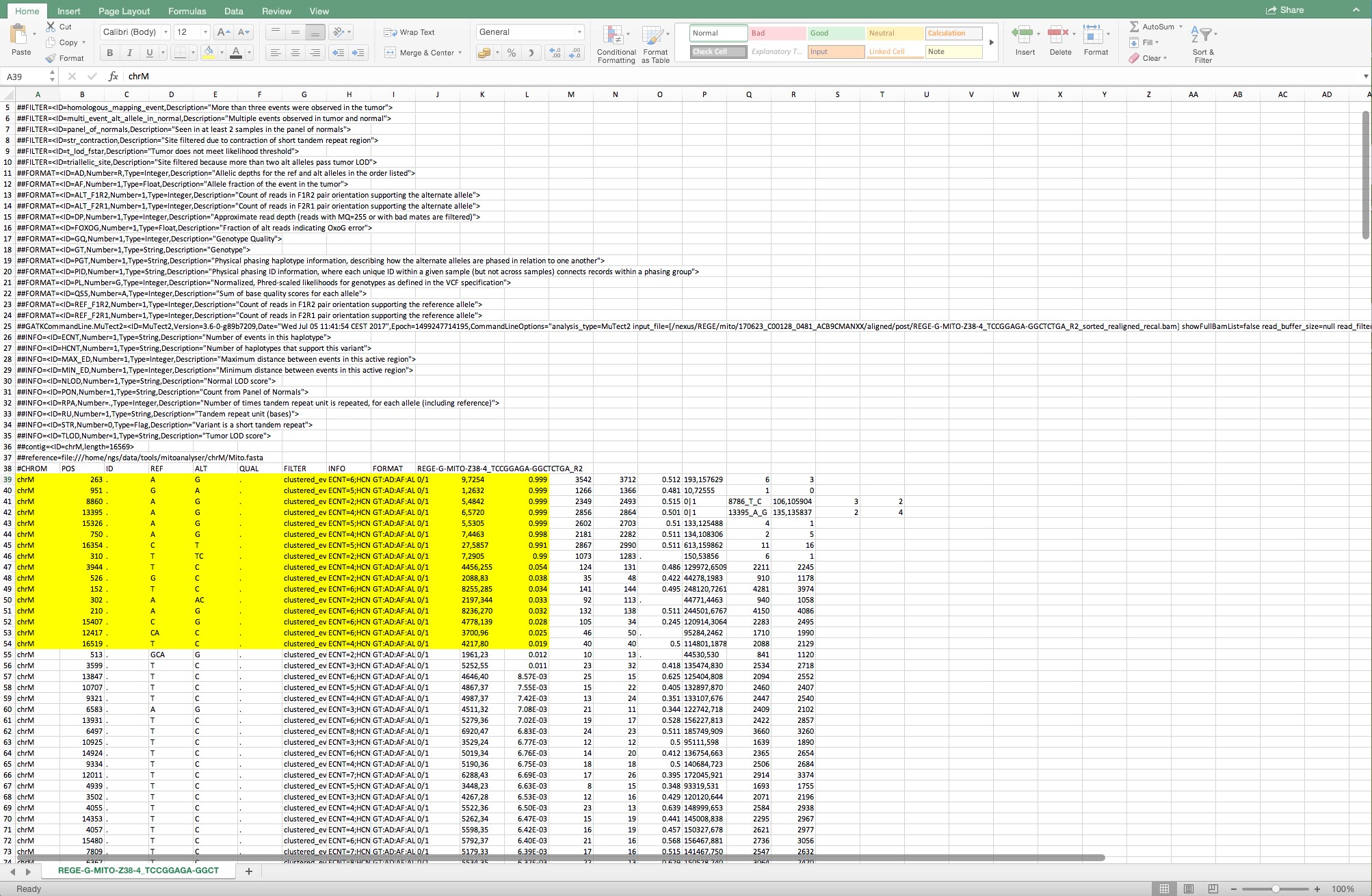
Figure 12. The cells shown in yellow are the variants that are considered for further analysis
- Open the Mutect file and follow the process shown in Figure 7:
- Crosscheck the frequency of the variants detected by MuTect with the variants of mtDNA server and report possible insertions/deletions.
- If there is a discrepancy, calculate the frequency given by the coverage file as follows:
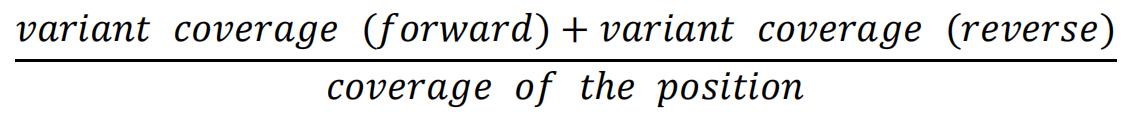
- Annotate the variant by looking up the region on http://mitowheel.org/mitowheel.html. An example is shown in Figure 13.
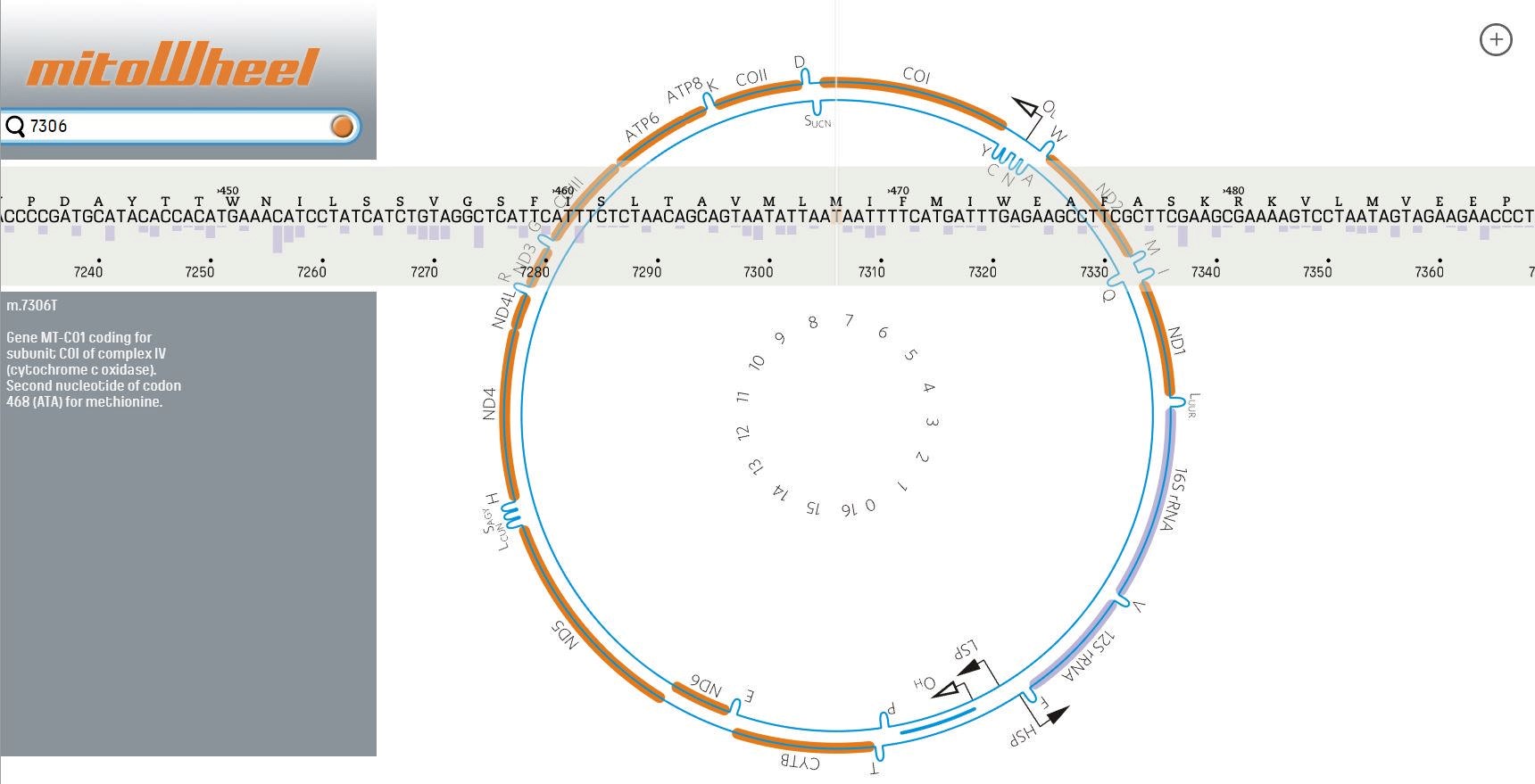
Figure 13. Results of the annotation provided by mitoWheel for the position 7306 - If the variant is in a protein-coding region, identify the potential amino acid change on http://mitimpact.css-mendel.it/. An example is shown in Figure 14.
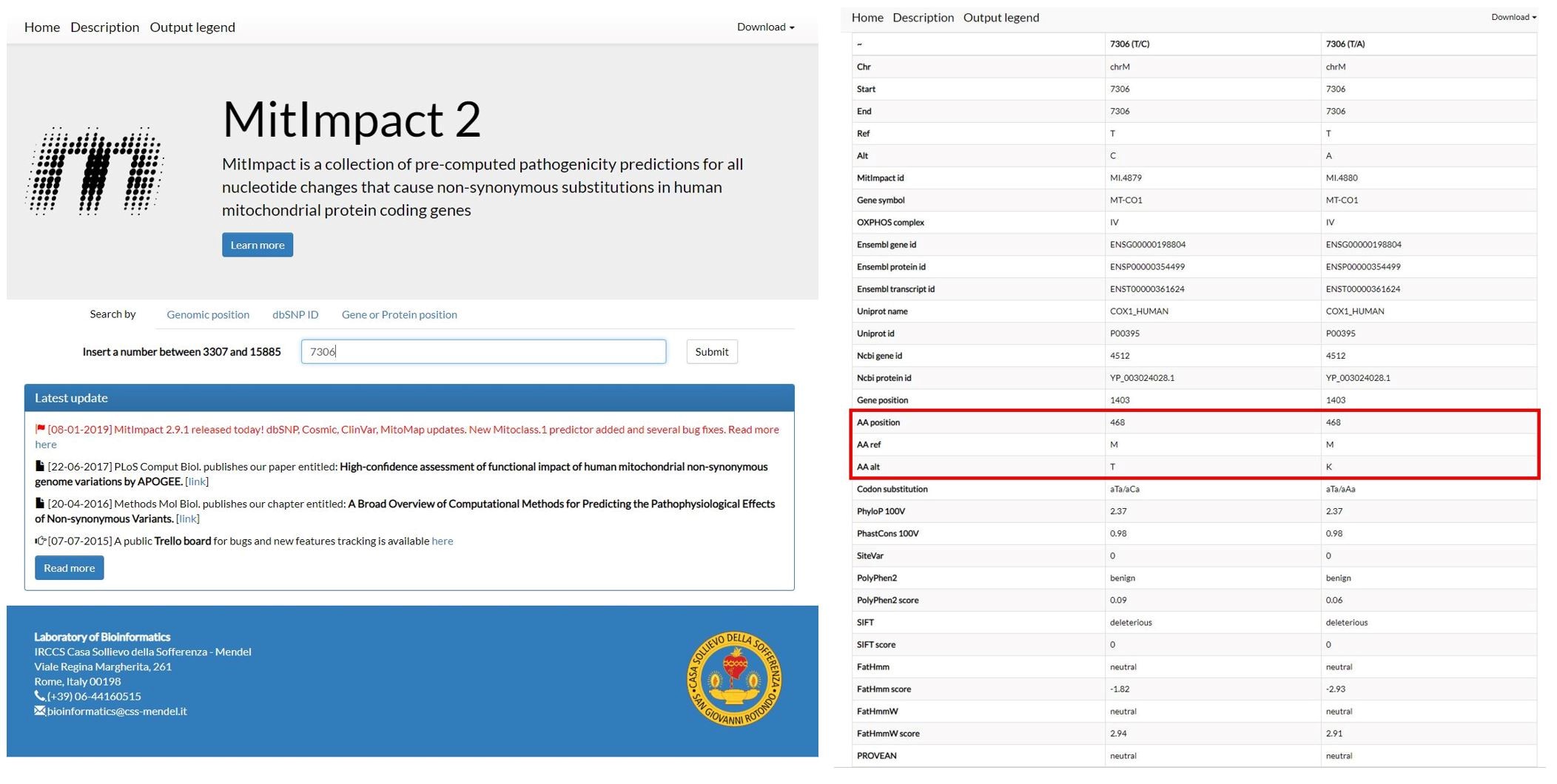
Figure 14. Results for the variants that cause an amino acid change on the position 7306 provided by MitImpact2 - Check if the amino acid change is pathogenic by uploading the amino acid sequence and amino acid changes on http://mutpred.mutdb.org/. An example is shown in Figure 15.
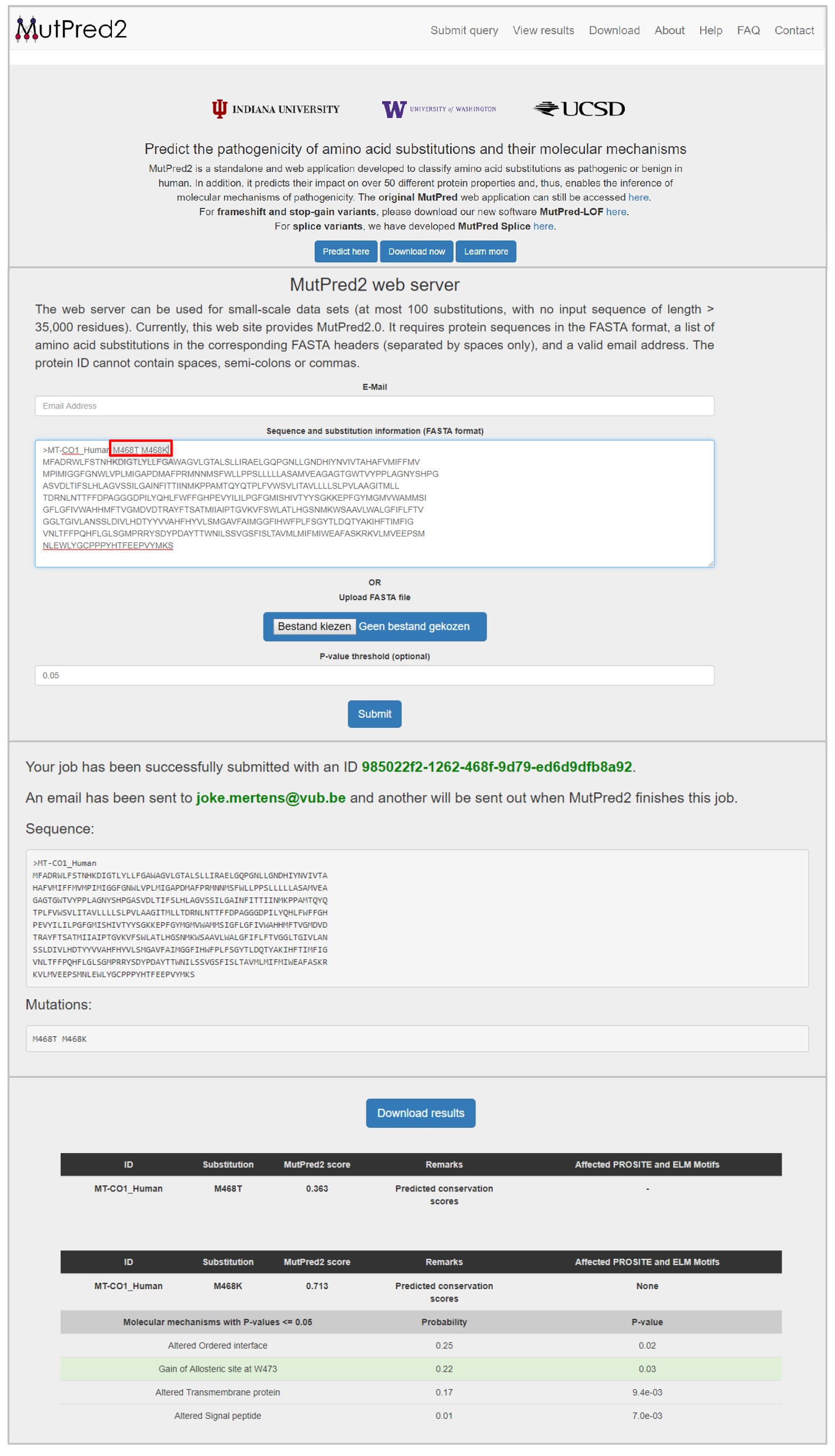
Figure 15. Results of the pathogenicity predictor MutPred2 for the amino acid changes M468T and M468K in the MT-CO1 gene - Certain regions were excluded from the analysis. These excluded regions can be found in the supplementary data of Zambelli et al., 2018 (https://www.cell.com/stem-cell-reports/fulltext/S2213-6711(18)30224-8#secsectitle0020).
Recipes
- Gel electrophoresis
50 ml of 1x TBE buffer
0.5 g agarose - Alkaline Lysis Buffer
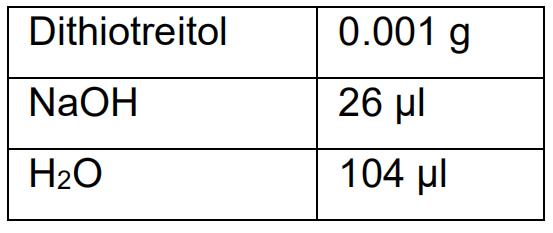
- EBT (Elution Buffer with Tris) buffer
10 mM Tris-HCl pH 8.5
1% Tween 20
Acknowledgments
This work was funded by the Wetenschappelijke Fonds Willy Gepts (UZ Brussel), the Fonds voor Wetenschappelijk Onderzoek (FWO, 1506616N) and the Methusalem grant of the Vrije Universiteit Brussel to Prof. Karen Sermon.
Competing interests
None.
Ethics
In this protocol, we have used human mtDNA for optimization only. All donors gave their informed consent and all studies related to this protocol received approval of appropriate local ethical committees.
References
- Bai, R. K. and Wong, L. J. (2004). Detection and quantification of heteroplasmic mutant mitochondrial DNA by real-time amplification refractory mutation system quantitative PCR analysis: a single-step approach. Clin Chem 50(6): 996-1001.
- Just, R. S., Irwin, J. A. and Parson, W. (2015). Mitochondrial DNA heteroplasmy in the emerging field of massively parallel sequencing. Forensic Sci Int Genet 18: 131-139.
- Rohlin, A., Wernersson, J., Engwall, Y., Wiklund, L., Bjork, J. and Nordling, M. (2009). Parallel sequencing used in detection of mosaic mutations: comparison with four diagnostic DNA screening techniques. Hum Mutat 30(6): 1012-1020.
- Spits, C., Le Caignec, C., De Rycke, M., Van Haute, L., Van Steirteghem, A., Liebaers, I. and Sermon, K. (2006). Whole-genome multiple displacement amplification from single cells. Nat Protoc 1(4): 1965-1970.
- Ye, F., Samuels, D. C., Clark, T. and Guo, Y. (2014). High-throughput sequencing in mitochondrial DNA research. Mitochondrion 17: 157-163.
- Zambelli, F., Mertens, J., Dziedzicka, D., Sterckx, J., Markouli, C., Keller, A., Tropel, P., Jung, L., Viville, S., Van de Velde, H., Geens, M., Seneca, S., Sermon, K. and Spits, C. (2018). Random mutagenesis, clonal events, and embryonic or somatic origin determine the mtDNA variant type and load in human pluripotent stem cells. Stem Cell Reports 11(1): 102-114.
- Zambelli, F., Vancampenhout, K., Daneels, D., Brown, D., Mertens, J., Van Dooren, S., Caljon, B., Gianaroli, L., Sermon, K., Voet, T., Seneca, S. and Spits, C. (2017). Accurate and comprehensive analysis of single nucleotide variants and large deletions of the human mitochondrial genome in DNA and single cells. Eur J Hum Genet 25(11): 1229-1236.
- Zhang, W., Cui, H. and Wong, L. J. (2012). Comprehensive one-step molecular analyses of mitochondrial genome by massively parallel sequencing. Clin Chem 58(9): 1322-1331.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Mertens, J., Zambelli, F., Daneels, D., Caljon, B., Sermon, K. and Spits, C. (2019). Detection of Heteroplasmic Variants in the Mitochondrial Genome through Massive Parallel Sequencing. Bio-protocol 9(13): e3283. DOI: 10.21769/BioProtoc.3283.
Category
Molecular Biology > DNA > DNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.