- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Live-cell Migration Assays to Study Motility of Neural and Glial (Oligodendrocyte) Progenitor Cells
Published: Vol 9, Iss 12, Jun 20, 2019 DOI: 10.21769/BioProtoc.3275 Views: 5951
Reviewed by: Giusy TornilloSubhamoy DasZe Yang
Original research article
The authors used this protocol in:

Aug 2016
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Cell motility has been extensively studied in in vitro models using fibroblasts and keratocytes, but the cell type-specific mechanisms underlying migration of lineage- or disease-specific cells, such as neural and glial progenitor cells, remain an active field for investigation. The migrating neural and glial progenitor cells contribute to the development, tissue repair and tumor invasion in the central nervous system (CNS). Cell migration is a highly dynamic process which relies on membranous protrusions to assemble, extend, disassemble and retract. In the CNS, the motility of neural and glial progenitor cells is affected by various cell-autonomous and non-cell-autonomous mechanisms such as signaling molecules, actin and microtubule interactions, and environmental cues. Here, we described a live-cell migration assay for use in the assessment of neural and glial progenitor cell migration. We first will demonstrate the procedures for isolating and culturing neural and glial progenitor cells. Next, we will demonstrate the acquisition of time-lapse images using phase contrast microscopy, the methods for quantification and the analyses of various motility parameters including speed, velocity, straightness and leading-edge dynamics. This method allows researchers to dissect the mechanisms of cell motility in response to different environmental cues, such as chemoattractive and repulsive signals, matrix adhesiveness and stiffness. This assay also allows researchers to study migration of pharmacologically and genetically manipulated cells.
Keywords: Cell migration assaysBackground
Cell migration plays a key role in many physiological (morphogenesis, tissue repair, regeneration) and pathological (tumor metastasis, atherosclerosis) processes. It is a highly orchestrated multistep process, which requires dynamic interactions between cells and the extracellular cues such as chemokines and signals from the extracellular matrix. In response to the extracellular cues, cells acquire a polarized morphology. Cell polarization requires delicate regulation of microtubule and actin polymerization for asymmetrical distribution of signaling molecules, cytoskeletal proteins, and directional vesicle trafficking. At the leading edge, actin polymerization drives the extension of membrane protrusions, such as lamellipodia (facilitating focal adhesion formation to anchor the protrusion) and filopodia (sensing the environment). In order to move forward, cells have to disassemble adhesions on the trailing edge and retract the trailing end, which require coordination of microtubule and actin networks (Palazzo and Gundersen, 2002; Wehrle-Haller and Imhof, 2003; Etienne-Manneville, 2004). Disruption of cell migration and polarization would affect the process of neurogenesis, and may lead to neurodevelopmental disorders or brain tumor progression (Rakic, 2003; Götz and Huttner, 2005; Taylor et al., 2005; Wang et al., 2016).
Neural and glial progenitor cells are the major proliferating and migrating cells in the CNS, and are crucial for the development of neurological functions of an organism. Neural and glial progenitor cells have also been implicated as the cell origin of gliomas. Indeed, glioma migration and infiltration recapitulate key aspects of glial progenitor cell migration during development. Specifically, glioma cells, similar to glial progenitors in the developing CNS, bear a unipolar or a bipolar morphology with a leading process and migrate along white matter tracts and blood vessels (Cayre et al., 2009).
In this report, we will introduce a live-cell migration assay and the analytical methods which allow us to study cell motility in the presence of inhibitors or different environmental cues. We will first demonstrate how to isolate and culture neural and glial progenitor cells. Next, we will demonstrate our approach to capturing time-lapse images, and to tracking migrating cells for quantification. The live-cell migration assay can be applied to neural and glial progenitor cells as well as to tumor spheres. This assay can also be used to study cell motility in response to inhibitors, different extracellular substrates and stiffness, attractive or repulsive cues, as well as changes in the electrical field (Li et al., 2015) without optimizing the duration of migration and final detection.
Materials and Reagents
- 10 cm Petri dish (Thermo Fisher Scientific, catalog number: 172931)
- 15 cm Petri dish (Thermo Fisher Scientific, catalog number: 168381)
- 15 ml conical tube (Thermo Fisher Scientific, catalog number: 339651)
- 40 μm cell strainer (Argos Technologies, catalog number: TC1040-A)
- 6-well plate (Thermo Fisher Scientific, catalog number: 140675)
- Glass bottom dish (MatTek, catalog number: P35G-1.5-14-C)
- 1 ml pipette
- Serological pipet
- Timed-pregnant mouse embryos
Note: Neural and glial progenitor cells can be isolated from embryonic day (E) 13.5 to E15.5 embryos, a stage where early and active neurogenesis occurs. Note that, during this stage, progenitor cells evolve rapidly between proliferative expansion and cell lineage differentiation, Therefore, it is critical to select embryos from similar developmental time points in experiments where comparison between progenitor cells from different embryos (e.g., certain genetic models, different treatment groups) are unavoidable. - Ethanol
- Hanks’ balanced salt solution (HBSS) (Thermo Fisher Scientific, catalog number: 14025092)
- DMEM/F12 (Thermo Fisher Scientific, catalog number: 11330032)
- Insulin (Sigma, catalog number: I0516)
- N2 supplement (Thermo Fisher Scientific, catalog number: 17502048)
- B27 supplement (Thermo Fisher Scientific, catalog number: 17504044)
- Epithelial growth factor (EGF) (Peprotech, catalog number: AF-100-15)
- Basic fibroblast growth factor (bFGF) (Peprotech, catalog number: AF-100-18B)
- DMSO (EMD Millipore, catalog number: S-002-D)
- Dulbecco’s PBS (DPBS), no calcium, no magnesium (Thermo Fisher Scientific, catalog number: 14190144)
- Accutase (Thermo Fisher Scientific, catalog number: A1110501)
- Platelet-Derived Growth Factor AA (PDGF-AA) (Peprotech, catalog number: AF-100-13A)
- Poly-L-ornithine (PLO) (Sigma, catalog number: P4957)
- Laminin (Thermo Fisher Scientific, catalog number: 23017015)
- Dulbecco’s PBS, with calcium and magnesium (Thermo Fisher Scientific, catalog number: 14040133)
- Neural Growth Medium (NGM) (see Recipes)
- Oligosphere Medium (OM) (see Recipes)
Equipment
- Pipettes
- Micro scissor (Ted Pella, catalog number: 1346)
- Tweezer (Ted Pella, catalog number: 525)
- Dressing forceps (Ted Pella, catalog number: 5002-42)
- Dissection sharp/blunt scissors (Ted Pella, catalog number: 1329)
- Hemocytometer
- Centrifuge
- Inverted live-cell imaging system, connected to an incubator with a controlled temperature (temperature module) and CO2 module such as EVOSTM FL Auto (Thermo Fisher Scientific)
Software
- Chemotaxis software (ibidt, https://ibidi.com/44-software-and-image-analysis)
- ImageJ (https://imagej.nih.gov/ij/index.html)
Procedure
- Isolation and culture of mouse neural stem cells (NSCs)
- Isolation of neural stem cells from E14.5 mouse brain
Day 1- Prepare several 10 cm Petri dishes filled with Hanks’ balanced salt solution (HBSS), place on ice.
- Euthanize an E14.5 timed-pregnant mouse with carbon dioxide following institutional animal protocols.
- Spray the abdomen with 70% ethanol to wet the fur and open the abdomen in Y-shape using standard dissection scissors and dressing forceps (Video 1).
 Video 1. Isolation of mouse cortices for NSC culture. (The University of Kansas Medical Center Animal Care and Use Committee approved all protocols performed (ACUP number:2016-2359, August 16, 2016-August 1, 2019).)
Video 1. Isolation of mouse cortices for NSC culture. (The University of Kansas Medical Center Animal Care and Use Committee approved all protocols performed (ACUP number:2016-2359, August 16, 2016-August 1, 2019).) - Dissect out the connective tissues and vessels between the uterine horns, remove the uterus and place it on a 10 cm Petri dish filled with ice-cold HBSS.
- Use micro-scissors (Ted Pella) and tweezers (Ted Pella) to separate the embryos from the placenta, transfer the embryos to a new 10 cm Petri dish filled with ice-cold HBSS.
- Decapitate, remove skin and skull bones, and carefully collect embryonic brains under a dissecting microscope.
Note: Place a 15 cm dish filled with ice under the 10 cm Petri dish to keep the embryos cold. - Remove the meninges and isolate the cortices, followed by dissecting the cortices into small pieces.
- Transfer the tissues to a 15 ml conical tube filled with ice-cold HBSS, centrifuge at 100 x g for 3 min at 4 °C.
- Remove supernatant, add ice-cold neural growth medium (NGM) (0.5 ml per brain), triturate with a 1 ml pipette, followed by incubation on ice for 2 min.
Note: Gently pass through a 1 ml pipette for approximately 20 times and avoid bubble formation during trituration. - Pass cell suspension through a 40 μm cell strainer to obtain single cells. Count the cells with a hemocytometer or cell/particle counter.
- Dilute single-cell suspension with NGM to a final concentration of 5 x 104 cells per ml with NGM.
- Culture 4 ml (5 x 104 cells per ml) of cells in each 6-well plate. Usually one embryo is sufficient to isolate around 106 total cells.
- On the following day, discard the differentiated cells that have been attached to the bottom of the dish by transferring suspension cells to a new well.
Days 3-6- Replace half medium with fresh NGM every other day for 4-5 days to allow neurosphere formation.
Note: Cautiously remove the 6-well plate from the incubator to avoid disturbing the neurospheres that are settled at the bottom of the well. Remove 2 ml of medium using P1000 piptetman from the top of each well gently to avoid accidental suctioning of the neurospheres during this time. Discard removed medium and replenish the well with 2 ml fresh medium pre-warmed to 37 °C. Cells may be cryopreserved in NGM with 10% DMSO at this step.
- Subculturing of neurospheres when the spheres reach a size of 100-150 μm under a microscope
- Transfer neurospheres with culturing medium to a 15 ml conical tube using serological pipet, centrifuge at 100 x g for 1 min. Remove supernatant.
- Resuspend neurospheres in 5 ml Dulbecco’s PBS (DPBS) without calcium and magnesium.
- Centrifuge at 100 x g for 1 min. Remove supernatant.
- Add 1 ml Accutase (Thermo Fisher Scientific) at room temperature for 3-5 min, followed by gentle trituration using a 1 ml pipette until no large clumps are visible.
Note: Gently pass through 1 ml pipette for approximately 20 times and avoid bubble formation. - Add 5 ml NGM to neutralize Accutase, leave the tube on the stand for 5 min to allow remaining cell clumps to settle at the bottom, collect the solution that contains dissociated single cells for cell counting. Centrifuge at 200-300 x g for 4 min. Remove supernatant.
Note: Cells may be cryopreserved in NGM with 10% DMSO at this step. - Resuspend cells with NGM to a final concentration of 2-3 x 104 cells per ml. Transfer 4 ml of cell suspension into each well of a 6-well plate.
- Replace half medium with fresh NGM every other day as described in Step A1n, repeat the subculturing process when the spheres reach a size of 100-150 μm (typically in 3-4 days).
- Isolation of neural stem cells from E14.5 mouse brain
- Induction and culture of glial progenitor cells
- Culture single-cell suspension 2-3 x 104 cells per ml from passages 2 to 6 of neurospheres in Oligosphere Medium (OM) to induce glial lineage differentiation.
Note: We found that induction efficiency is higher when using single-cell suspension from neurospheres rather than using undissociated neurospheres. Avoid adding ciliary neurotrophic factor (CNTF) to the medium to prevent astrocyte lineage differentiation. - Replace half medium every other day with fresh OM as described in Step A1n.
- Subculture spheres every 5-7 days for at least 12 days. The spheres should consist of > 95% glial progenitor cells at this time (Li et al., 2015).
- Culture single-cell suspension 2-3 x 104 cells per ml from passages 2 to 6 of neurospheres in Oligosphere Medium (OM) to induce glial lineage differentiation.
- Setting up the in vitro migration assay
- Preparation of poly-L-ornithine (PLO)-coated glass-bottomed dishes (MatTek).
- Add 200 μl PLO solution (Sigma) to glass-bottomed dishes. Place the dishes in the tissue culture incubator at 37 °C for 1 h.
- Remove PLO solution, allow the dishes to dry at the tissue culture hood for 5-10 min.
- Rinse the dishes with sterile water twice.
Note: PLO-coated glass-bottomed dishes can be stored at room temperature for 2 weeks.
- Seed cells onto the PLO-coated glass-bottomed dishes.
- Dilute mouse laminin (Thermo Fisher Scientific) with sterile water to a final concentration of 2 μg per ml. Add 150 μl diluted laminin onto the glass-bottomed dishes. Place the dishes in the tissue culture incubator for 1 h, followed by removing laminin and rinsing the dishes with calcium- and magnesium-containing DPBS once in the tissue culture hood.
- Add 100 μl NGM at the bottom of the PLO/laminin-coated dishes.
- Carefully transfer 100-150 μm spheres using 10 μl pipette under the microscope to the PLO/laminin-coated glass-bottomed dishes.
Note: We typically seed at least 8 spheres and image 6-8 spheres simultaneously. - Allow the spheres to attach to the bottom of the dishes in the tissue culture incubator at 37 °C for 15 min, followed by carefully adding 2 ml pre-warmed NGM.
Note: Avoid disturbing the spheres by adding NGM from the edge of the dish slowly.
- Preparation of poly-L-ornithine (PLO)-coated glass-bottomed dishes (MatTek).
- Microscope setup for time-lapse imaging
Note: Here we demonstrate how to take time-lapse images using the Invitrogen EVOSTM FL Auto Imaging System with the EVOSTM Onstage Incubator. For detailed information regarding microscope installation and operation procedures, please refer to the EVOSTM user manual.- Microscope setup
- Turn on the EVOSTM FL inverted microscope and the connected computer, select the appropriate stage holder, attach the environmental chamber to the microscope, followed by connecting the heated hose and the sensor cable to the environmental chamber.
- Add water to the “max fill” line in the water reservoir, and turn on the Onstage Incubator along with the air and CO2 tanks.
- Open the EVOSTM FL Auto software, set temperature at 37 °C, humidity > 80% and CO2 at 5%. Warm up for approximately 2 h.
Note: To prevent water from accumulating around the objective, an empty dish/well should be used during system warm-up. Also, please ensure sufficient gas supply during the experiment.
- Live-cell imaging
- Open the software. Choose the 10x objective and turn on the Trans light.
- Place a glass-bottomed dish with neurospheres attached to the bottom on the stage. Select 6-8 neurospheres with a similar size ranging between 100 and 150 μm by clicking “Add Beacon”.
Note: Please make sure the dish is tightly sealed around the rim of the stage. Any space would allow water to evaporate, causing blurry images and damaging the objective. - The duration of time-lapse image acquisition is approximately 2 h with a capturing interval of 2 min (for cells migrating on the surface coated with 20 μg per ml laminin).
Note: For statistical analysis, we use images taken every 4 min during a 2-h period. Total imaging duration partly depends on the speed of cell migration. When the concentration of laminin is low (e.g., 0.5 μg per ml), cells migrate more slowly. Therefore, the duration and interval of time-lapse imaging should be adjusted accordingly.
- Microscope setup
Data analysis
In the following section, we will demonstrate procedures for the analyses of three cell motility parameters–velocity, speed and straightness. Additionally, we will also demonstrate the use of kymography to assess leading edge dynamics (Figure 1). 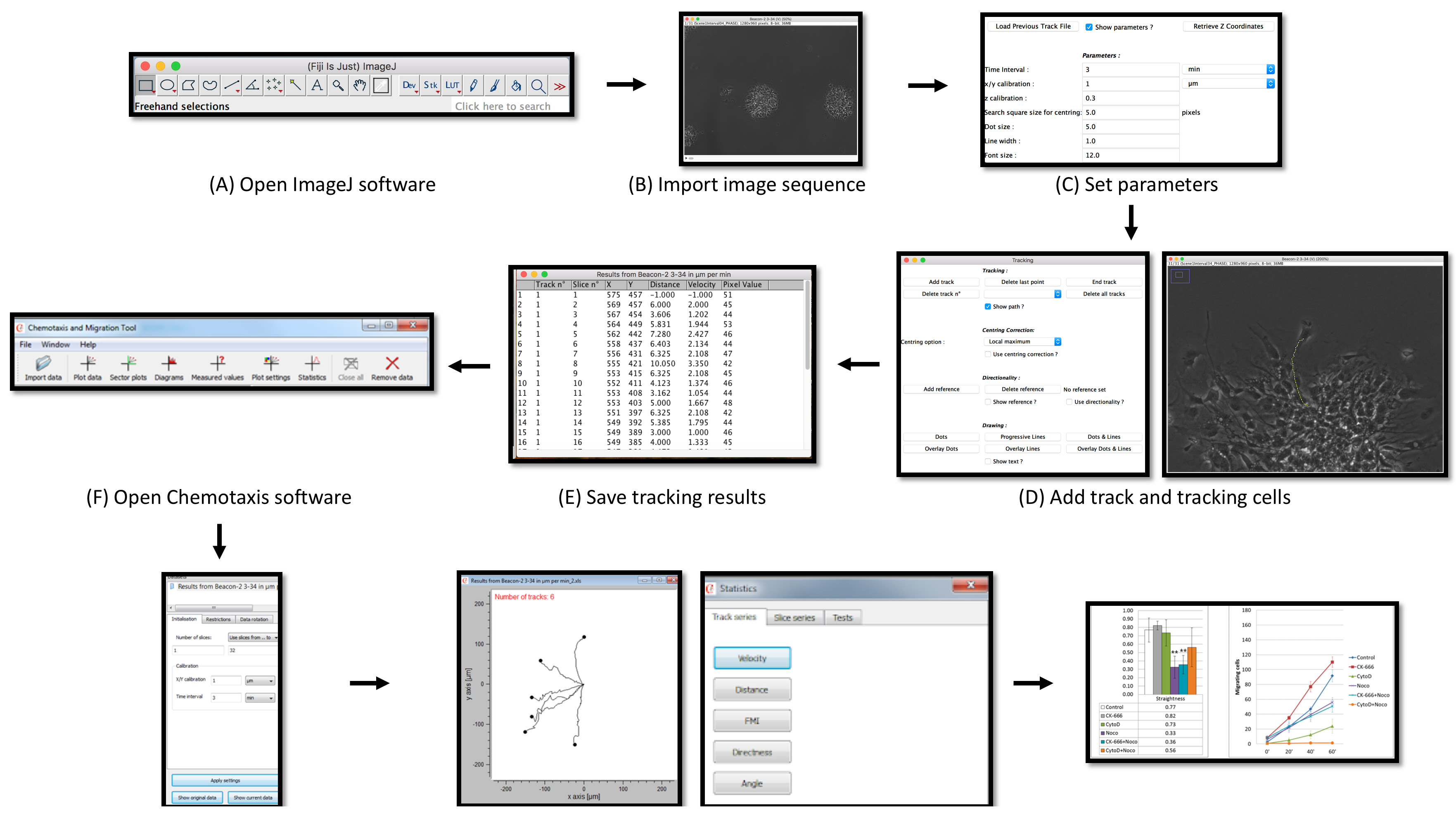
Figure 1. Schematic representation of cell motility analysis for neural and glial progenitor cells. (A-B) Following time-lapse image acquisition, image stacks were loaded onto ImageJ. (C-E) Paths of migrating cells were tracked. (F-H) Tracking data were opened in the ibidi Chemotaxis software for plotting and analysis. (I) Results were exported to a spreadsheet for graphing and statistical comparison.
- Velocity: Velocity is a vector quantity which is evaluated by displacement per time.
- Open ImageJ (freeware), click “Import” in the “File” pull-down menu and then click “Image Sequence”. Select the file that contains the time-lapse images (Video 2). Video 2. ImageJ setting up for migration assay
- Under “Plugins”, click “Tracking” and then “Manual Tracking”. Set parameters the same as those used for acquiring time-lapse images.
- Check “Show path?” and click “Add track”, followed by marking the cell of interest along its migration path. Tracking details will show up in a new window.
Note: We typically select cells that are on the edge of the spheres and we avoid selecting cells that undergo cell division during migration. - Click “Add track” again and mark the next cell of interest along its migration path. At least six cells from each neurosphere are typically tracked. Save the tracking results for all cells in the spreadsheet format.
- Open the ibidi Chemotaxis software, click “Import data” from the navigation bar to import the tracking result.
Note: ibidi Chemotaxis is a freeware. Please visit the ibidi website (https://ibidi.com/44-software-and-image-analysis) and follow the user instructions. - Set the parameters the same as the parameter used for acquiring time-lapse images under the “Initiation” tab. Click “Apply settings”.
- Click “Plot data” from the navigation bar. A window showing the tracking paths will pop up.
- Click “Statistics” from the navigation bar and then the “Velocity” tab to show the velocity results.
- Open ImageJ (freeware), click “Import” in the “File” pull-down menu and then click “Image Sequence”. Select the file that contains the time-lapse images (Video 2).
- Speed: Speed is a scalar quantity which is quantified as distance per time
- Repeat steps A1 to A7, if needed.
- Click “Statistics” from the navigation bar. Click “Distance” to display the migration distance for each tracked cell in a new window.
- Paste and save the result to a spreadsheet. Divide the distance by total time to obtain the speed for each cell.
- Straightness: Straightness of migration can be referred to as directness in ibidi Chemotaxis. If cells migrate without turning, the straightness would be 1. On the other hand, if cells keep turning around, the straightness would approach 0.
- Repeat steps A1 to A7, if needed.
- Click “Directness” from the “Statistics” window to show the directionality of each tracked cell.
- Paste and save the results to Excel for further analysis.
- Kymography: A kymograph is a graphic representation of spatial position over time. Kymography is a method to show leading edge dynamics during migration (Figure 2 and Video 3). Video 3. Kymography analysis by ImageJ
- Import phase-contrast images to ImageJ.
Note: We typically use images taken from a 20x objective (or higher) for Kymography analysis. - Use the line tool to draw a line on the edge of the migrating cell.
Note: The line should cover the entire span of the leading edge from the beginning to the end of the time-lapse images. - Under the pull-down menu “Plugins”, click “Kymograph” and then “KymographBuilder”.
Note: An alternative method is to click “Stacks” from the “Image” pull-down menu, then click “Reslice.”
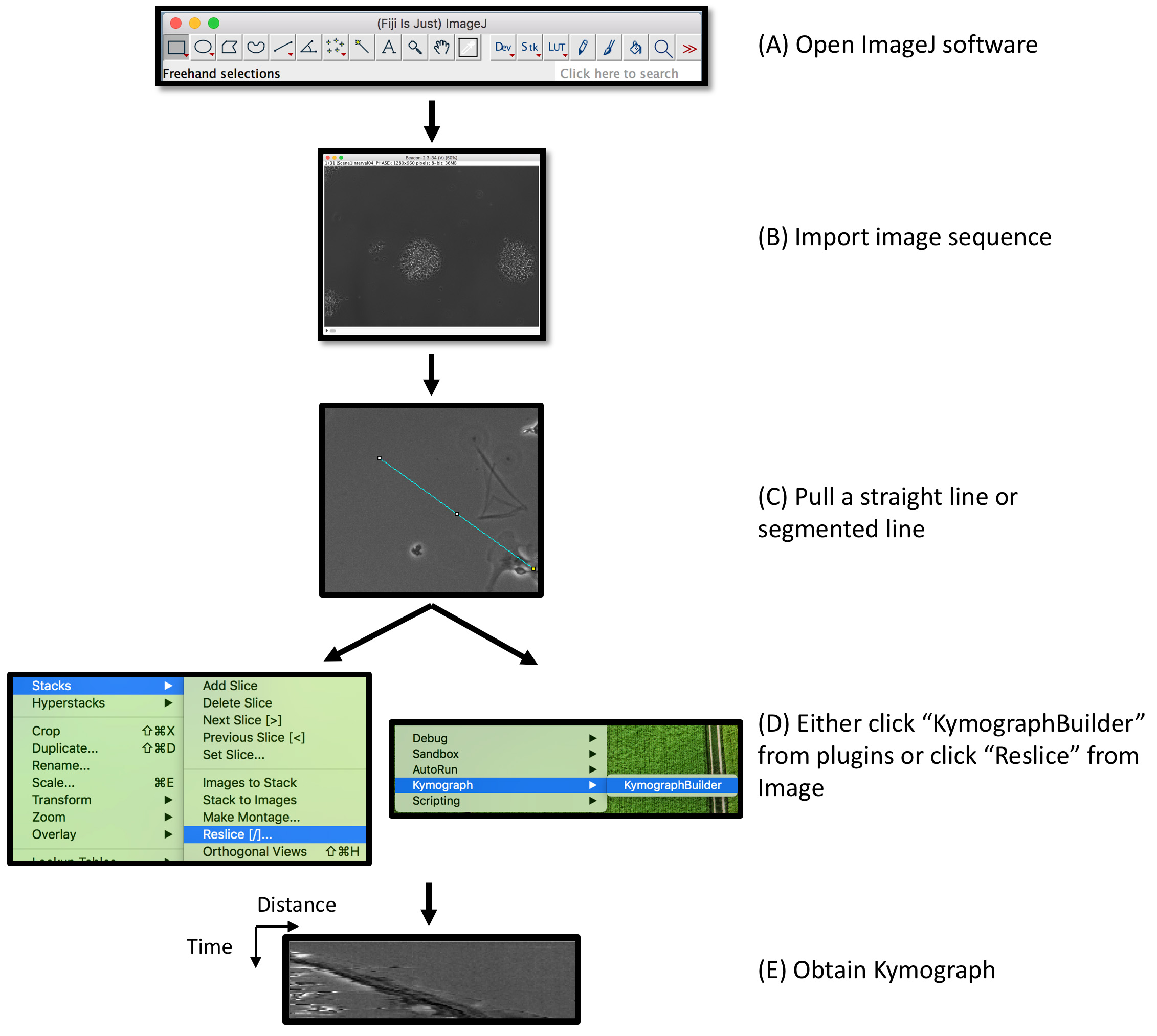
Figure 2. Schematic representation of kymography analysis. (A, B) Following time-lapse imaging acquisition, image stacks were loaded on ImageJ. (C, D) A straight line or a segmented line is drawn along the track of migrating leading edge, followed by obtaining a kymograph by either using the KymographBuilder or the Reslice function. (E) In a 2-dimentional kymograph, x-axis represents the distance of the leading edge, and the y-axis represents time. - Import phase-contrast images to ImageJ.
Recipes
- Neural Growth Medium (NGM)
DMEM/F12 with the final concentration of the following supplements and growth factors
1% N2
2% B27
20 ng/ml epithelial growth factor (EGF)
20 ng/ml basic fibroblast growth factor (bFGF)
After adding EGF and bFGF, the shelf life of NGM at 4 °C is approximately one week - Oligosphere Medium (OM)
DMEM/F12 with the final concentration of the following supplements and growth factors
1% N2
2% B27
20 ng/ml epithelial growth factor (EGF)
20 ng/ml Platelet-Derived Growth Factor AA (PDGF-AA)
After adding EGF and PDGF-AA, the shelf life of OM at 4 °C is approximately one week
Acknowledgments
This work was supported by funding from Children’s Mercy Children’s Research Institute.
Competing interests
The authors have no conflict of interest to disclose.
Ethics
The University of Kansas Medical Center Animal Care and Use Committee approved all protocols performed (ACUP number:2016-2359, August 16, 2016-August 1, 2019).
References
- Cayre, M., Canoll, P. and Goldman, J. E. (2009). Cell migration in the normal and pathological postnatal mammalian brain. Prog Neurobiol 88(1): 41-63.
- Etienne-Manneville, S. (2004). Actin and microtubules in cell motility: which one is in control? Traffic 5(7): 470-477.
- Götz, M. and Huttner, W. B. (2005). The cell boilogy of neurogenesis. Nat Rev Mol Cell Biol 6(10): 777-788.
- Li, Y., Wang, P. S., Lucas, G., Li, R. and Yao, L. (2015). ARP2/3 complex is required for directional migration of neural stem cell-derived oligodendrocyte precursors in electric fields. Stem Cell Res Ther 6: 41.
- Palazzo, A. F. and Gundersen, G. G. (2002). Microtubule-actin cross-talk at focal adhesions. Sci STKE 2002(139): pe31.
- Rakic, P. (2003). Developmental and evolutionary adaptations of cortical radial glia. Cereb Cortex 13(6): 541-549.
- Taylor, M. D., Poppleton, H., Fuller, C., Su, X., Liu, Y., Jensen, P., Magdaleno, S., Dalton, J., Calabrese, C., Board, J., Macdonald, T., Rutka, J., Guha, A., Gajjar, A., Curran, T. and Gilbertson, R. J. (2005). Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 8(4): 323-335.
- Wang, P. S., Chou, F. S., Ramachandran, S., Xia, S., Chen, H. Y., Guo, F., Suraneni, P., Maher, B. J. and Li, R. (2016). Crucial roles of the Arp2/3 complex during mammalian corticogenesis. Development 143(15): 2741-2752.
- Wehrle-Haller, B. and Imhof, B. A. (2003). Actin, microtubules and focal adhesion dynamics during cell migration. Int J Biochem Cell Biol 35(1): 39-50.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Chen, C., Chou, F. and Wang, P. (2019). Live-cell Migration Assays to Study Motility of Neural and Glial (Oligodendrocyte) Progenitor Cells. Bio-protocol 9(12): e3275. DOI: 10.21769/BioProtoc.3275.
Category
Neuroscience > Cellular mechanisms > Cell isolation and culture
Cell Biology > Cell movement > Cell migration
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.