- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Probing Conformational States of a Target Protein in Escherichia coli Cells by in vivo Cysteine Cross-linking Coupled with Proteolytic Gel Analysis


Published: Vol 9, Iss 12, Jun 20, 2019 DOI: 10.21769/BioProtoc.3271 Views: 5298
Reviewed by: Manjula MummadisettiChristopher HineAnna Vangone
Original research article
The authors used this protocol in:

Jan 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Transporters are dynamic membrane proteins that are essential to the physiology of cells. To function, transporters must cycle between various conformational states, so to understand their mechanistic details, it is critical to characterize how their structure changes during the transport cycle. One approach to studying the dynamics of transporters takes advantage of the chemistry of cysteine by using sulfhydryl-reactive, bi-functional cross-linkers to probe changes in the distance between two specific residues that have been substituted to cysteine. This approach is mostly used to study transporters in vitro, not in their natural cellular environment. Here we describe a protocol based on structure-guided cysteine cross-linking and proteolysis-coupled gel analysis to probe conformational changes of a target transporter in live Escherichia coli cells. Although cross-linking approaches have been used to probe the proximity between transmembrane segments in membrane proteins in vivo, to our knowledge this protocol is the first to be used to interrogate transporter dynamics in cells. The use of this protocol is optimal for proteins with known or modeled structures to guide the replacement of specific residues with cysteines and the selection of cross-linking agents with various spacer arm lengths. This protocol allows for discriminating easily cross-linked and uncross-linked species and does not require the often difficult or unavailable reconstitution of transport activity in an in vitro system. In addition, this protocol could be used to probe the conformation of transporters in cells treated with transport inhibitors in order to better understand their mechanism of action, and potentially dynamic interactions between domains in proteins that are not transporters.
Keywords: Conformational dynamicsBackground
Transport of substrates across membranes is fundamental to all organisms. Membrane proteins that function as transporters constitute a very large and structurally diverse group of proteins that translocate a vast landscape of substrates through different mechanisms. Not surprisingly, approximately 25% of polypeptides encoded in fully sequenced genomes are predicted to be membrane proteins, and many of them are likely transporters (Elofsson and von Heijne, 2007). Their essential role in human health is underscored by the fact that genetic defects in specific transporters cause disease (e.g., cystic fibrosis), and that some transporters are targeted by pharmaceuticals (e.g., inhibitors of efflux pumps) (Meng et al., 2018; Wiese and Stefan, 2019). Studying transporters is therefore important, but it is also challenging given their dynamic nature and the inherent difficulty of working with membrane proteins in biochemical and structural studies. Especially difficult is obtaining high-resolution structures, particularly those representing different states in the transport cycle. Even when available, static structures must be validated and their role in the transport cycle elucidated by complementary in vitro and in vivo assays that might not be available for many transporters (Mulligan and Mindell, 2017).
Chemical biology approaches such as cysteine cross-linking can provide mechanistic details about transporters even in the absence of high-resolution structures (Mulligan and Mindell, 2017). Indeed, cysteine cross-linking has been used to probe the structure and conformational dynamics of many membrane proteins in vitro (Sun et al., 1997; Wu et al., 1999; Rimon et al., 2002; Hennon and Dalbey, 2014; Radchenko et al., 2016; Mulligan and Mindell, 2017). However, a concern when using this approach is that the in vitro system might not properly reflect the native environment of the target protein. The in vitro approach also requires the purification of multiple protein variants, which can be difficult for many transporters. More importantly, in vitro cysteine cross-linking relies on the functional reconstitution of the activity of purified membrane proteins, which can be extremely difficult or unavailable for many transporters. For example, developing an in vitro functional assay can be difficult for glycolipid transporters because their complex substrates are often not available. In our case, we study the bacterial transporter MurJ, which translocates the glycolipid lipid II from the inner leaflet to the outer leaflet of the cytoplasmic membrane, and for which we do not currently have an in vitro reconstitution assay to probe its function (Ruiz, 2008; Butler et al., 2013 and 2014; Sham et al., 2014; Ruiz, 2015 and 2016; Rubino et al., 2018; Kumar et al., 2019). As a result, we developed an in vivo cysteine cross-linking protocol that captures the different conformations the MurJ transporter adopts in growing Escherichia coli cells, when MurJ activity is required for viability (Kumar et al., 2019).
We designed this protocol to probe whether the V-shaped target transporter MurJ can adopt two states during the transport cycle in live bacteria: one in which the internal cavity is open to the cytoplasm (i.e., inward-open conformation) and another in which the cavity is open to the periplasm (i.e., outward-open conformation). Based on a crystal structure assumed to reflect a single state in the transport cycle, these conformations were predicted to result from the relative movement of two large domains or lobes that traverse the membrane and give the transporter its V shape (Kuk et al., 2017; Zheng et al., 2018). To test this model and obtain evidence that these states exist in cells, we first introduced a cysteine residue into either the periplasmic (out) or the cytoplasmic (in) side of each of the lobes (N-lobe and C-lobe) of our otherwise cysteine-less transporter. We then used homo-bifunctional cross-linking reagents of various lengths that could cross-link specific cysteine pairs when the two lobes adopted only one of the two predicted states, inward-open (Figure 1A) or outward-open (Figure 1B). To easily distinguish cross-linked and uncross-linked target proteins, we engineered a functional variant of the target protein containing a well-known protease site (thrombin) in the loop connecting the N and C lobes that could be cleaved after cells were treated with cross-linkers (Figure 2A). In addition to the protease site, a fusion tag (FLAG) was also added to the N terminus of the target protein (Figure 2A) for detection by immunoblotting (Figure 2B). Using this protocol, we demonstrated that, in vivo, the glycolipid MurJ transporter adopts both inward- and out-ward open conformations and hence uses the alternate-access mechanism. Furthermore, we also showed that the transporter stalls in the outward-open state in cells treated with ionophores that collapse the membrane potential (Kumar et al., 2019). We therefore showed that our protocol can be used 1) to test the mechanism of transport by monitoring the distance between specific sites in a transporter during the transport cycle in live cells, 2) to validate high-resolution structures and in silico structural predictions, and 3) to test the effects of small-molecule inhibitors of transporters. It should also be noted that our protocol could also be used to probe dynamic interactions between domains of proteins that are not transporters (Kumar et al., 2019).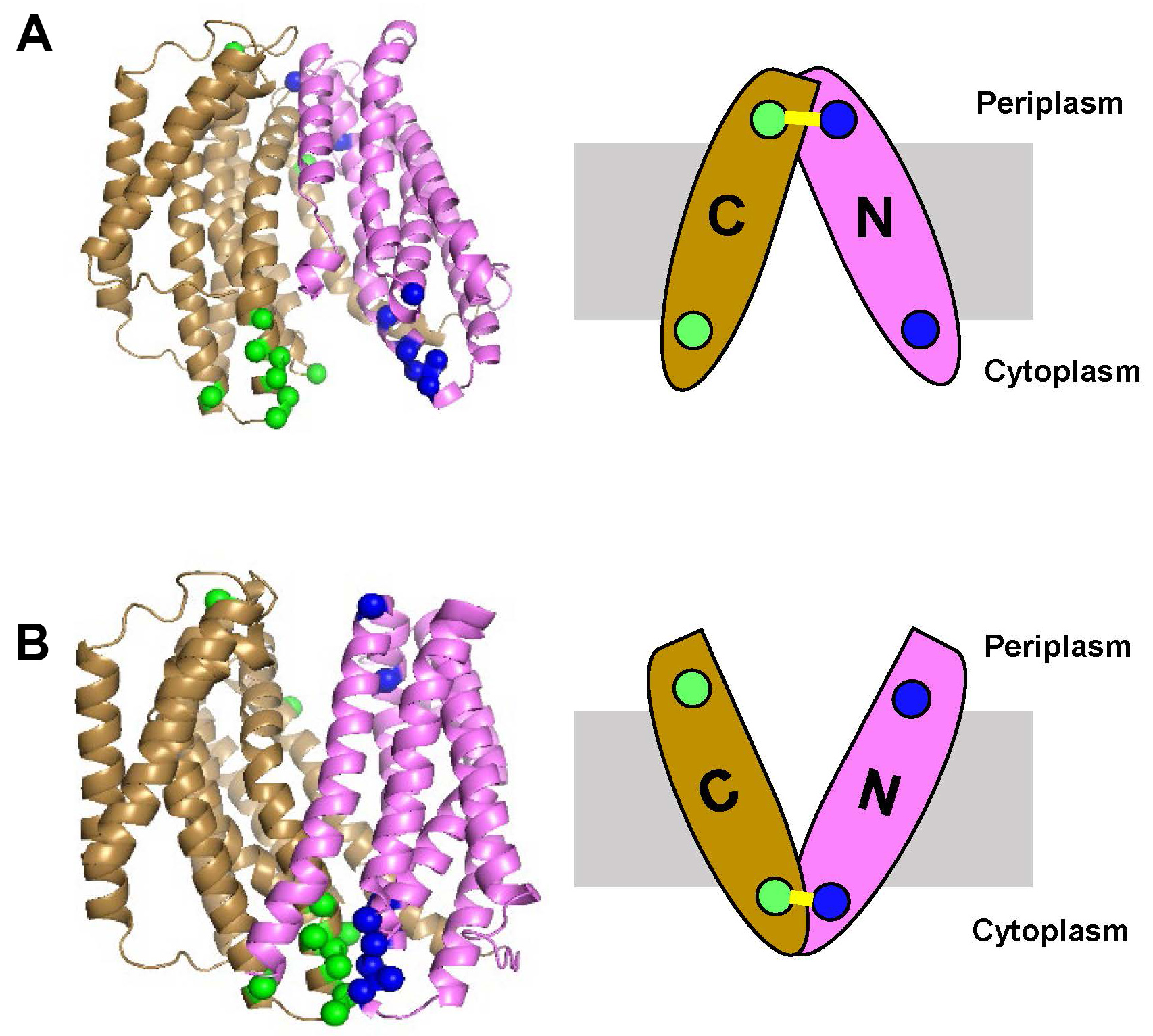
Figure 1. Selection of residues on target protein (E. coli MurJ) to be modified to cysteines for in vivo cross-linking. On the left, side view of the inward-open structure (panel A, Protein Data Bank ID code 6CC4) and outward-open homology model of MurJ (panel B). The N lobe (TM1-6) is colored pink and C lobe (TM7-14) is colored gold. The positions of amino acids to be substituted to cysteines in both periplasmic and cytoplasmic sides are denoted as blue spheres for those in the N lobe and green spheres for those in the C lobe. On the right, a cartoon representation of the structures showing that crosslinking (yellow line) can only occur in one conformation: between periplasmic cysteines closed enough to crosslink in the inward-open conformation (panel A) or between cytoplasmic cysteines closed enough to crosslink in the outward-open conformation (panel B). The membrane is represented with a grey box. PyMOL (version 2.0.1, Schrodinger) software was used to prepare this figure.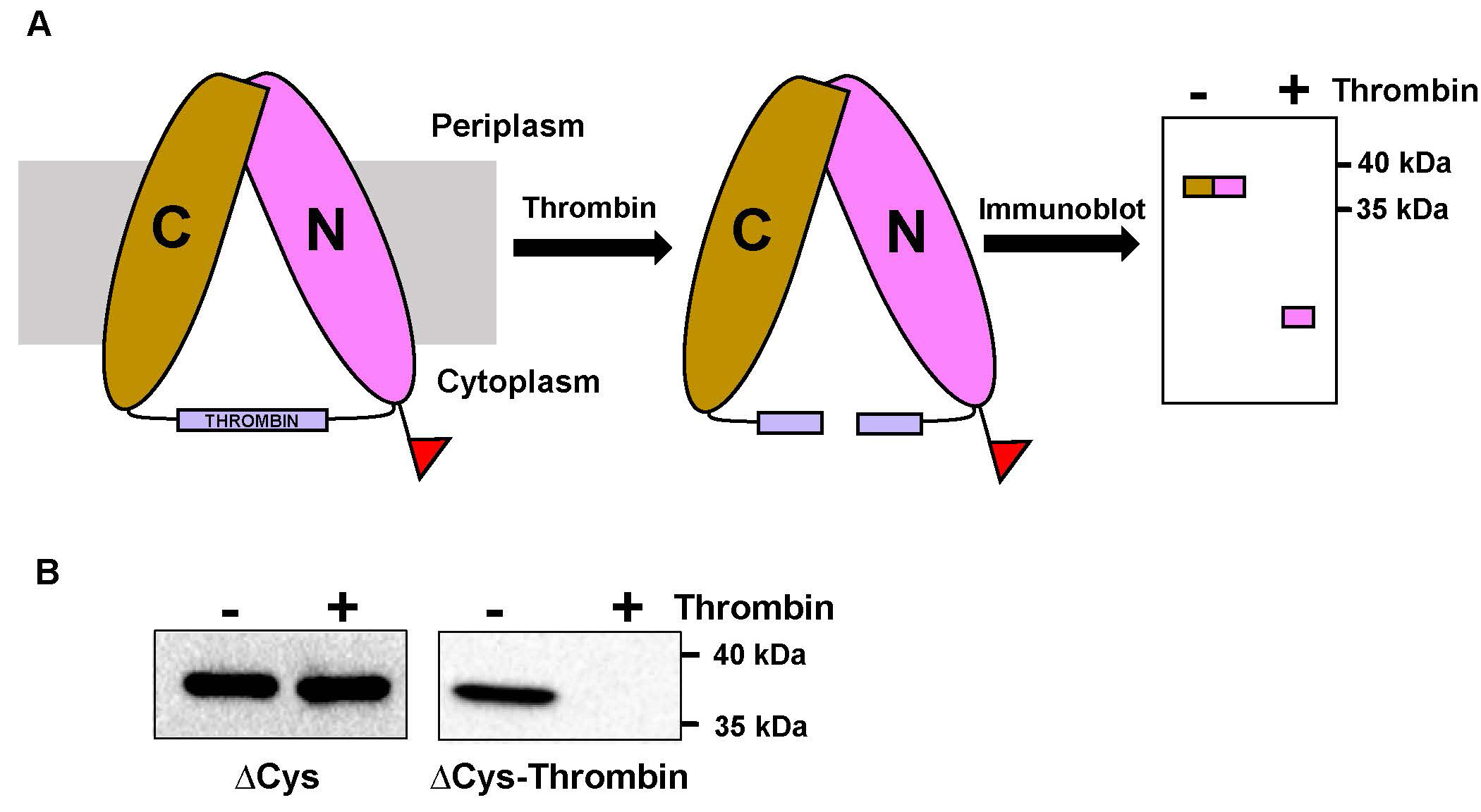
Figure 2. Proteolytic analysis of target protein variant utilized for probing the conformational states in vivo. A. Cartoon representation of the target protein (E. coli MurJ) variant and its proteolytic analysis. A thrombin protease site (purple box) was introduced between the N lobe (pink) and C lobe (gold) of cysteine-less target protein (∆Cys). An N-terminal FLAG tag used for detection by immunoblotting is shown as a red flag on the N lobe. In the absence of thrombin treatment, the full-length ∆Cys-thrombin variant should appear as a ~37 kDa band in a FLAG immunoblot. Thrombin treatment should cleave the two lobes, so only a fast-migrating band corresponding to the N lobe should be detected by FLAG immunoblotting. B. Result of thrombin treatment of full-length FLAG-MurJ∆Cys (∆Cys) and FLAG-MurJ∆Cys-thrombin (∆Cys-Thrombin) proteins. The small fragment resulting from thrombin digestion is not shown as it runs off the 10% SDS-polyacrylamide gel used for immunoblotting. We used this type of gel because it is optimal for the detection of the full-length target protein.
Materials and Reagents
- Nunc cryotube vials (Thermo Scientific, catalog number: 374502)
- Bacteriological Petri plates (VWR, catalog number: 25384-092)
- Sterile test tubes (17 x 100 mm, RPI, Research Products International, catalog number: 168599)
- 15-ml centrifuge tubes (VWR, catalog number: 10025-690)
- 50-ml centrifuge tubes (GeneMate, catalog number: C-33943-3)
- 1.7-ml microcentrifuge tubes (Axygen, catalog number: MCT-175-C)
- Ultracentrifuge tubes (Beckman Coulter, catalog number: 343778)
- Serological pipettes: 5-ml pipette (VWR, catalog number: 89130-896), 10-ml pipette (VWR, catalog number: 89130-910), 25-ml pipette (VWR, catalog number: 89130-910)
- 0.1-10 μl pipette tips (GeneMate, catalog number: P-1236-10)
- 20-200 μl pipette tips (GeneMate, catalog number: P-1236-200)
- 101-1,000 μl pipette tips (GeneMate, catalog number: P-1236-1250)
- 0.2 μm filter (Millipore, Model; MillexGP, catalog number: 202012)
- Aluminum foil
- All-purpose wrap (Fisher Brand, catalog number: 15-610)
- Nitrile or latex gloves (VWR, catalog number: 89038-270)
- Microscope slides (Corning, catalog number: 2947-75X25)
- Thick Blot paper (Bio-Rad, catalog number: 1703965)
- Escherichia coli crosslinking strains carrying derivatives of plasmid pET23/42FLAGMurJ∆cysThrombin (Dr. Natividad Ruiz strain database) (Kumar et al., 2019)
Note: These strains will vary depending on the target protein and plasmids available. - Immun-Blot PVDF membrane (Bio-Rad, catalog number: 1620177)
- Trans-Blot SD semidry transfer cell (Bio-Rad, catalog number 1703848)
- Ice
- Distilled sterile water (dH2O)
- LB Broth, Lennox (Fisher Scientific, catalog number: BP1427-2)
- Difco-Agar Bacteriological (BD, catalog number: BD-214510)
- Ampicillin sodium salt (Sigma-Aldrich, catalog number: A9518-5G) or other antibiotic appropriate for the selection of plasmids encoding other target proteins
- 1,6-bis(maleimido)hexane (BMH; Thermo Scientific Pierce, catalog number: 22330)
- 1,2-bis(maleimido)ethane (BMOE; Thermo Scientific Pierce, catalog number: 22323)
- o-PDM (Sigma-Aldrich, catalog number: 104590-5G)
- L-cysteine (Sigma-Aldrich, catalog number: 168149-5G)
- Lysozyme (Amresco, catalog number: 0663-5G)
- Benzonase (Sigma-Aldrich, catalog number: E1014 5KU)
- Thrombin from bovine plasma (Sigma-Aldrich, catalog number: T6634-100UN)
- Ethylenediaminetetraacetic acid disodium dihydrate (EDTA) (Sigma-Aldrich, catalog number: E5134-1KG)
- Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: 223506-500G)
- Ammonium persulfate (Amresco, catalog number: 0486-100G)
- N-Dodecyl-β-D-maltoside (DDM; Affymetrix, catalog number: D310S)
- Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: T6634-100UN)
- Sodium citrate tribasic dihydrate (Sigma-Aldrich, catalog number: C8532-1KG)
- Disodium hydrogen phosphate heptahydrate (Na2HPO4·7H2O) (Amresco, catalog number: 0348-1KG)
- Sodium dihydrogen phosphate (NaH2PO4) (Fischer Scientific, catalog number: S397-500)
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: 59888-2.5KG)
- PEG-8000 (Sigma-Aldrich, catalog number: 89510-250G)
- Sucrose (Amresco, catalog number: 0335-1KG)
- Dimethyl sulfoxide (DMSO) (Amresco, catalog number: 0231-500ml)
- Tris (GoldBioCom, catalog number: T-4005-5)
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: 62862-1KG)
- Urea (Ameresco, catalog number: 0568-1KG)
- Bromophenol blue sodium (Amresco, catalog number: 0312-50G)
- Glycerol (Sigma, catalog number: G7893)
- β-mercaptoethanol (Amresco, catalog number: 0482-250ml)
- Acrylamide (Protogel) (National diagnostic, catalog number: EC-890)
- Glycine (Fisher Scientific, catalog number: BP381-5)
- Tetramethylethylenediamine (TEMED) (Bio-Rad, catalog number: 161-0800)
- Instant non-fat dry milk powder
- Anti-FLAG M2 (Sigma-Aldrich, catalog number: F3165)
- Methanol (Fisher, catalog number: A412P-4)
- Tween 20 (Amresco, catalog number: 0777-1L)
- ECLTM anti-mouse IgG, horseradish peroxide-linked whole antibody (from sheep) (GE healthcare, catalog number: NA931V)
- Clarity Western ECL substrate (Bio-Rad, catalog number: 1705061)
- Spheroplast buffer (see Recipes)
- Thrombin-reaction buffer (see Recipes)
- Thrombin-reconstitution buffer (see Recipes)
- Benzonase dilution buffer (see Recipes)
- 2x AB Sample buffer (see Recipes)
- Phosphate Buffered Saline (1x PBS; see Recipes)
- 10x SDS-PAGE running buffer (see Recipes)
- 1x Transfer buffer (see Recipes)
- 10x TBS Buffer (see Recipes)
- 1x TBS + 0.1% Tween 20 (1x TBST) (see Recipes)
- 5% Milk TBST (see Recipes)
- 1 M L-cysteine (see Recipes)
- Maleimide homo-bifunctional cross-linkers (see Recipes)
Equipment
- Microscope (Nikon, model: Eclipse E200)
- pH meter (Thermo scientific, model: STARA1110)
- 37 °C Incubator (Fisher Scientific, model: isotemp)
- Roller drum (Eppendorf, New BrunswickTM, model: TC-7)
- 4 °C refrigerator
- -20 °C freezer
- -80 °C freezer
- 250-ml flasks (Pyrex USA, catalog number: 4980)
- Platform shaker
- Weighing balance (Mettler Toledo, model: ML32002E/03 and AE163)
- 0.5-10 μl Pipettor (USA Scientific, model: Ergo One, catalog number: 7100-0510)
- 2-20 μl Pipettor (USA Scientific, model: Ergo One, catalog number: 7100-0220)
- 20-200 μl Pipettor (USA Scientific, model: Ergo One, catalog number: 7100-2200)
- 100-1,000 μl Pipettor (USA Scientific, model: Ergo One, catalog number: 7110-1000)
- PIPETMAN® (JENCONS, model: Powerpippette plus)
- Table-top centrifuge (Eppendorf, model: 5810R)
- High-speed microcentrifuge (Eppendorf, model: 5418)
- Clay Adams Nutator (Marshal Scientific, catalaog number: CANU)
- Ultracentrifuge (Beckman Coulter, model: OptimaMax-TL ultra)
- Ultracentrifuge fixed angle rotor (Beckman Coulter, model: TLA120.2)
- ChemiDocTM XRS+ Imaging system (Bio-Rad)
- Biowave Personal Cell Density Meter (Biochrome, catalog number: 5300-06)
- Vortex Genie 2 (Scientific Industries, catalog number: SI-0236)
- Mini-PROTEAN® Tetra Handcast Systems (Bio-Rad, catalog number: 1658004)
- PowerPac Basic power supply (Bio-Rad, catalog number: 1645050)
Software
- ImageLab (Bio-Rad)
- PyMOL (version 2.0.1, Schrodinger)
Procedure
Note: The full protocol is described in Figure 3.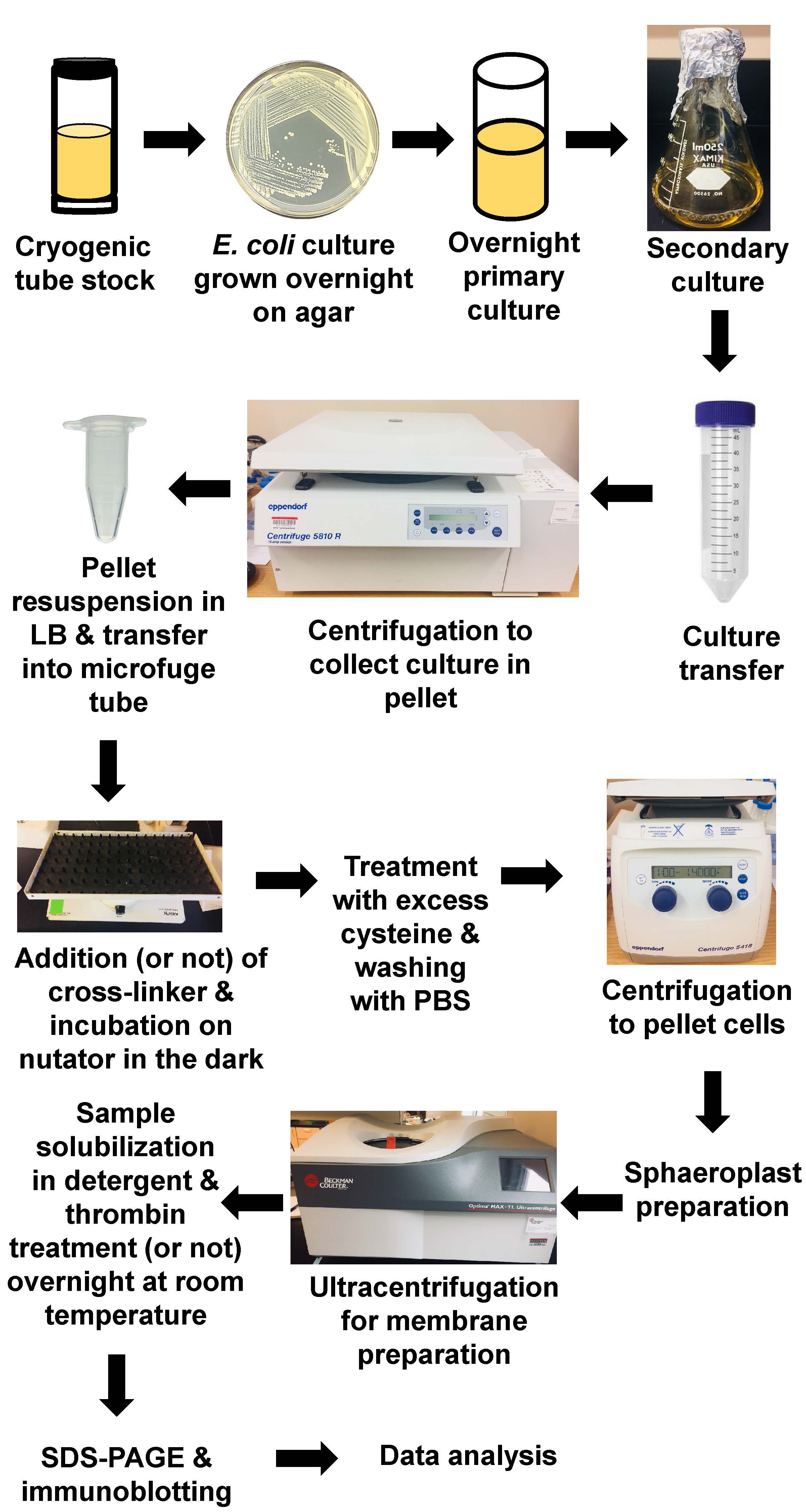
Figure 3. Schematic step-by-step protocol for in vivo cysteine cross-linking using maleimide homo-bifunctional cross-linkers
- Growth of E. coli cross-linking strains
Note: The crystal structure of the inward-open state and the in-silico homology model predicting the outward-open state of the target protein guided us in the selection of residues to be replaced with cysteine using site-directed mutagenesis. We selected pairs of residues so that the distance between their Cα positions would be within the arm-length of homo-bifunctional cross-linking reagents when the target protein adopted only one of the two conformational states. Based on this strategy, we built E. coli cross-linking strains, each harboring a plasmid encoding a double-cysteine variant of the target protein. The detailed experimental rationale for the selection of residues for cysteine substitution and homo-bifunctional cross-linking reagents, and the construction of double cysteine variants can be found in our recent study (Kumar et al., 2019).- Streak frozen stocks stored at -80 °C of E. coli cross-linking strains, each harboring a plasmid encoding a double-cysteine variant of the target protein (pET23/42FLAGMurJ∆cysThrombin derivatives were used in this protocol) (Kumar et al., 2019), on LB agar plates supplemented with 125 μg/ml of ampicillin.
Note: The antibiotic ampicillin selects for plasmid maintenance, so replace it according to the plasmid system used. - Incubate plates overnight at 37 °C.
- Use an individual colony to inoculate 5 ml of LB medium supplemented with 125 μg/ml of ampicillin and incubate overnight at 37 °C in a roller drum spinning at the top speed of 56 rpm.
- Inoculate 50 ml of LB medium (in a 250 ml Erlenmeyer flask) with 1 ml of culture from Step A2 and grow the culture at 37 °C shaking at 200 rpm until it reaches an OD600 of 1.
Note: Appropriate antibiotics can be used while growing this secondary culture to maintain the plasmid but they are not necessary because of the short incubation.
- Streak frozen stocks stored at -80 °C of E. coli cross-linking strains, each harboring a plasmid encoding a double-cysteine variant of the target protein (pET23/42FLAGMurJ∆cysThrombin derivatives were used in this protocol) (Kumar et al., 2019), on LB agar plates supplemented with 125 μg/ml of ampicillin.
- Preparation of samples for cross-linking with homo-bifunctional cross-linkers
- Transfer the required volume of E. coli culture from Step A4 into a 50-ml centrifuge tube.
Note: The required volume of E. coli culture (in ml) should equal to 10 multiplied by the number of treatments, where the number of treatments represents the number of different cross-linkers used plus one to be used without any cross-linker treatment (negative control). - Centrifuge culture samples for 5 min at 2,057 x g in a centrifuge at room temperature.
- Discard the supernatant and resuspend cell pellet with a volume of LB broth corresponding to 500 μl multiplied by the number of treatments (i.e., number of cross-linkers plus one).
- Vortex samples to ensure proper mixing and transfer 500 μl of each resuspended cell pellet into a new 1.7-ml microcentrifuge tube.
- Transfer the required volume of E. coli culture from Step A4 into a 50-ml centrifuge tube.
- Crosslinking with homo-bifunctional cross-linkers
Note: Prior to the experiment, remove stocks of all the cross-linkers from -20 °C and let them warm up to room temperature. In our experiments, we used 1,6-bis(maleimido)hexane (BMH; which has a flexible 16-Å spacer arm length), 1,2-bis(maleimido)ethane (BMOE; which has a flexible 8-Å spacer arm length), and N,N′-(o-phenylene)dimaleimide (o-PDM; which has a rigid 6-Å spacer arm length). We used maleimide cross-linkers because they are reactive in aqueous environments such as the periplasm and cytoplasm, and form thioether bonds with sulfhydryl groups of cysteines that are resistant to reversal by reducing agents (Figure 4) (Mulligan et al., 2017). Perform all steps at room temperature unless stated otherwise.- Add 10 μl of DMSO to the microcentrifuge tube containing resuspended cells that is labeled “negative control”.
Note: DMSO is used in this case because DMSO is the solvent used to dissolve the cross-linkers. - To each of the remaining microcentrifuge tubes containing resuspended cells, add 10 μl of the 50 mM stock of a cross-linker (BMH, BMOE, o-PDM) for a final concentration of 1 mM.
Note: The final concentration of cross-linkers should be optimized as it may vary depending on the target protein. - Vortex all samples.
- Place sample tubes on a nutator placed at room temperature and cover them with aluminium foil.
Note: Samples are covered with foil because some cross-linkers are light sensitive. - Incubate samples in the dark for 5 min rocking on the nutator at room temperature.
Note: The incubation time with the cross-linkers should be optimized as it may vary depending on the target protein. - Add 50 μl of 0.1 M L-cysteine (10 mM final concentration) to each sample.
Note: Providing cysteine in excess titrates any unused cross-linker. The L-cysteine stock solution stored at -20 °C should be thawed to room temperature within an hour prior to addition. - Vortex all samples.
- Incubate for 5 min rocking on a nutator at room temperature.
- Add 1 ml of 1x PBS to each sample.
- Centrifuge for 1 min at 15,777 x g in a tabletop microcentrifuge.
- Discard the supernatant and resuspend each cell pellet in 500 μl of 50 mM Tris (pH 8).
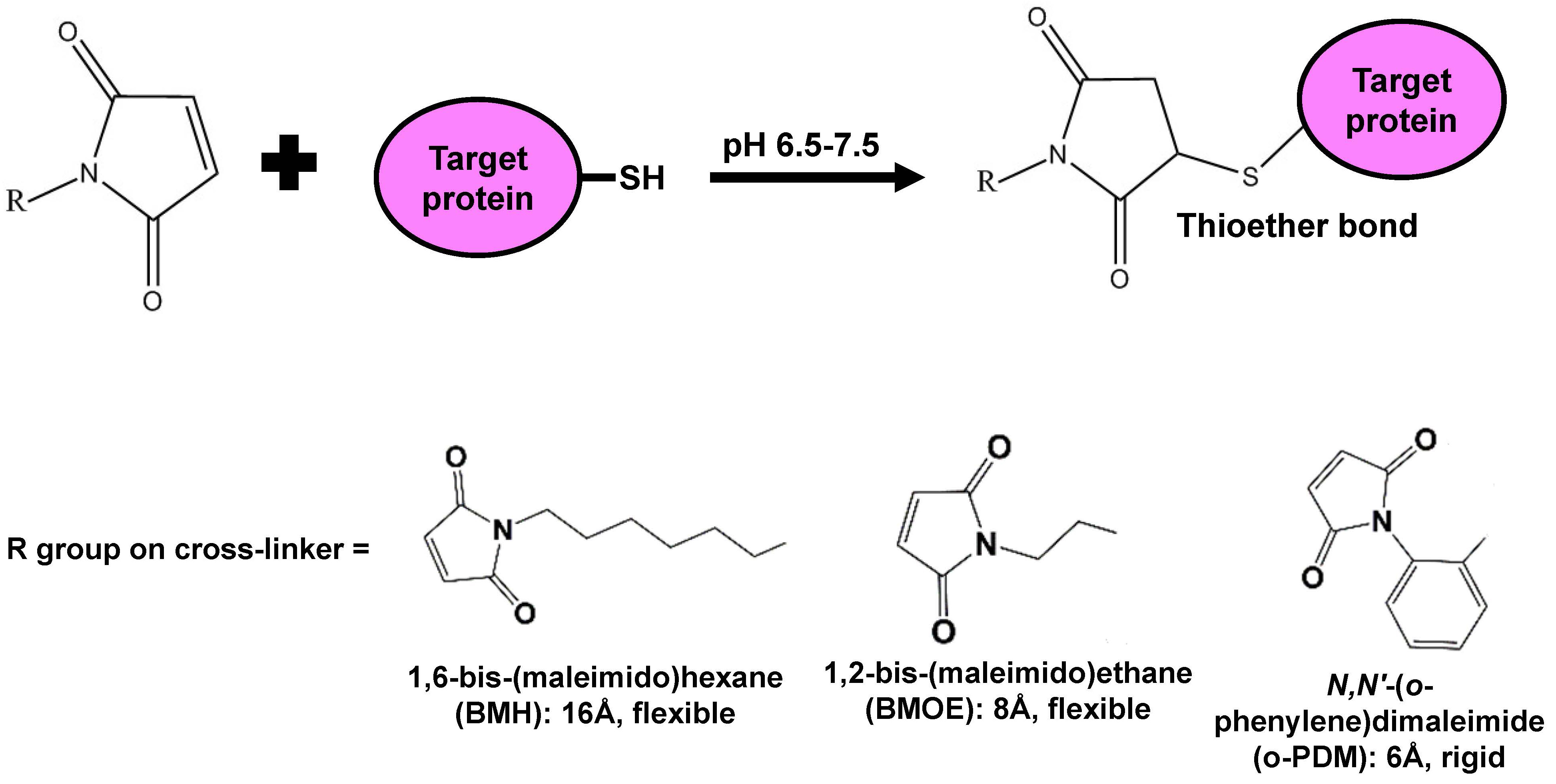
Figure 4. Reaction between a maleimide cross-linker and the sulfhydryl group of a cysteine on a target protein. The maleimide moiety of the cross-linker reacts with the sulfhydryl group (-SH) present on cysteine residues to form a stable thioether bond that cannot be reversed by reducing agents. The R group, which varies among cross-linking agents, includes a spacer arm and another maleimide group. - Add 10 μl of DMSO to the microcentrifuge tube containing resuspended cells that is labeled “negative control”.
- Isolation of cytoplasmic membranes containing the target protein
- For spheroplast formation, transfer each sample from Step C11 to a new 15-ml centrifuge tube.
Note: During spheroplast formation, the outer membrane and peptidoglycan cell wall of E. coli cells are destroyed, but their cytoplasmic membrane remains intact. - Add 500 μl of spheroplast buffer.
- Add 2.5 μl of 100 mg/ml lysozyme stock.
- Add 1 ml of dH2O.
- Incubate for 15 min at room temperature. Gently vortex at low setting a couple of times for uniform mixing during the 15-min incubation.
- Add 50 μl of 1 M MgCl2.
- Transfer 3 μl of each sample to microscope glass slides and view sample under 40x objective to confirm spheroplast formation.
Note: Spheroplasts appear as large bright round cells. Uniform spheroplast formation is necessary. If not, the incubation time at room temperature should be optimized. - Collect spheroplasts by centrifugation at 2,057 x g for 5 min at 4 °C.
- Discard the supernatant and resuspend spheroplasts in 500 μl of ice-cold 50 mM Tris (pH 8).
- Transfer each of the resuspended spheroplast samples into separate pre-chilled ultracentrifuge tubes.
Note: This step should be performed on ice to minimized degradation of the target protein. - Add 1 μl of 25 U/μl Benzonase stock.
- Mix well by carefully pipetting up and down.
- Centrifuge at 434, 902 x g for 1 h at 4 °C in an Optima-MAX-TL ultracentrifuge (Beckman Coulter) using a TLA120.2 rotor to pellet membranes.
- Discard the supernatant and resuspend membrane pellets in 150 μl of thrombin reaction buffer.
- Add 15 μl of 10% DDM stock (final concentration 1%).
Note: 1% DDM is used for solubilization of our target protein. However, choice of detergent can vary depending on the target protein. - Transfer resuspended membrane preparations to a pre-chilled 1.7-ml microcentrifuge tube.
- Centrifuge at 15,777 x g for 10 min at 4 °C in a tabletop microcentrifuge to remove unsolubilized or aggregated proteins.
- For each sample, transfer supernatant to a separate microcentrifuge tube.
- For spheroplast formation, transfer each sample from Step C11 to a new 15-ml centrifuge tube.
- Thrombin treatment
- For each sample in Step D18, transfer 50 μl into two separate microcentrifuge tubes.
- Add 4 μl of 0.1 U/μl thrombin stock (final concentration 0.4 U) to one of the microcentrifuge tubes of each treatment and leave the other one untreated.
Note: Lyophilized thrombin powder should be first reconstituted as 0.1 U/μl in thrombin reconstitution buffer before using. Store the stock at -20 °C. - Incubate all samples for 20 h at room temperature.
Note: Incubation time and temperature should be optimized to ensure cleavage of all target proteins. - Add 50 μl of 2x AB buffer to all samples.
Note: This step stops the proteolytic reaction and prepares samples for electrophoresis. We used 2x AB buffer for our protein samples; however, other types of SDS-PAGE loading buffers could be used depending on the target protein. - Briefly centrifuge at 15,777 x g for 10 s at room temperature in a tabletop centrifuge to collect samples at the bottom of the microcentrifuge tube.
- SDS-PAGE and immunoblotting
- Assemble a 10% SDS-PAGE gel in a Mini-PROTEAN® Tetra system.
Note: The % of SDS-PAGE gel should be decided based on the molecular weight of the target protein and its thrombin proteolytic cleave pattern. - Load 25 μl of each sample from Step E5 on a 10% SDS-PAGE gel. When doing so, load each thrombin treated/untreated pair adjacent to each other for each treatment with or without a particular cross-linker.
Note: Equivalent amounts of samples should be loaded on the gel because equal numbers of cells were collected at Step B4 after using equal volumes of cultures that had reached the same OD600. However, methods to measure protein concentration could also be used for normalization before loading the samples on a gel. - Start electrophoresis at 120 V using 1x SDS-PAGE running buffer and stop after the visible molecular weight markers have migrated the desired distance.
- Transfer proteins from the gel to a polyvinylidene difluoride (PVDF) membrane at 10 V for 2.5 h using a semidry transfer apparatus (Bio-Rad).
Note: Transfer time and voltage should be optimized according to the target protein and % SDS PAGE gel. - Block the PVDF membrane in TBST with 5% milk for 30 min at room temperature on a platform shaker.
- Probe the PVDF membrane with the primary antibody.
Note: For our target protein, we probed with anti-FLAG M2 (1:10,000; Sigma-Aldrich) in TBST with 5% milk by incubation at 4 °C on a shaker overnight. - Wash three times with 1x TBST with 5 min incubation on a shaker at room temperature.
- Probe the PVDF membrane with a secondary antibody.
Note: For our target protein, we probed with anti-mouse horseradish-peroxidase conjugated antibodies (1:10,000; GE Healthcare) in 1x TBST with 5% milk by incubating for 1 h on a shaker at room temperature. - Wash three times with 1x TBST with 5 min incubation on a shaker at room temperature.
- Develop the signal using the Clarity Western ECL substrate according to the manufacturer's instructions (Bio-Rad) and detected using Image Lab program in a ChemiDoc XRS+ system (Bio-Rad).
- Assemble a 10% SDS-PAGE gel in a Mini-PROTEAN® Tetra system.
Data analysis
The described in vivo cysteine cross-linking experiments using maleimide-containing cross-linkers with different spacer arms should probe the proximity of substituted cysteine residues in both periplasmic- and cytoplasmic-facing residues in the N-lobe and C-lobe of the target protein and test for the presence of inward- and outward-open conformations (Figures 5A and 5B). The results from the proteolytic gel analysis should be analyzed as follows:
- In any sample lacking protease treatment, only full-length protein variants should be detected.
- In protease-treated samples in which cross-linking did not occur because either no cross-linking agent was added or the two cysteines were too distant to be cross-linked, the target protein should be cleaved into two fragments. This should be reflected by the absence of the band corresponding to full-length protein after immunoblotting (Figures 5C and 5D). A gel with higher acrylamide percentage could be used to confirm the detection of the smaller tagged fragment (Figure 2A).
- In protease-treated samples prepared from cells in which cysteine pairs were cross-linked by homobifuntional crosslinking reagents, the full-length protein should be detected even after protease cleavage because the two lobes should remain covalently linked by the cross-linkers (Figures 5C and 5D). This result should be interpreted to mean that the two cysteine residues came within the cross-linkable distance of a particular cross-linker during the transport cycle. The intensity of the band corresponding to the full-length protein should be proportional to the amount of cross-linking. Cross-linking efficiency between two cysteines could vary between cross-linkers, since it will depend on the distance between these residues and the length of the spacer-arm of the various cross-linkers.
In our experiment, the presence or absence of full-length protein after cross-linking and protease treatment of variants containing specific cysteine substitutions in the periplasmic or the cytoplasmic side demonstrated the existence of two conformations: inward- and out-ward open conformations during the transport cycle for the target protein.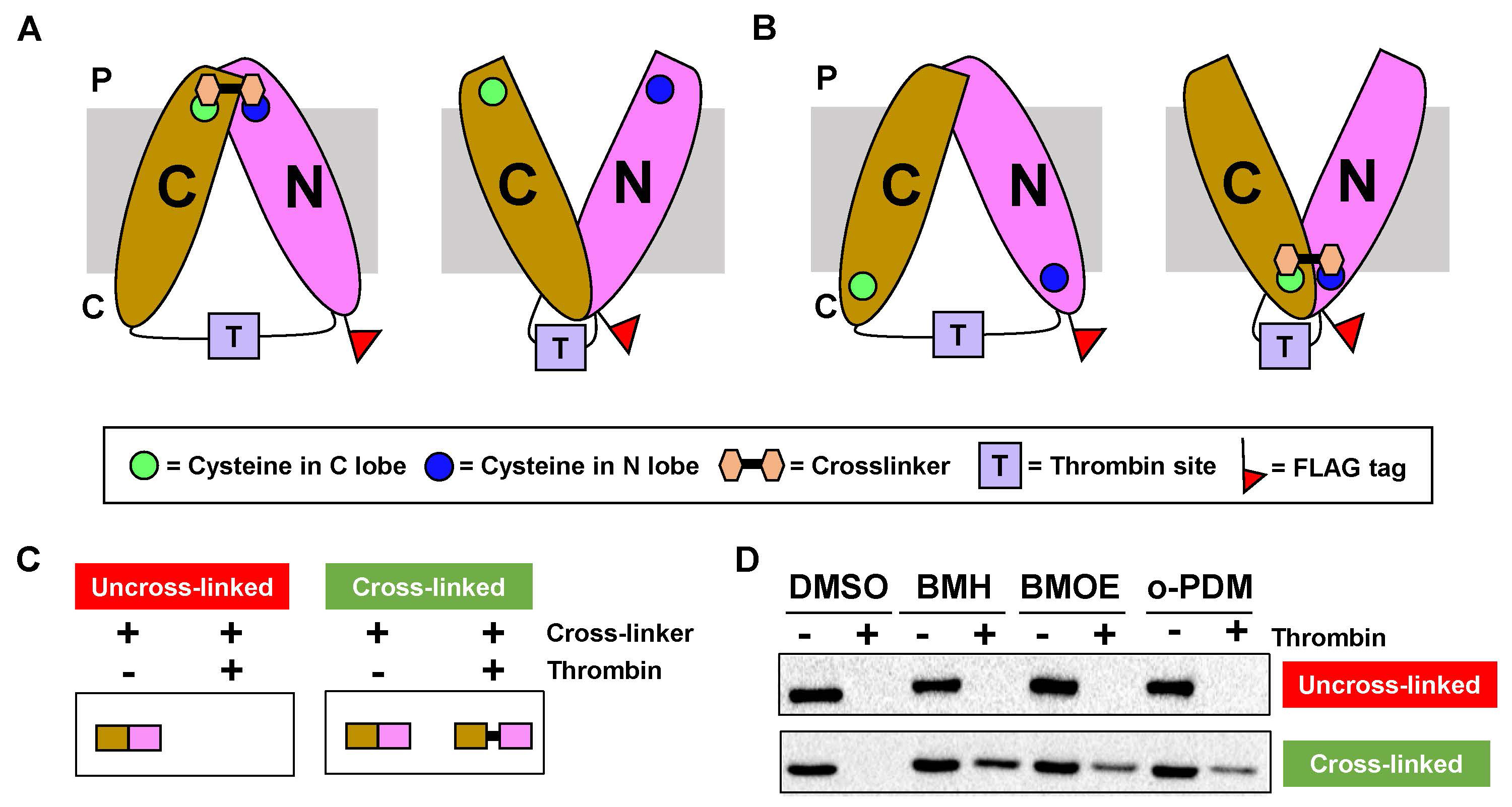
Figure 5. In vivo double-cysteine cross-linking results. The two cysteines each located in the N lobe and C lobe of the target protein facing either the periplasm (P) or the cytoplasm (C) are cross-linked with maleimide homo-bifunctional cross-linkers only in one of two conformations: inward-open conformation for cysteines facing the periplasm (panel A) or outward-open conformation for cysteines facing the cytoplasm (panel B). C. After thrombin treatment, the full-length protein will not be visible after immunoblotting if cross-linking did not occur (mock blot on the left). If cross-linking occurred (mock blot on the right), the full-length protein will be detected even after the thrombin treatment. D. Results of the cross-linking experiments of the target protein with dimethyl sulfoxide (DMSO) as a negative control and three different maleimide homo-bifunctional cross-linkers of various spacer arm lengths (1,6-bis(maleimido)hexane (BMH, which has a flexible 16-Å spacer arm length), 1,2-bis(maleimido)ethane (BMOE, which has a flexible 8-Å spacer arm length), and N,N′-(o-phenylene)dimaleimide (o-PDM, which has a rigid 6-Å spacer arm length).
Notes
- Our protocol could be applied to other transporter proteins that utilize the alternate-access mechanism. Moreover, this protocol could also be used with proteins that are not transporters in order to probe dynamic interactions between domains that might be subject to regulation.
- Demonstration of the functionality of all the target protein variants should be tested prior to use since it is crucial to validate the cross-linking results.
- It is necessary to check the expression level of the target protein variants by suitable methods before cysteine cross-linking.
- It is highly recommended that negative controls are included to validate the technical details of the procedure. For example, a cysteine-less variant of the target protein, use of mono-cysteine variants, and double cysteine mutants facing opposite side to each other are recommended (Kumar et al., 2019).
- Try to prepare the stocks of maleimide homo-bifunctional cross-linker fresh each time for best results (repeated freezing and thawing of cross-linker may impact results).
- We engineered a functional variant of the target protein containing a well-known protease site (thrombin). Another protease site such as factor Xa protease (Sun and Kaback, 1997) could also be used. However, care should be taken that the protease treatment does not cause non-specific cleavage of the target protein.
- To ensure reproducibility, assays should be performed in triplicate.
- Our protocol could also be used to test the effects of small-molecule inhibitors of transporters as we did to study the effect of protonophores on MurJ (Kumar et al., 2019).
Recipes
- Spheroplast buffer
50 mM Tris-HCl, pH 8.0
1 M sucrose
2 mM EDTA - Thrombin-reaction buffer(store the buffer at 4 °C)
50 mM Tris-HCl, pH 8.0
150 mM NaCl
2.5 mM CaCl2 - Thrombin-reconstitution buffer
50 mM sodium citrate, pH 6.5
200 mM NaCl, 0.1%PEG-8000, 50% glycerol
Note: Use this buffer to reconstitute thrombin by generating a 0.1 U/μl thrombin stock (store stock at -20 °C). - Benzonase dilution buffer (store the buffer at -20 °C)
50 mM Tris-HCl, pH 8.8
2 mM MgCl2
50% glycerol
Note: Dilute Benzonase in this buffer to 25 U/μl before using. - 2x AB Sample buffer
6.84 mM Na2HPO4
3.16 mM NaH2PO4
50 mM Tris-HCl, pH 6.8
6 M urea
1% β-mercaptoethanol
3% SDS
10% glycerol
0.1% bromophenol blue - 1x PBS
8 mM Na2HPO4·12H2O, 2 mM NaH2PO4·2H2O, 131 mM NaCl in dH2O - 10x SDS-PAGE running buffer
30.0 g of Tris base, 144.0 g of glycine, and 10.0 g of SDS in 1,000 ml of dH2O - 1x Transfer buffer (1 L)
5.82 g Tris
2.93 g glycine
200 ml methanol
800 ml dH2O - 10x TBS Buffer
a.Dissolve 24 g Tris and 88 g NaCl in 900 ml distilled water
b.Adjust pH to 7.6 with 12 N HCl
c.Add dH2O to a final volume of 1 L
Note: Dilute to 1x when using. - 1x TBST (1x TBS + 0.1% Tween 20) (1 L)
50 ml 20x TBS
1 ml Tween 20
950 ml dH2O - 5% Milk TBST (25 ml)
1.25 g dry milk
25 ml 1x TBS + 0.1% Tween 20 - 1 M L-cysteine
121 mg L-cysteine in 1 ml dH2O
Note: Store stock at -20 °C in aliquots that should not be refrozen after thawing. - Maleimide homo-bifunctional cross-linkers
50 mM BMH: 6.875 mg BMH in 500 μl of DMSO
50 mM BMOE: 5.5 mg BMOE in 500 μl of DMSO
50 mM o-PDM: 6.7 mg o-PDM in 500 μl of DMSO
Note: Store stocks solutions at -20 °C wrapped in aluminum foil.
Acknowledgments
We thank Rebecca M. Davis for helping in the preparation of this manuscript. This work was supported by grant GM1000951 from the National Institute of General Medical Sciences to N.R. Parts of some figures are adapted and modified from our previous study (Kumar et al., 2019).
This protocol is adapted from Kumar, S., Rubino, F. A., Mendoza, A. G., and Ruiz, N. (2019). The bacterial lipid II flippase MurJ functions by an alternating-access mechanism. The Journal of biological chemistry 294, 981-990.
Competing interests
The authors declare there are no potential conflicts of interest.
References
- Butler, E. K., Davis R. M., Bari, V., Nicholson, P. A. and Ruiz, N. (2013). Structure-function analysis of MurJ reveals a solvent-exposed cavity containing residues essential for peptidoglycan biogenesis in Escherichia coli. J Bacteriol 195(20): 4639-4649.
- Butler, E. K., Tan, W. B., Joseph, H. and Ruiz, N. (2014). Charge requirements of lipid II flippase activity in Escherichia coli. J Bacteriol 196(23): 4111-4119.
- Elofsson, A. and von Heijne, G. (2007). Membrane protein structure: prediction versus reality. Annu Rev Biochem 76: 125-140.
- Hennon, S. W. and Dalbey, R. E. (2014). Cross-linking-based flexibility and proximity relationships between the TM segments of the Escherichia coli YidC. Biochemistry 53(20): 3278-3286.
- Kuk, A. C., Mashalidis, E. H. and Lee, S. Y. (2017). Crystal structure of the MOP flippase MurJ in an inward-facing conformation. Nat Struct Mol Biol 24(2): 171-176.
- Kumar, S., Rubino, F. A., Mendoza, A. G. and Ruiz, N. (2019). The bacterial lipid II flippase MurJ functions by an alternating-access mechanism. J Biol Chem 294(3): 981-990.
- Meng, X., Clews, J., Martin, E. R., Ciuta, A. D. and Ford, R. C. (2018). The structural basis of cystic fibrosis. Biochem Soc Trans 46(5): 1093-1098.
- Mulligan, C. and Mindell, J. A. (2017). Pinning down the mechanism of transport: probing the structure and function of transporters using cysteine cross-linking and site-specific labeling. Methods Enzymol 594: 165-202.
- Radchenko, M., Nie, R. and Lu, M. (2016). Disulfide cross-linking of a multidrug and toxic compound extrusion transporter impacts multidrug efflux. J Biol Chem 291(18): 9818-9826.
- Rimon, A., Tzubery, T., Galili, L. and Padan, E. (2002). Proximity of cytoplasmic and periplasmic loops in NhaA Na+/H+ antiporter of Escherichia coli as determined by site-directed thiol cross-linking. Biochemistry 41(50): 14897-14905.
- Rubino, F. A., Kumar, S., Ruiz, N., Walker, S., and Kahne, D. E. (2018). Membrane potential is required for murJ function. Journal of the American Chemical Society 140(13):4481-4484.
- Ruiz, N. (2008). Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli. Proc Natl Acad Sci U S A 105(40): 15553-15557.
- Ruiz, N. (2015). Lipid flippases for bacterial peptidoglycan biosynthesis. Lipid Insights. 8 (Suppl 1): 21-31.
- Ruiz, N. (2016). Filling holes in peptidoglycan biogenesis of Escherichia coli. Curr Opin Microbiol 34: 1-6.
- Sham, L. T., Butler, E. K., Lebar, M. D., Kahne, D., Bernhardt, T. G., and Ruiz, N. (2014). Bacterial cell wall. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 345(6193): 220-2.
- Sun, J. and Kaback, H. R. (1997). Proximity of periplasmic loops in the lactose permease of Escherichia coli determined by site-directed cross-linking. Biochemistry 36(39): 11959-11965.
- Wiese, M. and Stefan, S. M. (2019). The A-B-C of small-molecule ABC transport protein modulators: From inhibition to activation-a case study of multidrug resistance-associated protein 1 (ABCC1). Med Res Rev. doi: 10.1002/med.21573.
- Wu, J., Hardy, D. and Kaback, H. R. (1999). Site-directed chemical cross-linking demonstrates that helix IV is close to helices VII and XI in the lactose permease. Biochemistry 38(6): 1715-1720.
- Zheng, S., Sham, L. T., Rubino, F. A., Brock, K. P., Robins, W. P., Mekalanos, J. J., Marks, D. S., Bernhardt, T. G. and Kruse, A. C. (2018). Structure and mutagenic analysis of the lipid II flippase MurJ from Escherichia coli. Proc Natl Acad Sci U S A 115(26): 6709-6714.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Kumar, S. and Ruiz, N. (2019). Probing Conformational States of a Target Protein in Escherichia coli Cells by in vivo Cysteine Cross-linking Coupled with Proteolytic Gel Analysis. Bio-protocol 9(12): e3271. DOI: 10.21769/BioProtoc.3271.
- Kumar, S., Rubino, F. A., Mendoza, A. G. and Ruiz, N. (2019). The bacterial lipid II flippase MurJ functions by an alternating-access mechanism. J Biol Chem 294(3): 981-990.
Category
Microbiology > Microbial biochemistry > Protein
Biochemistry > Protein > Activity
Biochemistry > Protein > Structure
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.