- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Detailed Protocol for Large-scale Recombinant Expression and Validation of Human FGFR2 with Its Transmembrane and Extracellular Domains in Escherichia coli

Published: Vol 9, Iss 11, Jun 5, 2019 DOI: 10.21769/BioProtoc.3261 Views: 5970
Reviewed by: Alessandro DidonnaMichael Nguyen TrinhAli Asghar Kermani
Original research article
The authors used this protocol in:

Jun 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Receptor tyrosine kinases (RTKs) are an important class of transmembrane receptors that mediate some of the most crucial biochemical pathways essential to the growth, differentiation, and survival of a cell and thus, are highly involved in cancers. Due to the complexity of RTKs having biochemically different domains including a transmembrane domain, an intact crystal structure of any of these proteins remain elusive as it is difficult to produce milligram amounts of intact functional RTKs for crystallography studies. A heavily studied RTK is fibroblast growth factor receptor 2 (FGFR2), which plays a key role in fibroblast growth regulation, differentiation, and oncogenesis. Previous studies have focused on expressing FGFR2’s extracellular, transmembrane, and intracellular domains individually. For this protocol, we have focused on the extracellular and transmembrane domains of the FGFR2 protein. The function of the expressed protein is validated by demonstrating its ability to bind heparin and fibroblast growth factor 1 (FGF1). The primary contribution of our protocol is expressing two RTK domains together, including the transmembrane domain, in milligram quantities. Being able to express RTKs to define its crystal structures would enable pharmacologists to design cancer drugs that selectively target active conformations.
Keywords: Receptor tyrosine kinaseBackground
Receptor tyrosine kinases have eluded scientists in the quest for an intact fully functional crystal structure. In general, transmembrane proteins have been highly challenging to conduct structural studies especially due to challenges with expressing large quantities of fully functional protein. While most studies of FGFR expression focus on crystallizing the extracellular receptor or tyrosine kinase domain separately, working with the transmembrane domain linked to either of the two domains can result in multiple biochemical challenges (Mohammadi et al., 1996; Plotnikov et al., 1999; Plotnikov et al., 2000; Schlessinger et al., 2000; Yeh et al., 2002; Zhang et al., 2009). These eukaryotic transmembrane proteins, when expressed in high quantities in E. coli are usually highly unstable and often precipitate out of solution or aggregate (Rosano and Ceccarelli, 2014b). Moreover, RTKs have three functionally and structurally distinct domains, making it difficult to optimize expression because each domain may function optimally in different conditions. Detergent must always be used to keep the protein soluble due to the hydrophobic nature of the transmembrane domain (Rosano and Ceccarelli, 2014a). Host cells heavily regulate the expression of protein kinases due to their roles in activating important pathways. As a result, overexpression of these proteins to produce significant quantities may be toxic to leading to inclusion bodies and degradation. We chose FGFR2, a receptor responsible for activating complex mechanisms that result in angiogenesis, skeletal formation, and cell proliferation, and growth, as the best candidate for structural studies based on preliminary small-scale expression experiments (Turner and Grose, 2011).
A primary goal of expressing functional FGFR in significant quantities (milligrams) is for X-ray crystallography. Two to fifty milligrams of protein is typically necessary for X-ray crystallography (Dessau, and Modis, 2011). A crystal structure would greatly facilitate the design of that selectively target the receptor. While there are studies that have determined the crystal structure of the FGFR2 ECD, TM, and intracellular kinase domains separately, they do not tell us the mechanism by which the external signal is transduced to the intracellular kinase domain. While the approach described in this paper does not describe expression of the entire protein, it does provide an important starting point by describing the expression of two linked domains including the technically challenging transmembrane domain. Recently, there have been a few studies that have demonstrate successful recombinant expression of EGFR, EphA2, PDGFR, and Kit (Mi et al., 2008 and 2011; Lu et al., 2012; Paavilainen et al., 2013; Opatowsky et al., 2014; Chen et al., 2015). However, none of these studies employed E. coli as a host and described production of protein quantities sufficient for X-ray crystallography studies. We hope our methods will contribute to the ongoing effort towards crystallizing the structure of an intact receptor tyrosine kinase.
The determination of the crystal structure for intact FGFR2 would greatly facilitate drug discovery. FGFR drugs may potentially be used to treat a multitude of cancers including breast, stomach, lung, bladder, osteosarcoma, and intrahepatic cholangiocarcinoma (Babina and Turner, 2017). In addition, there is the potential for pharmacological in-utero treatment of FGFR2 associated craniosynostosis syndromes such as Apert Syndrome, Pfieiffer syndrome, and Bear-Stevenson syndrome (Robin et al., 2011). Overall, crystallizing an intact FGFR2 may also set a precedent for the crystallization of intact receptor tyrosine kinases, including other FGFRs, which are key drug targets.
Materials and Reagents
- Pipette tips (VWR, Next Generation Low-Binding, catalog numbers: 89076-476, 89076-486)
- SnakeskinTM membrane (Thermo Fisher Scientific, catalog numbers: 68035, 88244)
- Superdex S200 10/300 GL size exclusion chromatography column (GE Healthcare)
- HiTrap Heparin Affinity Chromatography Column (GE Healthcare, catalog number: 17040601)
- SnakeSkinTM Dialysis Clips (Thermo Fisher Scientific, catalog number: 68011)
- PVDF membrane (Thermo Fisher Scientific, catalog number: 88518)
- 1.5 ml centrifuge tubes (Fisher Scientific, catalog number: 05-408-129)
- 3 ml syringes, Luer lock (Becton Dickinson, catalog number: BD309657)
- Petri Dishes for LB Plates, 100 x 10 mm (VWR, catalog number: 25384-324)
- pSpeedET vector, DNASU plasmid repository (EvNO00285791)
Note: pSpeedET must have N-terminal E. coli maltose binding protein (MBP) fusion tag. - Lemo21 (DE3) Competent E. coli (New England Biolabs® Inc., catalog number: 2528J)
- Primers
31F: 5’ ctgtacttccagggcACCACATTAGAGCCAGAAGGAGCAC 3’
406R_noGFP: 5’ gtttaattaagtcgcgtta CTTCTTGGTCGTGTTCTTCATTCGGC 3’ - Phusion® High-Fidelity PCR Kit (New England Biolabs® Inc., catalog number: C2528J)
- ThermoScientific GeneJET Gel Extraction Kit (Thermo Fisher ScientificTM, catalog number: K0691)
- General Electric Healthcare illustraTM plasmidPrep Mini Spin Kit (GE Healthcare, catalog number: GE28-9042-69)
- Dimethylsulfoxide (DMSO) (Sigma-Aldrich, catalog number: D650-5X5 ML)
- Agarose (Lonza SeaKem LE, catalog number: 50004)
- SOC recovery media (Thermo Fisher Scientific, catalog number: 15544034)
- Kanamycin (Research Productions International, catalog number: K22000)
- Chloramphenicol (Research Productions International, catalog number: C61000)
- Carbenicillin (Research Productions International, catalog number: C46000)
- Terrific broth (TB) (Sigma Aldrich, catalog number: T0918)
- Beta-D-1-thiogalactopyranoside (IPTG) (Sigma-Aldrich, catalog number: l6758-1G)
- DNase I (New England Biolabs, catalog number: M0303S)
- CaCl2 (Fisher Scientific, catalog number: AC349610250)
- Benzamidine (Sigma-Aldrich, catalog number: 449709)
- PMSF (Sigma-Aldrich, catalog number: 10837091001)
- Protease Inhibitors E-64 (Sigma-Aldrich, catalog number: E3132)
- Pepstatin (Sigma-Aldrich, catalog number: P5318-5MG)
- Bestatin (Sigma-Aldrich, catalog number: B8385-1MG)
- β-mercaptoethanol (BME) (Sigma-Aldrich, catalog number: M6250)
- e2TAK polymerase (Takara Bio, catalog number: RF001A)
- Rapid Transfer Buffer, 10x (VWR® Amresco® Life Science, catalog number: N789-1L)
- Anti-MBP Antibody (New England Biolabs, catalog number: E-8038)
- Tris-glycine gels 8-16% (NuSep, catalog number: NG15-816)
- n-dodecyl-β-D-maltoside DDM (Anatrace, catalog number: D310)
- KPL TMB Membrane Peroxidase Substrate System Kit (Sera Care, catalog number: 5420-0025)
- Amresco RapidBlockTM solution (VWR, catalog number: 97064-124)
- NaCl (Sigma-Aldrich, catalog number: S9888)
- HEPES at pH 7.5 (VWR, catalog number: 97061-822)
- MgSO4 (Sigma-Aldrich, catalog number: M7506)
- Glycerol (Sigma-Aldrich, catalog number: G9012)
- Tris-HCl pH 7.5 (Sigma-Aldrich, catalog number: 10812846001)
- Tween 20 (Sigma-Aldrich, catalog number: P9416)
- Guanidinium-HCl (Sigma-Aldrich, catalog number: G3272)
- Dithiothreitol (DTT) (Sigma Aldrich, catalog number: DTT-RO)
- L-cysteine (Sigma-Aldrich, catalog number: 168149)
- Imidazole (Alfa Aesar, catalog number: AA47274)
- Gel Green® (Biotium, catalog number: 41005)
- LB agar (VWR, catalog number: 97064-106)
- NEB® 5-alpha Competent E. coli (High Efficiency) DH5α cells (New England Biolabs, catalog number: C2987l)
- Coomassie Brilliant Blue (Thermo Fisher Scientific, catalog number: 20279)
- FGF1 pMCSG7 plasmid (DNASU plasmid repository, catalog number: HsCD00343156)
- HRP-conjugated anti-MBP antibody (New England Biolabs, catalog number: E8038)
- Milk Powder
- Lysis Buffer (see Recipes)
- TBST (see Recipes)
- Denaturing buffer dialysis solution #1 (see Recipes)
- Dialysis Solution #2 (see Recipes)
- Binding buffer for heparin affinity chromatography (see Recipes)
- Elution buffer for heparin affinity chromatography (see Recipes)
- Elution buffer for FGF1 (see Recipes)
- Running Buffer for SEC (see Recipes)
- Blocking Buffer (see Recipes)
Equipment
- Baffled, 125 ml Erlenmeyer flasks (Sigma-Aldrich, catalog number: CLS4444125)
- Pipettes (Gilson, Pipetman Classic)
- Centrifuge for collecting liters of cell culture (Thermo Fisher, Sorvall, catalog number: ST16)
- Tabletop microcentrifuge (Eppendorf, model: 5415)
- PCR Thermal cycler (Thermo Scientific, model: Hybaid PX2 Gradient Thermal Cycler)
- Sonicator (Thermo Fisher, model: 550 Sonic Dismembrator)
- Akta Purifier or FPLC (GE Healthcare, AKTA Purifier 100 FPLC, Frac-950)
- Refrigerated shaker/incubator for growing cells (New Brunswick, Innova 4230 Refrigerated Incubator)
- Vortexer (Fisher Scientific, catalog number: 02215365)
- JA20 Fixed-Angle Aluminum rotor Biosafety Lid- 8 x 50 ml, 20,000 rpm, 48,400 x g (Beckman Coulter, catalog number: 334831)
- Magnetic stir bar (Fisher Scientific, catalog numbers: 14-513-83, 14-513-93, 14-513-95, 14-513-94)
- Peristaltic pump (Gilson, Rabbit Plus)
- Visible/UV spectrophotometer (Cary 50)
- Test tube rotator (Scientific Equipment, model: 60448 Test Tube Rotator)
- Western Blot apparatus (Bio-Rad)
- Clare Dark Reader Transilluminator (Clare Chemicals, DR46)
Software
- ApE (A plasmid Editor) program for nucleotide analysis (http://jorgensen.biology.utah.edu/wayned/ape/)
- NCBI BLASTn for sequence comparison (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch&BLAST_SPEC=blast2seq&LINK_LOC=align2seq)
Procedure
Part I: Large Scale Expression and Purification of FGFR2’s Extracellular and Transmembrane Domains
- Synthesizing FGFR2 ECD + TM construct via polymerase incomplete primer extension (PIPE) cloning
Polymerase Incomplete Primer Extension (PIPE) cloning is a more efficient alternative to cloning with restriction and ligation enzymes. Instead of adding flanking restriction sites and using restriction enzymes to digest the sites followed by phosphorylation and ligation, PIPE cloning uses ligation independent cloning from annealing overhanging PCR ends and requires less time and components. Overhangs are added on to the sequences by using one half of the primers for the FGFR sequence and another half for the vector. The single-stranded overhangs that are now flanking the FGFR sequences are complementary to the vector sequence. As a result, by combining the two with subsequent transformation, a recombinant plasmid is formed (Klock and Lesley, 2009).- PCR
- Prepare the PCR reaction according to the instructions specified by the Phusion® High-Fidelity PCR kit using primers for generating the FGFR2 ECD + TM (31-406) construct.
- Add 6% DMSO to each reaction along with the proper PCR components for 25 μl reactions for the FGFR2 and vector pSpeedET + Maltose Binding Protein (MBP).
- PCR Reaction components for a 25 μl reaction are:
FGFR/pSpeedET + MBP vector template: 20 ng each
Phusion Hi Fidelity buffer: 5 μl
Phusion Hi Fidelity DNA polymerase: 0.25 μl
0.2 μM dNTP (0.5 μl of 10 mM dNTP)
0.5 μM forward and reverse primers (1.25 μl of 10 μM stocks)
6% DMSO: 1.5 μl of DMSO
Type 1 nuclease-free water: add to a total volume of 25 μl - Adjust the settings on the thermocycler to:
98 °C for 6 min;
Followed by a cycle of 35 times with the settings 98 °C for 30 s;
50 °C for 30 s;
72 °C for 2 min;
Finally 72 °C for 10 min followed by a 4 °C holding period. - Run the PCR product on a 1% agarose gel.
- Extract the PCR products of expected lengths using the Thermo Scientific GeneJET® Gel Extraction Kit.
- Expected Lengths are:
FGFR2 Primers: PIPE overhangs are in lower case. Sequences matching FGFR2 are in upper case.Sequence in question Expected size (bp) FGFR2 31-406 (ECD + TM) 783 pSpeedET + MBP 6642
pSpeedET-R2: cctcgagtgaagaacc TTCCTCATTGGTTGTGAGAGTGAGAATTCG;
MBP-F: ctgtacttccagggc ACCACATTAGAGCCAGAAGGAGCAC

Figure 1. FGFR construct in pSpeedET plasmid. The TEV protease cleavage site is in the overhang for the forward primers for the FGFR2 construct.
Figure 2. Process of polymerase incomplete primer extension (PIPE) cloning- Transformation for cloning–Immediately after the PIPE annealing, transfer into E. coli DH5α cells for cloning
- Thaw 1 vial of DH5α competent cells on ice.
- Add 2 μl of the PIPE reaction to the cells and incubate on ice for 10 min.
- Heat shock the cells at 42 °C for 30 s and place the cells back on ice for 2 min.
- Add 500 μl of SOC recovery media (at RT) to the cells and place them in a 37 °C shaker for one hour.
- Spread 100 μl of the transformation on LB + kanamycin plates and incubate them at 37 °C overnight. Kanamycin final concentration is at 50 μg/ml.
- Colony polymerase chain reaction, DNA extraction, and sequencing–Verify success of PIPE cloning by size and sequence using colony PCR and sequencing
- Use a pipette tip to pick a colony and resuspend it in 13.7 μl of nuclease-free water (the volume of water needed for a 25 μl PCR reaction).
- Add 13.7 μl water, 2.5 μl of 2.5 mM dNTPs, 2.5 μl of 2 mM forward primer, 2.5 μl of 2 μM reverse primer, 0.3 μl e2TAK polymerase (Takara Bio), and 5 μl polymerase 5x Buffer. The thermocycler conditions are as follows: 94 °C for 6 min followed by 35 cycles of 94 °C for 30 s, 50 °C for 30 s, and 72 °C for 2 min, and finally 72 °C for 10 min followed by a 4 °C holding period.
- Run on a 1% agarose gel to confirm the size of the expected insert.
- Grow the selected colonies in 3 ml LB + kanamycin overnight at 37 °C in a shaker at 230 rpm in 5 ml culture tubes.
- Isolate the recombinant plasmid using the General Electric Healthcare illustra plasmidPrep Mini Spin Kit.
- Determine the presence and accuracy of the construct by sequencing.
- Verify the sequence is correct by analysis with ApE program for nucleotide analysis and BLASTn comparison.
- PCR
- Large scale expression of the FGFR2 construct
- Transformation and inoculation for protein expression in Lemo21 DE3 cells
- Using the protocol as specified from above (Step A2), positively select using LB + kanamycin agar plates.
- Chose a colony and grow overnight between 12 h and 16 h in 100 ml of LB with 50 μg/ml kanamycin + 25 μg/ml chloramphenicol.
- After 16 h, prepare the inoculation: 100 ml overnight culture, 100 μg/ml kanamycin, 50 μg/ml chloramphenicol, 0.5 mM L-rhamnose, and 2 mM of MgSO4.
Note: The concentrations of the reagents are based on a volume of 1,000 ml of Terrific Broth (TB). - Place the inoculated TB culture and antibiotics into the shaker at 37 °C. Monitor the OD600 as it approaches 0.6. Once the culture reaches an OD600 of 0.4-0.5, cool the cells to 18 °C in the shaker to slow the growth of cells.
Note: OD should be checked every 10 min. The bacterial culture reaches OD = 0.6 after 30-45 min. - Take 1 ml of each construct culture to serve as a negative control for later experiments. In addition, take a glycerol stock (50 μl of 50% glycerol in ddH2O and 50 μl of culture) for archival purposes. Store the glycerol stock at -80 °C. The glycerol should be sterile.
- Add isopropyl beta-D-1-thiogalactopyranoside (IPTG) at a 0.1 mM final concentration to each culture to induce expression.
- Incubate the cells in the shaker at 18 °C overnight at 230 rpm.
- The next day, harvest the cells as followed: centrifuge the culture at 4 °C at 4,000 x g for 10 min.
- Discard the supernatant and wash the pellet by resuspending it in 100 ml 4 °C lysis buffer (Recipe 1).
- Centrifuge again at 4,000 x g for 10 min and discard the lysis buffer.
- Lysing cells
- Resuspend the post induction Lemo21 cell in 40 ml lysis buffer by vortexing and pipetting up and down to ensure homogeneity in a 50 ml conical tube. This must all be done on ice.
- Add 1 μl of 2000 U/ml DNase I, 1 μM CaCl2, and protease inhibitors (1 mM benzamidine, 1 mM PMSF, 1 μM E-64, 0.5 mM TCEP, 1 μM pepstatin, and 1 μM bestatin) to lysis buffer prior to lysis by sonication.
- Sonicate the cells. Sonication conditions: 6 cycles; 1 cycle: 20 s sonication ON followed by 40 s OFF.
- Following sonication, place the suspension in a heavy-duty centrifuge tube and centrifuge at 48,384 x g using a JA20 rotor for 30 min. Separate and store both supernatant and cell pellet at -80 °C.
Note: Optional stopping point. Ideally, better if the procedure is continued.
- Transformation and inoculation for protein expression in Lemo21 DE3 cells
- Detergent extraction
- Prepare a lysis buffer that includes 1% n-dodecyl β-D-maltoside (DDM) detergent.
- For every 100 μg of cell pellet or 100 μl of supernatant, add 500 μl of 1% detergent solution and resuspend along with PMSF.
- Invert the suspension continuously for 2 h at 4 °C.
- Centrifuge the suspension at 23,708 x g for 15 min at 4 °C.
- Separate the supernatant and the cell pellet and store both at -80 °C. (Both the cell pellet and supernatant should possess FGFR2, but the cell pellet should contain more).
- Running samples through NuSep 8-16% Tris-Glycine Gels via polyacrylamide gel electrophoresis (PAGE) to determine if the protein of interest is present
- Resuspend the pellet in 300 μl MilliQ water and 100 μl of 4x loading dye with β-mercaptoethanol (BME) while 100 μl of the same loading dye was added to the 300 μl supernatant/portion of cell pellet.
- Centrifuge at 23,708 x g for 10 min in preparation for loading into a polyacrylamide gel.
Note: Steps C1 and C2 are to ensure that the loaded sample is not too viscous for electrophoresis. - Load the samples (20 μl per well) along with a ladder and run at 200 V for about 45 min.
- Run 2 gels so that one can be stained and the other can be used for Western blotting.
- Western blotting and antibody detection using anti-MBP monoclonal antibody HRP conjugated
- Western blot the gel of interest using the VWR Amresco® Buffers.
- Soak the Tris-glycine gel in Amresco rapid transfer buffer for 10 min. Use just enough buffer to cover the entire gel.
- Soak the PVDF membrane for 30 s in methanol, then subsequently soak in water for 5 min, then finally soak in rapid transfer buffer until it no longer floats. Use just enough solution to cover the membrane.
- Assemble the Western block stack according to Amresco’s wet transfer protocol.
- Place the stack in the gel electrophoresis chamber with Amresco rapid transfer buffer and run at 75 V for 20 min. The rapid transfer buffer must be filled to the top of the chamber so that the stack is completely submerged in the buffer.
- Disassemble the stack and incubate the membrane in Amresco RapidBlockTM solution for 5 min.
- Incubate the PVDF membrane in 20 ml of blocking buffer for 1 h at room temperature.
- Incubate the PVDF Membrane in 20 ml of blocking buffer with 10 μl of anti-MBP monoclonal antibody (HRP Conjugated) from New England Biolabs overnight at 4 °C.
- The next day, use 25 ml of TBST (Recipe 2) to wash the PVDF membrane three times for 5 min each.
- Detect the presence of the protein using the KPL TMB Membrane Peroxidase Substrate System Kit. The volumes needed for the reaction to occur are listed in the kit’s instructions.
- Refolding FGFR2 by dialysis
- Washing inclusion bodies
Resuspend the pellets in 0.5% guanidinium-HCl (with just enough volume to get the pellets resuspended homogenously), and subsequently centrifuge at 4,000 x g for 20 min at 4 °C. After washing, the inclusion bodies should have a colorless appearance. - Solubilization and denaturation of FGFR recombinant protein
- Denature and solubilize the original inclusion bodies in 10 ml dialysis solution #1 (Recipe 3).
- To facilitate solubilization, warm the cell pellet and dialysis solution mixture to 40 °C temporarily in a water bath followed by gentle vortexing. The solution should be colorless and clear. Incubate the mixture prior to dialysis for 30-40 min at 40 °C.
- Assembly of apparatus and incubation
- Load the solubilized inclusion bodies in the dialysis solution #1 into a snakeskin membrane with clips.
- Suspend the filled membrane in a container with 700 ml dialysis solution #2 (Recipe 4).
- Place the apparatus in the cold room (4 °C) with gentle constant stirring using a magnetic stir bar overnight.
- The next day, place the sample from the snakeskin into heavy-duty centrifuge tubes and centrifuge at 48,384 x g for 30 min. Separate the pellet and supernatant and store at -80 °C. The protein of interest is in the supernatant.
- Washing inclusion bodies
- Purification and functionality testing via heparin binding chromatography
- To test isolate and begin testing the functionality of FGFR2, use the 1 ml HiTrapTM Heparin Affinity Chromatography column (GE Healthcare) with a syringe pump.
Note: GE Healthcare provides instructions for how to use the column. The following is additional details for this procedure.- Load 10 column volumes (CVs) of binding buffer (Recipe 5) onto the column with a syringe to equilibrate the column at a rate of 1 ml/min. 1 CV = 1 ml
- Syringe filter the FGFR2 supernatant from dialysis to prevent clogging and load it onto the column.
Note: Make sure no air bubbles get into the column. - Load the FGFR2 Supernatant unto the column.
- Wash the column of any debris with 5 CVs of binding buffer.
- Run 10 CVs of elution buffer (Recipe 6). Use sterile 1.5 ml centrifuge tubes to collect all flow through, wash, and elution fractions. The rate of loading and running is at 1 ml/min. The volume of each fraction should be 1 ml. 1 CV = 1 ml.
- Run a PAGE and Western Blot analysis to confirm FGFR2 isolation. For Western blotting, use the anti-MBP HRP-conjugated antibody from New England Biolabs to detect FGFR2 (Figure 3).
- Run size exclusion chromatography (SEC) of the purified FGFR2 combined with FGF1, heparin, and FGFR2 to verify dimerization (Figure 4). Follow by Western blotting of the SEC elution fractions with the anti-MBP antibody to identify the size of the eluted complex.
- The first step in testing functionality is to determine whether or not FGFR2 can bind to heparin and FGF1. This step is essential since heparin and FGF1 binding is a necessary step to ternary complex formation and dimerization. To do this, repeat Procedure F above along with loading 1 ml of FGF1’s elution from SEC, followed by 10 CVs (10 ml) of binding buffer between Steps F2 and F3.
- Critical notes have been included throughout the protocol at important steps. Overall, this is a lengthy procedure with few stopping points. As an additional note, we have included how we express, isolate and purify FGF1 to test for FGFR2 functionality.
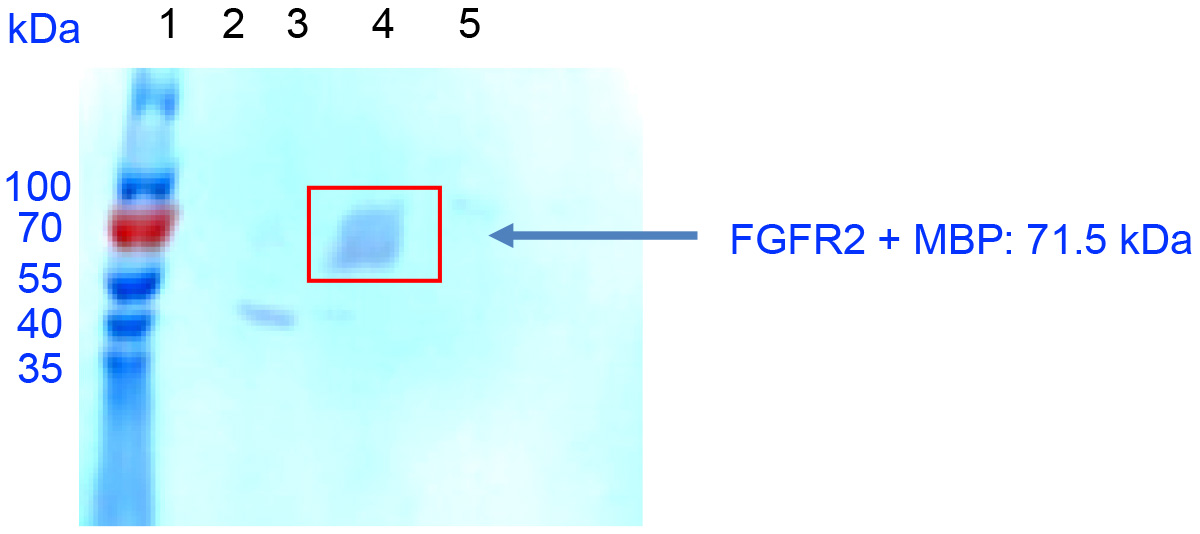
Figure 3. Western blot shows elution of FGFR2 + MBP Construct (red rectangle) from HiTrap heparin affinity chromatography demonstrating recombinant protein’s ability to bind to heparin. Lane 1: Protein standard. Lanes 2-3: Wash fractions. Lanes 4-5: Elution fraction.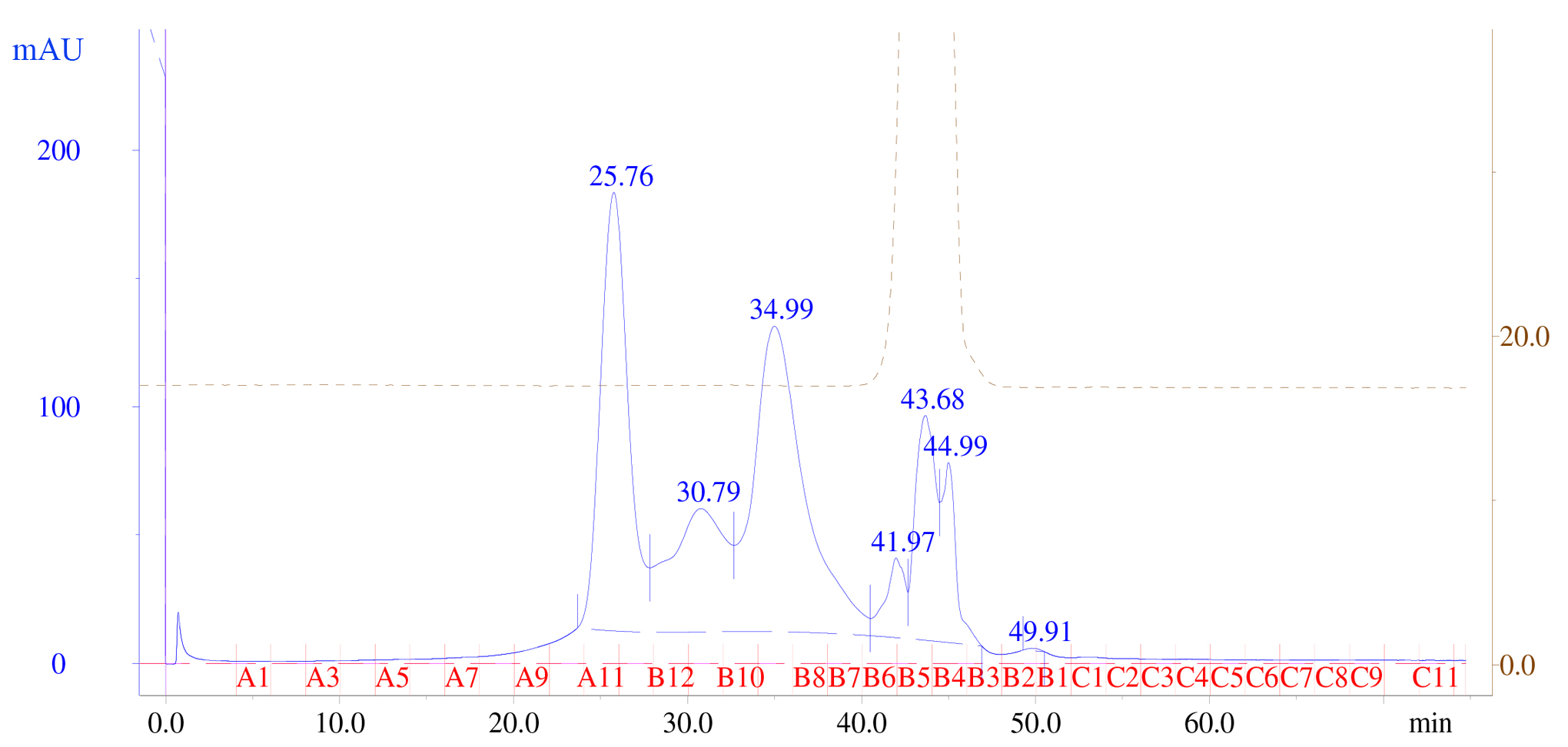
Figure 4. Size exclusion chromatography profile. Elution fractions are in red and range from A9-12, followed by B12-B7. Elution fractions A11 and A12 correspond to inactive FGFR2 containing the ECD + TM domains in dimers in micelles. Elution fraction B11 corresponds to FGFR2 containing the ECD + TM domains. Elution fractions B11-B8 corresponds to FGF1. Each elution fraction has a volume of 1 ml.
- FGF1 expression
- Transform DH5α cells using the FGF1 expression vector in pMCSG7 from the DNASU plasmid repository. To clone it, use the same procedure as detailed under Step A2 but use carbenicillin instead of kanamycin as the selection marker.
- Grow a colony in 3 ml of LB with 100 μg/ml carbenicillin overnight.
- Isolate DNA from the culture using the GE Healthcare illustra plasmidPrep Mini Spin Kit.
- Confirm FGF1 sequence using T7 forward and reverse primers.
- Large scale expression
- Transform the FGF1 expression plasmid into competent E. coli Lemo21 (DE3) cells and positively select with carbenicillin.
- Pick a colony and grow in 100 ml LB with chloramphenicol and carbenicillin overnight.
- The next day, use this culture to inoculate 1,000 ml of TB with 100 μg/ml carbenicillin, 50 μg/ml chloramphenicol, 0.5 mM rhamnose, and 2 mM of MgSO4 following Steps B1c above.
- Continue to cell harvesting, cell lysis by sonication and ultra-centrifugation as discussed above in Step B2, all at 4 °C. The protein will be in the supernatant.
- FGF1 Purification
Note: Purify the FGF1 using a 1 ml HiTrapTM Column (GE Healthcare) with a syringe.- Fill the syringe with 1 ml of binding buffer which is the same lysis buffer used for the large-scale expression of FGFR2. Making sure the air bubbles are out of the syringe, run the same lysis buffer through the column at a rate of 1 ml/min.
- Before reaching the end of the syringe, it was refilled with binding buffer, and again pushed through the syringe. This was done with 9 more CVs to equilibrate the column.
- Run the entire FGF1 sample (supernatant from ultracentrifugation) through the column using the syringe.
- Wash column with 10 CVs (10 ml total) of lysis buffer to elute any contaminating proteins that may have non-specifically bound to FGF1 and heparin.
- Elute FGF1 with elution buffer (Recipe 7).
- Determine the success of purification by PAGE and Western blot analysis as discussed above under Procedure C and Procedure D, but instead of using an anti-MBP antibody (HRP conjugated), an anti-His antibody (HRP conjugated) was used from Pierce.
- Use size exclusion chromatography (SEC) to further purify the protein. Load 2 ml of the elution onto the column (this was the maximum volume allowed and its concentration was determined to be acceptable to subsequent functionality experiments with FGFR: 1.756 μg/ml). The running buffer (Recipe 8) used for size exclusion chromatography (SEC) was 25 mM HEPES, 0.1% DDM, and 150 mM NaCl at pH 7.5.
Recipes
- Lysis Buffer
300 mM NaCl
50 mM HEPES at pH 7.5
0.1 mM MgSO4
5% glycerol - TBST
20 mM Tris-HCl pH 7.5
150 mM NaCl
0.1% Tween 20 - Denaturing buffer dialysis solution #1
6 M Guanidinium-HCl
0.1% DDM
10 mM DTT
1 μM Protease inhibitors E-64
1 mM benzamidine
1 mM PMSF
1 μM bestatin
1 μM pepstatin at pH 8.0 - Dialysis Solution #2
25 mM HEPES
150 mM NaCl
10% glycerol
1 mM L-cysteine at pH 7.5 - Binding buffer for heparin affinity chromatography
150 mM NaCl
25 mM HEPES
1 mM benzamidine
0.1% DDM at pH 7.5 - Elution buffer for heparin affinity chromatography
25 mM HEPES
1.5 M NaCl
0.1% DDM
1 mM benzamidine
1 mM PMSF at pH 7.5 - Elution buffer for FGF1
1.0 M NaCl
10% glycerol
25 mM HEPES
10 mM imidazole
1 mM benzamidine at pH 7.5 - Running Buffer for SEC
25 mM HEPES
0.1% DDM
150 mM NaCl at pH 7.5 - Blocking Buffer
5% Milk Powder
TBST
Acknowledgments
This project was funded by NSF CAREER Award 1350555 (HLN), the University of Hawaii at Manoa, and the University of Hawaii Undergraduate Research Opportunities Program (AB).
Competing interests
AB: No competing interests to disclose. HLN: No competing interests to disclose.
References
- Chen, P. H., Unger, V. and He, X. (2015). Structure of full-length human PDGFRβ bound to its activating ligand PDGF-B as determined by negative-stain electron microscopy. J Mol Biol 427(24): 3921-3934.
- Dessau, M. A. and Modis, Y. (2011). Protein crystallization for X-ray crystallography. J Vis Exp(47).
- Klock, H. E. and Lesley, S. A. (2009). The Polymerase Incomplete Primer Extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol Biol 498: 91-103.
- Klock, H. E., Koesema, E. J., Knuth, M. W. and Lesley, S. A. (2008). Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins 71(2): 982-994.
- Lu, C., Mi, L. Z., Schurpf, T., Walz, T. and Springer, T. A. (2012). Mechanisms for kinase-mediated dimerization of the epidermal growth factor receptor. J Biol Chem 287(45): 38244-38253.
- Mi, L. Z., Grey, M. J., Nishida, N., Walz, T., Lu, C. and Springer, T. A. (2008). Functional and structural stability of the epidermal growth factor receptor in detergent micelles and phospholipid nanodiscs. Biochemistry 47(39): 10314-10323.
- Mi, L. Z., Lu, C., Li, Z., Nishida, N., Walz, T. and Springer, T. A. (2011). Simultaneous visualization of the extracellular and cytoplasmic domains of the epidermal growth factor receptor. Nat Struct Mol Biol 18(9): 984-989.
- Mohammadi, M., Schlessinger, J. and Hubbard, S. R. (1996). Structure of the FGF receptor tyrosine kinase domain reveals a novel autoinhibitory mechanism. Cell 86(4): 577-587.
- Opatowsky, Y., Lax, I., Tome, F., Bleichert, F., Unger, V. M. and Schlessinger, J. (2014). Structure, domain organization, and different conformational states of stem cell factor-induced intact KIT dimers. Proc Natl Acad Sci U S A 111(5): 1772-1777.
- Paavilainen, S., Grandy, D., Karelehto, E., Chang, E., Susi, P., Erdjument-Bromage, H., Nikolov, D. and Himanen, J. (2013). High-level expression of a full-length Eph receptor. Protein Expr Purif 92(1): 112-118.
- Plotnikov, A. N., Hubbard, S. R., Schlessinger, J. and Mohammadi, M. (2000). Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell 101(4): 413-424.
- Plotnikov, A. N., Schlessinger, J., Hubbard, S. R. and Mohammadi, M. (1999). Structural basis for FGF receptor dimerization and activation. Cell 98(5): 641-650.
- Rosano, G. L. and Ceccarelli, E. A. (2014a). Recombinant protein expression in microbial systems. Front Microbiol 5: 341.
- Rosano, G. L. and Ceccarelli, E. A. (2014b). Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol 5: 172.
- Schlessinger, J., Plotnikov, A. N., Ibrahimi, O. A., Eliseenkova, A. V., Yeh, B. K., Yayon, A., Linhardt, R. J. and Mohammadi, M. (2000). Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell 6(3): 743-750.
- Turner, N. and Grose, R. (2010). Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 10(2): 116-129.
- Yeh, B. K., Eliseenkova, A. V., Plotnikov, A. N., Green, D., Pinnell, J., Polat, T., Gritli-Linde, A., Linhardt, R. J. and Mohammadi, M. (2002). Structural basis for activation of fibroblast growth factor signaling by sucrose octasulfate. Mol Cell Biol 22(20): 7184-7192.
- Zhang, F., Zhang, Z., Lin, X., Beenken, A., Eliseenkova, A. V., Mohammadi, M. and Linhardt, R. J. (2009). Compositional analysis of heparin/heparan sulfate interacting with fibroblast growth factor·fibroblast growth factor receptor complexes. Biochemistry 48(35): 8379-8386.
- Babina, I. S. and Turner, N. C. (2017). Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer 17 (5):318-332.
- Robin, N. H., Falk, M. J., Haldeman-Englert, C. R. (1993). FGFR-Related Craniosynostosis Syndromes. In: Adam, M. P., Ardinger, H. H., Pagon, R. A. et al. (eds) GeneReviews ((R)). Seattle (WA).
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Bajinting, A. and Ng, H. L. (2019). A Detailed Protocol for Large-scale Recombinant Expression and Validation of Human FGFR2 with Its Transmembrane and Extracellular Domains in Escherichia coli. Bio-protocol 9(11): e3261. DOI: 10.21769/BioProtoc.3261.
Category
Biochemistry > Protein > Expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.