- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Protocol to Quantitatively Assess the Structural Integrity of Perineuronal Nets ex vivo
Published: Vol 9, Iss 10, May 20, 2019 DOI: 10.21769/BioProtoc.3234 Views: 6852
Reviewed by: Arnau Busquets-GarciaMarta MiquelJoshua Brumberg
Original research article
The authors used this protocol in:

Nov 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Perineuronal nets (PNNs) are extracellular matrix assemblies of highly negatively charged proteoglycans that wrap around fast-spiking parvalbumin (PV) expressing interneurons in the cerebral cortex. PNNs play important roles in neuronal plasticity and modulate biophysical properties of the enclosed interneurons. Various central nervous system diseases including schizophrenia, Alzheimer disease and epilepsy present with qualitative alteration in PNNs, however prior studies failed to quantitatively assess such changes at single PNN level and correlate them with functional changes in disease. We describe a method to quantify the structural integrity of PNNs using high magnification image analysis of Wisteria Floribunda Agglutinin (WFA)-labeled PNNs in combination with cell-type-specific marker such as PV and NeuN. A polyline intensity profile of WFA along the entire perimeter of cell shows alternate segments with and without WFA labeling, indicating the intact chondroitin sulfate proteoglycan (CSPG) and holes of PNN respectively. This line intensity profile defines CSPG peaks, where intact PNN is present, and CSPG valleys (holes) where the PNN is missing. The average number of peaks reflect the integrity of the lattice assembly of PNN. The average size of PNN holes can be readily computed using image analysis software. Furthermore, degradation of PNNs using a bacterial-derived enzyme, Chondroitinase ABC (ChABC), allows to experimentally manipulate PNNs in situ brain slices during which biophysical properties can be assessed by patch-clamp recordings. We describe optimized experimental parameters to degrade PNNs in brain slices before as well as during recordings to study the possible change in function in real time. Our protocols provide effective and appropriate methods to modulate and quantify the PNN’s experimental manipulations.
Keywords: Perineuronal netsBackground
The extracellular spaces of the central nervous system are filled by an amorphous interstitial matrix that is composed of dense network of proteoglycans, hyaluronic acid (HA), tenascins and link proteins, and is contiguous with well-organized lattice-like structures called PNNs that enclose the somata, axons and dendrites of PV expressing interneurons in the cerebral cortex (Härtig et al., 1999). The lattice of PNNs is composed of chondroitin sulfate proteoglycans (CSPGs), HA, tenascins, various link proteins (Deepa et al., 2006), etc. Owing to the high density of sulfated proteoglycans, PNNs are highly negatively charged (Morawski et al., 2015). The development of PNN coincides with the closure of ocular dominance critical period and studies have shown reinstatement of ocular dominance plasticity upon removal of the PNNs thereby suggesting inhibitory role of PNNs on neuronal plasticity (Pizzorusso et al., 2002). PNNs are also suggested to modulate the biophysical properties PV interneurons (Balmer, 2016, Favuzzi et al., 2017, Tewari et al., 2018), and are prone to degradation by ECM remodeling enzymes. Most notably, under pathological conditions including epilepsy (McRae et al., 2012, Rankin-Gee et al., 2015, Dubey et al., 2017, Tewari et al., 2018, Patel et al., 2019), schizophrenia and Alzheimer disease (Testa et al., 2018) ECM remodeling is common.
One of the major challenges in reporting on the status of PNNs is the quantification of the structural integrity of their lattice-like assembly. Essentially all published studies use the CSPG binding lectin, WFA, as non-specific PNN marker to visualize PNNs, and use WFA intensity to quantify the overall CSPG levels without detailed analysis of individual PNN’s architecture (Slaker et al., 2015 and 2016, Lensjø et al., 2017, Ueno et al., 2018). We developed a method to quantify the structural integrity of individual PNNs using high-magnification fluorescence imaging. This method also minimizes the errors due to procedure-related variations in the WFA intensity. After acquiring multichannel high magnification images (40x objective lens with 5 digital zoom) of individual PNNs, a polyline is drawn around the periphery of the cell. The fluorescence intensity of WFA along this line shows a discrete pattern of peaks separated by low-intensity valleys. Each peak represents the continuous CSPG and the gap between the two consecutive peaks represent a hole in the PNN (Tewari et al., 2018). The average number of peaks and average width of holes can be derived from the WFA intensity data points and are reflective of the PNN’s structural integrity. The peaks in the line profile can be counted manually or by using clampfit program (Molecular Devices).
A vast majority of studies, which explore the function of PNNs and their influence on the neuronal physiology, use ChABC, which is a bacterial enzyme that cleaves the chondroitin sulfate side chains thereby dismantling the PNN assembly (Dityatev et al., 2007, Balmer, 2016, Testa et al., 2018). The most common approach in such studies involves degradation of PNN in-vivo/in-situ brain slices/primary cultures and studying the function afterward by comparing two different populations of samples. However, it would be advantageous to record from PNN bearing neurons during the real-time degradation of PNNs to evaluate the physiological role of PNN on neuronal function. Such studies are largely absent in the literature. Here, we report two variants of experimental PNN degradation in the brain slices, which reliably degrade PNNs to allow studying their function. The first variant includes degradation of PNNs by incubating brain slices with ChABC in an incubation chamber followed by performing experiments to compare their properties with non-treated controls. The other variant is to study the baseline physiology in the presence of intact PNNs followed by superfusing ChABC solution and recording the functional activity during and after PNN depletion in the experimental set-up. Both the variants involve controlled degradation of PNNs and identifiable traces of degraded PNNs around the cells can be observed to confirm the presence/absence of PNNs on the experimental cell/tissue after post-experimental WFA staining of the samples. These methods provide unparalleled advantages to study the physiological functions associate with the PNNs.
Materials and Reagents
Materials
- PNN degradation in brain slices outside experimental setup
- Carbogen inlet and outlet needles (BD PrecisionGlide needle, catalog number: 305198)
- Perfusion Tubes (Becton Dickinson, PE-160, catalog number: 427431)
- Carbogen bubbling tubes (Fisher Scientific, Tygon, catalog number: S3 E-3603)
- Multichannel splitter (Luner valve assortment, catalog number: WPI-14055)
- Transfer pipettes (Fisher brand, catalog number: 13-711-7M)
- Glass Petri dish (Cole-Parmer Instrument, catalog number: EW-34551-06)
- Polystyrene foam (from any commercial source)
- PNN degradation in brain slices in experimental setup
- 15 ml tube (Falcon, catalog number: 352097)
- Thin perfusion lines (PVC pump tubes-1.52 mm ID, catalog number: 116-0549-19)
- Bubbling tubes (Fisher Scientific, Tygon, catalog number: S3 E-3603)
- Perfusion Tubes (Becton Dickinson, PE-160, catalog number: 427431)
- PNN’s structural integrity analysis
- High magnification (40 x 5 or higher) images of WFA with PV or NeuN
- High magnification (40 x 5 or higher) images of WFA with PV or NeuN
- WFA staining of brain slices
- 24-well plate (Falcon, catalog number: 35-3226)
- Paint Brush (Any fine tipped clean brush)
- Coverslips (Fisher Finest, catalog number: 12-548-5M)
- Glass slide (Micro slides, VWR, catalog number: 48311-703)
- Mounting medium (Thermo Fisher, Invitrogen, catalog number: S36936)
- Marker (Lab marker, VWR, catalog number: 52877-310)
- Transfer pipettes (Fisher brand, catalog number: 13711-7M)
Reagents
- ChABC (Sigma-Aldrich, catalog number: C3667-10U)
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A8806-1G)
- 4% Paraformaldehyde (PFA) (Electron Microscopy Science, catalog number: 15714-S)
- PBS (Fisher Bioreagent, catalog number: BP661-10)
- Biotinylated Wisteria Floribunda Agglutinin/Lectin (WFA/WFL) (Vector Laboratories, catalog number: B-1355)
- Streptavidin, Alexa FluorTM 555 Conjugate (Thermo Fisher, Invitrogen, catalog number: S32355)
- 4',6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) (Thermo Fisher, Life Tech, catalog number: D1306)
- Mounting medium (Thermo Fisher, Invitrogen, catalog number: S36936)
- Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, catalog number: D8418)
- ChABC (see Recipes)
- PBS (see Recipes)
- 4% PFA (see Recipes)
- ACSF (standard ringer’s ACSF for acute brain slices) (Papouin and Haydon, 2018, Tewari et al., 2018) (see Recipes)
- DAPI stock (see Recipes)
Equipment
- Measurement pipettes (Gilson, catalog number: F123602)
- Timer (from any commercial source)
- Sterile empty glass vials, 30 ml (such as Hospire, catalog number: 5816-31)
- Slice incubation assembly: assemble as shown in Figures 2B-2D
- Bath heater (Fisher Scientific, model: Isotemp 205)
- Peristaltic Perfusion pump (Gilson, model: mini-plus 3)
- In-line heater (Warner instrument, catalog number: TC324B)
- High resolution imaging microscope, such as Confocal microscope (Nikon Elements)
- 95% O2/5% CO2 tank (such as AirGas)
- -20 °C freezer (such as Norlake)
Software
- Nikon NIS-Elements
- Nikon NIS-Elements AR analysis
- (Optional) Clampfit (Molecular Devices)
- (Optional) Microsoft Excel/Origin (OriginLab)
Procedure
- Structural integrity of PNNs in high-magnification images
- Acquire high-magnification (40x objective lens with 5 digital zoom) multichannel (with PV or NeuN) single plane fluorescence images of individual PNNs in different experimental groups. The images should be taken from the optical plane that covers the largest perimeter of the cell.
- Draw a manual/automated (depending on the used software) polyline along the periphery of the cell (stained with NeuN/PV) with PNN (Figures 1A and 1B, large images).
- If using Nikon elements program, then use the auto ROI function to determine the perimeter of the cell followed by drawing a polyline along with this perimeter.
- If using Fiji/ImageJ, use the freehand line tool to draw a line along the cell perimeter followed by clicking on “Analyze” then “Plot profile” buttons.
- WFA peaks can be counted from the peak profile graph itself or numerical data for the plot profile can be exported to plot a graph in different software and count the peaks.
- After establishing the line profile, the software generates a line profile graph that shows the fluoresce intensity of WFA along the line (Figures 1A and 1B, bottom panels). The line profile graph and its numerical data can be exported in a Microsoft Excel file that has length (in µm) and fluorescence intensity as x and y-axis respectively. Peaks can be counted either from this graph generated by the acquisition program (as shown in Figures 1A and 1B, lower panels) or data can be plotted in Excel/analysis software such as Sigma plot or Origin for peak counting and representation.
- Mark and count the number of peaks in the line profile by setting a threshold of half the maximum intensity (Figures 1A-1B, lower panels). To determine the size of the biggest hole in the PNN, measure the distance between two consecutive peaks that appear to be farthest from each other (white double-headed arrows in lower panels in Figures 1A-1B). (Alternately, plot the data in Clampfit result window and save it as an .atf file. Open this file later and use the peak detection function to count the number of peaks. The parameters of peak detection function may detect few false peaks, which can be removed from the analysis. In addition, placing two cursers covering the gap and remarking the distance between them can be used to determine the size of the largest hole.)
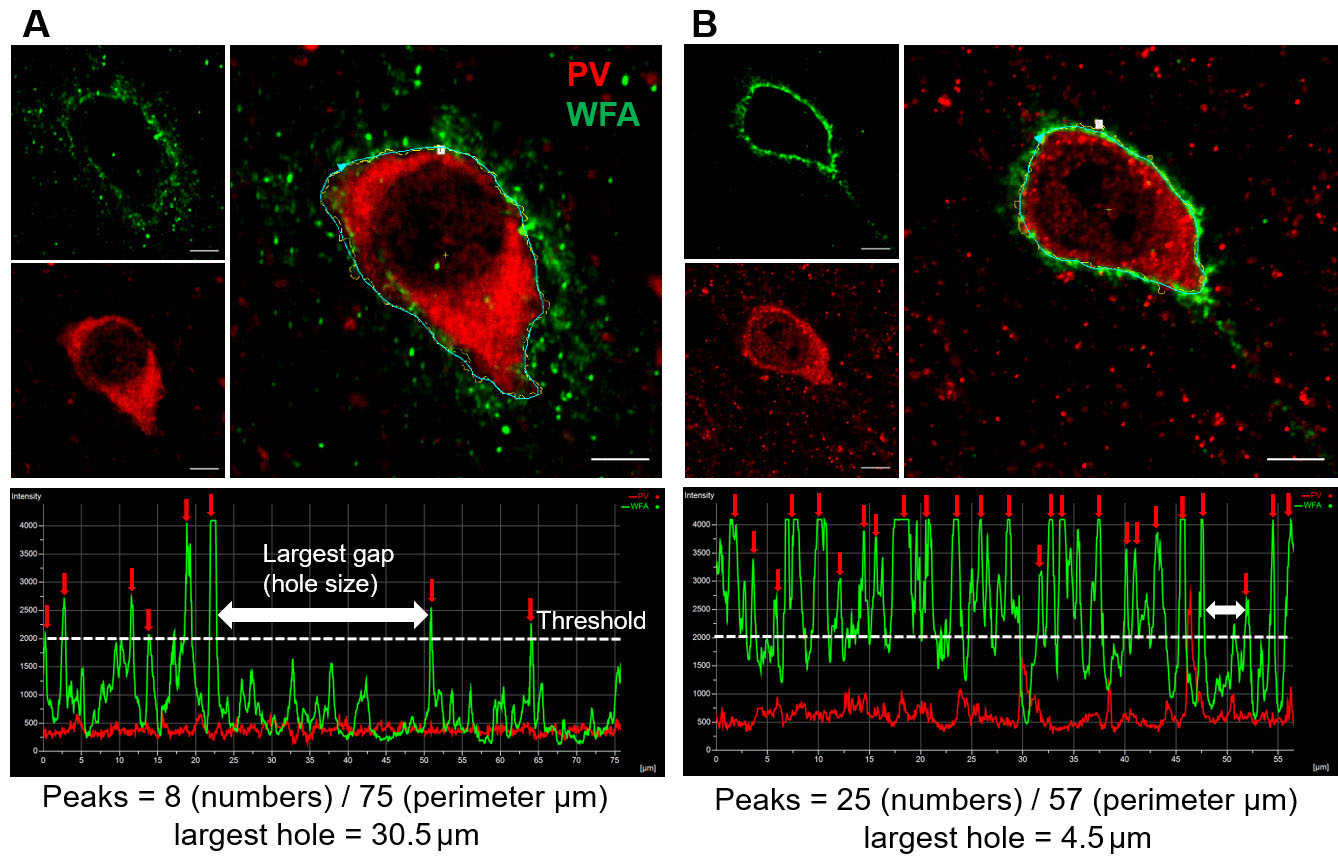
Figure 1. Analysis of WFA intensity peaks of PNN in two experimental conditions. A and B. High magnification (40x objective lens with 5 digital zoom) immunohistochemical staining images of WFA (green), PV (red) and overlay (large images on the right) showing single PNN in the cerebral cortex of a mouse model of glioma-associated epilepsy (A), and corresponding sham control (B). The polyline (right) along the perimeter of the PV, encompasses WFA staining and shows discrete WFA intensity peaks (marked by red arrows) separated by low WFA intensity baseline. White dotted line indicates the threshold (Maximum WFA intensity/2) to determine the WFA intensity peak. The two-headed white arrows show the highest apparent distance between two consecutive WFA peaks and indicate the size of the largest holes in the PNN. The total number of peaks are divided by the perimeter to account for the different size of cells and their PNNs. Scale bars: 5 µm.
- Pre-experimental degradation of PNNs in brain slices
- Prepare brain slices according to the standard brain slicing protocols and after completion of the recovery use for the experiments (Papouin and Haydon, 2018, Tewari et al., 2018).
- Prepare the slice incubation assembly using two glass vials; make two small holes on each cap to insert the inlet and outlet needles of carbogen gas supply. Insert an appropriate length piece of tube on the tip of needles to extend the gas supply close to the surface of the buffer. Do not dip these gas tubes into the solution (it can damage the slices).
- Make one needle inlet by connecting it to the carbogen supplying tube and leave the other needle open to function as outlet (Figure 2B).
- Next, fill 3 ml well-oxygenated ACSF in each vial and add ChABC stock to make a final concentration of 0.5 U/ml of ACSF in one of the vials labeled as treated.
- Transfer 2-3 brain slices in each vial using a modified transfer pipette (cut the tip of transfer pipette to broaden the tip for the ease of slice transfer as shown in Figure 2C).
- Set timer for 45 min from the time of ChABC addition and ensure that the carbogen gas supply through the inlet is working fine.
- Place both the vials into a custom-made floating polystyrene foam (Figure 2D) that allows vials to stay straight while floating in the water bath during incubation.
- Transfer both vials into the water bath, preset at 32 °C (Figure 2E).
- On completion of 45 min incubation, carefully take out vials, rinse slices with ACSF, and transfer slices from both the vials back to the recovery chamber (Figure 2G). The slices can be used for experimentation now.
- After completion of the experiment, carefully take out slices from the experimental setup and fix overnight with 4% PFA. Stain the fixed slices with WFA (and DAPI, if needed) to confirm the PNN degradation effect of ChABC as described in Procedure D.

Figure 2. Degradation of PNNs in the acute brain slices using pre-experiment incubation method. A. Brain slice recovery chamber with cortical slices in the ACSF with continuous carbogen (95% O2, 5% CO2) bubbling. B. Fill the incubation vials (for control and ChABC-treated slices) with 3 ml ACSF (with/without ChABC) and assemble the carbogen gas supply tubes with an inlet and one outlet. C. Add 2-3 brain slices from (A) to each vial by modified transfer pipette (B on Petri dish) and subsequently insert the vials in the polystyrene foam to held them straight in the water bath (E) while incubating at 32 °C for 45 min (F). G. On completion of incubation, transfer slices back into the recovery chamber at room temperature for experimental purposes.
- PNN degradation during real time experimentation in brain slices
- Record the baseline activity of cells/slices for 5-10 min (depending on the specific experiment) in the recording setup.
- Superfuse 1 U/ml ChABC solution for 50 min while strictly maintaining the bath temperature at 32-33 °C (Figure 3A).
- After 50 min of recording, perfuse ACSF again and record the activity in the presence of ACSF for 5-10 min (Figure 3B).
- Stop the experiment and transfer slices to a 24-well plate and add 500 µl, 4% PFA to fix the slice and keep at room temperature for overnight.
- Perform WFA staining next day as described in Procedure D.

Figure 3. Degradation of PNNs in acute brain slices during real time data acquisition. A. ACSF containing ChABC with continuous carbogen bubbling is superfused and re-circulated at a speed of 2-3 ml/min to the experimental set-up using a peristaltic pump. B. Recording setup with an inline heater to keep the bath temperature constant at 32 °C.
- WFA and DAPI staining in fixed acute brain slices
- Rinse slices 4 times with PBS with 5 min interval between each wash.
- Add 0.5 ml PBS that contains Biotinylated-WFA solution (1:300 in PBS), in each slice and incubate for 1 h at room temperature followed by 4 rinses with PBS at every 5 min interval.
- Add 0.5 ml PBS that contains Streptavidin-conjugated Alexa fluor-555 solution (1:300 in PBS), in each slice and incubate for 1 h at room temperature followed by 4 rinses with PBS at every 5 min interval.
- Incubate slices with PBS that contains DAPI (1:1,000 from 10 mg/ml stock in DMSO) for 4 min and rinse 3 times with PBS at every 5 min.
- Mount slices in between 2 coverslips that allows user to access both the surfaces of the slice for imaging. Use mounting medium and do not press hard the coverslips to prevent tissue distortion.
- Keep mounted tissue on a glass slide and take images of recorded cells/or regions of brain to confirm the degradation of PNNs.
Data analysis
- PNN’s structural integrity
Average the number of peaks/unit perimeter length of the cell. Use > 5 PNNs per mouse and pool data from all mice of the same experimental group. Compare the number of peaks in different experimental groups, apply appropriate statistical tests and plot the graph. Average the lengths of biggest gaps in the individual PNNs and pool data from all mice of the same experimental group. Compare the average size of holes (gap length) in different experimental groups, apply appropriate statistical tests, and plot the graph. - Confirmation of experimental PNN degradation
Take images from both sides of the slices using a fluorescence microscope. In case of pre-experimental degradation (Procedure B), both the surfaces of slice do not show any intact PNNs. However, magnified images show faint outlines of WFA around the PNN expressing cells (Inset image in Figure 4B). Only ACSF treated slices show intact PNNs (Figure 4A). In case of PNN degradation in experimental setup (Procedure C), only the top or exposed surface of slice shows the degradation effect of ChABC (Figure 4D). The high magnification images (20x) show faint outline of PNNs around the PNN expressing neurons (Inset image in Figure 4D). In case of whole cell patch clamp experiments, a traces dye can also be added to the pipette buffer to label the recorded cell, and presence of faint WFA outline around the recorded cells suggests that cells expressed PNN before the ChABC-treatment (Tewari et al., 2018).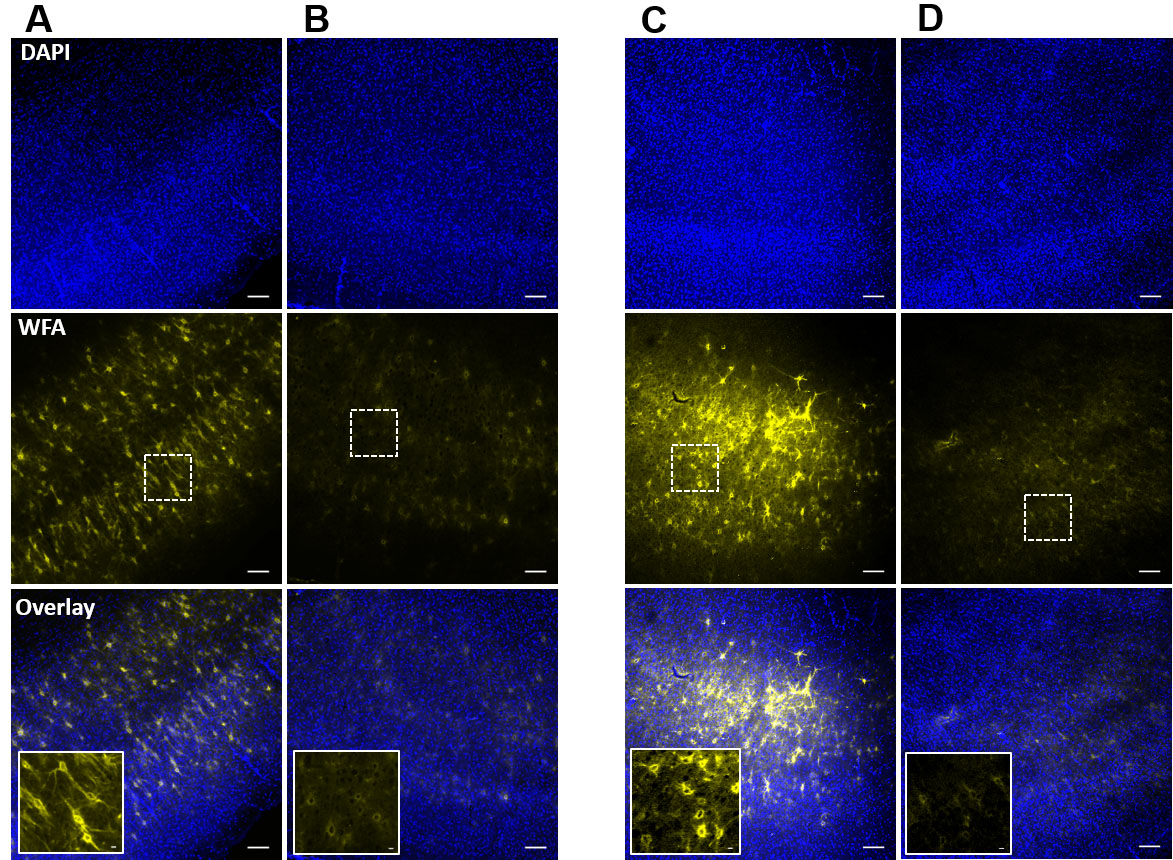
Figure 4. Confirmation of PNN degradation after ChABC-treatment of acute slices in incubation chamber and in recording setup. A and B. Confocal images of DAPI (top) and WFA (middle) fluorescence of acute cortical slices after post-experimental fixation from control (A), and ChABC-treated (B) groups. Inset images in overlay show the traces of controlled degradation of PNNs. C and D. Confocal images of DAPI (top) and WFA (middle) fluorescence from the ChABC-unexposed bottom surface (C), and ChABC-exposed top surface (D) of an acute brain slice in which PNNs were degraded in the experimental setup simultaneously with the experimental recording. The unexposed surface (C) shows relatively intact PNNs compared to the exposed surface (D), which shows degraded PNNs due to direct exposure of ChABC. Inset images in overlay are from the marked rectangular areas in the slices and show the traces of controlled PNN degradation. Scale bars: 100 µm in main images, 10 µm in inset images.
Notes
- In Procedures B and C, bath temperature is crucial to maintain at 32-33 °C for reliable PNN degradation.
- In Procedure B, the incubation time should not exceed 45 min otherwise; PNNs will be completely degraded making it difficult to find the traces of the degraded PNNs.
- In Procedure C, recirculating the ChABC solutions into the recording setup is recommended to minimize the quantity of the enzyme.
- In Procedure C, the total length of perfusion tubing for recirculation of ChABC should be kept minimum to reduce the time lag and the volume of total ChABC solution.
- For more precise comparison of the change in function upon PNN degradation, brain slices can be cut into 2 halves separating the cerebral hemispheres and one half can be treated with ChABC and other can be used as a control.
- For PNN’s structural integrity analysis in immunohistochemistry (IHC), freely accessible software including ImageJ/Fiji can also be utilized, provided all the experimental groups are analyzed with the same software.
- The high magnification images for PNNs structural integrity analysis should have the whole range of fluorescence signal with minimal saturation to minimize the variations in WFA intensity due to IHC procedure.
Recipes
- ChABC
- Reconstitute ChABC from Proteus vulgaris in a 0.01% BSA aqueous solution according to the manufacturer’s instruction to make 1 U/40 µl stock solution
- Prepare aliquots of 2 U and store at -20 °C until used
- Just before experiment, add appropriate amount directly into the bubbling ACSF to make the working concentration
- PBS
Dissolve PBS salts mixture in deionized water (DI) to make PBS solution - 4% PFA
Dilute the supplied 32% PFA to 1(PFA):7(PBS) to make 4% working solution - ACSF
Standard ringer’s buffer (125 NaCl, 3 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1.3 MgSO4, 25 glucose (all in mM); pH 7.3; 310 ± 5 mOsm) used for acute slice recordings (Tewari, Chaunsali et al., 2018) - DAPI stock
Dissolve DAPI in DMSO to make 10 mg/ml stock and store at -20 °C until use
Acknowledgments
This work was supported by NIHRO1-NS036692, NIH-RO1-NS082851, and NIH-RO1-NS052634.
Competing interests
The authors declare no competing interests
Ethics
All animal procedures were approved and performed in accordance with the ethical guidelines set by Virginia Tech Institutional Animal Care and Use Committee (IACUC) under protocols 15-241 (02/11/2016-02/09/2019), 15-106 (07/31/2015-07/30/2018), and 18-126 (07/18/2018-07/15/2021).
References
- Balmer, T. S. (2016). Perineuronal nets enhance the excitability of fast-spiking neurons. eNeuro 3(4): ENEURO.0112-16.2016.
- Deepa, S. S., Carulli, D., Galtrey, C., Rhodes, K., Fukuda, J., Mikami, T., Sugahara, K. and Fawcett, J. W. (2006). Composition of perineuronal net extracellular matrix in rat brain: a different disaccharide composition for the net-associated proteoglycans. J Biol Chem 281(26): 17789-17800.
- Dubey, D., McRae, P. A., Rankin-Gee, E. K., Baranov, E., Wandrey, L., Rogers, S. and Porter, B. E. (2017). Increased metalloproteinase activity in the hippocampus following status epilepticus. Epilepsy Res 132: 50-58.
- Dityatev, A., Bruckner, G., Dityateva, G., Grosche, J., Kleene, R. and Schachner, M. (2007). Activity-dependent formation and functions of chondroitin sulfate-rich extracellular matrix of perineuronal nets. Dev Neurobiol 67(5): 570-588.
- Favuzzi, E., Marques-Smith, A., Deogracias, R., Winterflood, C. M., Sanchez-Aguilera, A., Mantoan, L., Maeso, P., Fernandes, C., Ewers, H. and Rico, B. (2017). Activity-dependent gating of parvalbumin interneuron function by the perineuronal net protein brevican. Neuron 95(3): 639-655 e610.
- Härtig, W., Derouiche, A., Welt, K., Brauer, K., Grosche, J., Mader, M., Reichenbach, A. and Bruckner, G. (1999). Cortical neurons immunoreactive for the potassium channel Kv3.1b subunit are predominantly surrounded by perineuronal nets presumed as a buffering system for cations. Brain Res 842(1): 15-29.
- Lensjø, K. K., Christensen, A. C., Tennoe, S., Fyhn, M. and Hafting, T. (2017). Differential expression and cell-type specificity of perineuronal nets in hippocampus, medial entorhinal cortex, and visual cortex examined in the rat and mouse. eNeuro 4(3): ENEURO.0379-16.2017.
- McRae, P. A., Baranov, E., Rogers, S. L. and Porter, B. E. (2012). Persistent decrease in multiple components of the perineuronal net following status epilepticus. Eur J Neurosci 36(11): 3471-3482.
- Morawski, M., Reinert, T., Meyer-Klaucke, W., Wagner, F. E., Troger, W., Reinert, A., Jager, C., Bruckner, G. and Arendt, T. (2015). Ion exchanger in the brain: Quantitative analysis of perineuronally fixed anionic binding sites suggests diffusion barriers with ion sorting properties. Sci Rep 5: 16471.
- Papouin, T. and Haydon, P. G. (2018). Obtaining acute brain slices. Bio Protoc 8(2): e2699.
- Patel, D. C., Tewari, B. P., Chaunsali, L. and Sontheimer, H. (2019). Neuron-glia interactions in the pathophysiology of epilepsy. Nat Rev Neurosci. DOI: 10.1038/s41583-019-0126-4.
- Pizzorusso, T., Medini, P., Berardi, N., Chierzi, S., Fawcett, J. W. and Maffei, L. (2002). Reactivation of ocular dominance plasticity in the adult visual cortex. Science 298(5596): 1248-1251.
- Rankin-Gee, E. K., McRae, P. A., Baranov, E., Rogers, S., Wandrey, L. and Porter, B. E. (2015). Perineuronal net degradation in epilepsy. Epilepsia 56(7): 1124-1133.
- Slaker, M. L., Harkness, J. H. and Sorg, B. A. (2016). A standardized and automated method of perineuronal net analysis using Wisteria floribunda agglutinin staining intensity. IBRO Rep 1: 54-60.
- Slaker, M. L, Churchill, L., Todd, R. P., Blacktop, J. M., Zuloaga, D. G., Raber, J., Darling, R. A., Brown, T. E. and Sorg, B. A. (2015). Removal of perineuronal nets in the medial prefrontal cortex impairs the acquisition and reconsolidation of a cocaine-induced conditioned place preference memory. J Neurosci 35(10): 4190-4202.
- Testa, D., Prochiantz, A. and Di Nardo, A. A. (2019). Perineuronal nets in brain physiology and disease. Semin Cell Dev Biol 89: 125-135.
- Tewari, B. P., Chaunsali, L., Campbell, S. L., Patel, D. C., Goode, A. E. and Sontheimer, H. (2018). Perineuronal nets decrease membrane capacitance of peritumoral fast spiking interneurons in a model of epilepsy. Nat Commun 9(1): 4724.
- Ueno, H., Suemitsu, S., Murakami, S., Kitamura, N., Wani, K., Matsumoto, Y., Okamoto, M., Aoki, S. and Ishihara, T. (2018). Juvenile stress induces behavioral change and affects perineuronal net formation in juvenile mice. BMC Neurosci 19(1): 41.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Tewari, B. P. and Sontheimer, H. (2019). Protocol to Quantitatively Assess the Structural Integrity of Perineuronal Nets ex vivo. Bio-protocol 9(10): e3234. DOI: 10.21769/BioProtoc.3234.
Category
Neuroscience > Nervous system disorders > Epilepsy
Neuroscience > Cellular mechanisms > Extracellular matrix
Cell Biology > Cell staining > Extracellular matrix
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.