- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Improved HTGTS for CRISPR/Cas9 Off-target Detection
Published: Vol 9, Iss 9, May 5, 2019 DOI: 10.21769/BioProtoc.3229 Views: 6883
Reviewed by: Arif Md. Rashedul KabirMario RuizAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Precise genome editing is essential for scientific research and clinical application. At present, linear amplification-mediated high-throughput genome-wide translocation sequencing (LAM-HTGTS) is one of most effective methods to evaluate the off-target activity of CRISPR-Cas9, which is based on chromosomal translocation and employs a “bait” DNA double-stranded break (DSB) to capture genome-wide “prey” DNA DSBs. Here, we described an improved HTGTS (iHTGTS) method, in which size-selection beads were used to enhance reaction efficiency and a new primer system was designed to be compatible with Illumina Hiseq sequencing. Compared with LAM-HTGTS, iHTGTS is lower cost and has much higher sensitivity for off-target detection in HEK293T, K562, U2OS and HCT116 cell lines. So we believe that iHTGTS is a powerful method for comprehensively assessing Cas9 off-target effect.
Keywords: CRISPR-Cas9Background
The CRIPSR-Cas9 (clustered regularly interspaced short palindromic repeats and CRISPR-associated proteins) has been widely used as a powerful genome editing tool (Cong et al., 2013; Jinek et al., 2013; Mali et al., 2013). However its off-target activity causes DNA DSBs at imperfectly matched loci. During the past few years, several methods based on next-generation sequencing have been published to detect off-target sites. LAM-HTGTS (Frock et al., 2015; Hu et al., 2016), which is based on chromosomal translocation, makes use of a “bait” DSB to capture “prey” DSBs to sensitively identify off-target hotspots; GUIDE-seq inserts a specific DNA oligo into break site and applies PCR to enrich DSBs (Tsai et al., 2015). In vitro methods such as Digenome-seq (Kim et al., 2015) and CIRCLE-seq (Tsai et al., 2017) are more sensitive but often need to be verified in vivo. Though LAM-HTGTS is very sensitive, there is still room for improvement.
In the improved HTGTS (iHTGTS), we applied size-selection beads to deplete surplus biotinylated primer for the bridge adapter ligation efficiency enhancement. iHTGTS showed 4 times greater sensitivity than LAM-HTGTS. Also a new primer system was employed which can accommodate 150 bp x 2 Hiseq sequencing instead of 250 bp x 2 Miseq, so that the sequencing cost is much saved. Taken together, iHTGTS is a cost-effective and high efficient method. We believe iHTGTS can give researchers deeper insights into the off-target activity of CRIPSR/Cas9.
Materials and Reagents
- Pipette tips (Quality Scientific Plastics)
- 1.5 ml tube (Axygen, catalog number: MCT-150-C)
- 0.22 μm syringe filter (MILLEX, catalog number: PR03683)
- 200 μl PCR tubes (Axygen, catalog number: 14-222-261)
- Agarose (Thermo Fisher, catalog number: R0492)
- 1 kb DNA plus ladder (Transgen Biotech, catalog number: BM211)
- Streptavidin C1 beads (Thermo Fisher, catalog number: 65001)
- AMPure XP beads (Axygen)
- Protease K (Sigma, catalog number: P8044)
- FastPfu (Transgen Biotech, catalog number: AP221-02)
- dNTPs (Transgen Biotech, catalog number: AD101-11)
- AxyPrep MAG PCR Clean-Up (Axygen, catalog number: MAG-PCR-CL)
- NaCl (VWR Life Sciences, catalog number: 97061-266)
- EDTA (Amresco, catalog number: BDH9232)
- Tris (VWR Life Sciences, catalog number: 97062-420)
- PEG 8000 (Sigma, catalog number: 89510-250G-F)
- T4 ligase (Thermo Fisher, catalog number: EL0011)
- EasyTaq (Transgen Biotech, catalog number: AP111-01)
- Gel extraction kit (Thermo Fisher, catalog number: K0691)
- 75% Ethanol (Beijing Chemical Works)
- Isopropyl (Beijing Chemical Works)
- EDTA-Na2·2H2O (Amresco, catalog number: BDH9232)
- NaOH (Sigma, catalog number: 221465)
- HCl (Sigma, catalog number: 7647-01-0)
- SDS (Sigma, catalog number: 72455)
- TAE (see Recipes)
- Proteinase K (see Recipes)
- 5 M NaCl (see Recipes)
- 0.5 M EDTA (pH 8.0) (see Recipes)
- 1 M Tris-HCl (pH 7.4) (see Recipes)
- Cell lysis buffer (see Recipes)
- TE buffer (see Recipes)
- 50% (wt/vol) PEG 8000 (see Recipes)
- 2x B&W buffer (see Recipes)
- Annealing buffer (see Recipes)
- 50 mM bridge adapter (see Recipes)
Equipment
- Foam floating tube rack
- 8-well magnet stand
- Pipettes (GILSON)
- Thermomixer C (Eppendorf)
- PCR Thermal Cyclers (Applied Biosystems)
- Covaris (M220 Focused-ultrasonicator)
- Centrifuge (Eppendorf, model: 5418R)
- NanoDrop (DeNOVIX, DS11)
- Incubator (Thermo Fisher)
- Autoclave
Procedure
- Design the primers used for iHTGTS
Choose Cas9-generated on-target site DSB as the “bait” to capture other genome-wide “prey” DSBs. To be compatible with 2 x 150 bp Hiseq sequencing, biotinylated primer for LAM-PCR was designed to bind 150 bp upstream of Cas9 binding site; nested primer was designed annealling to the downstream of the biotinylated primer about 90 bp away from the cut site. (Figure 1, Table 1)
Note: Avoid designing primers at DNA repetitive region.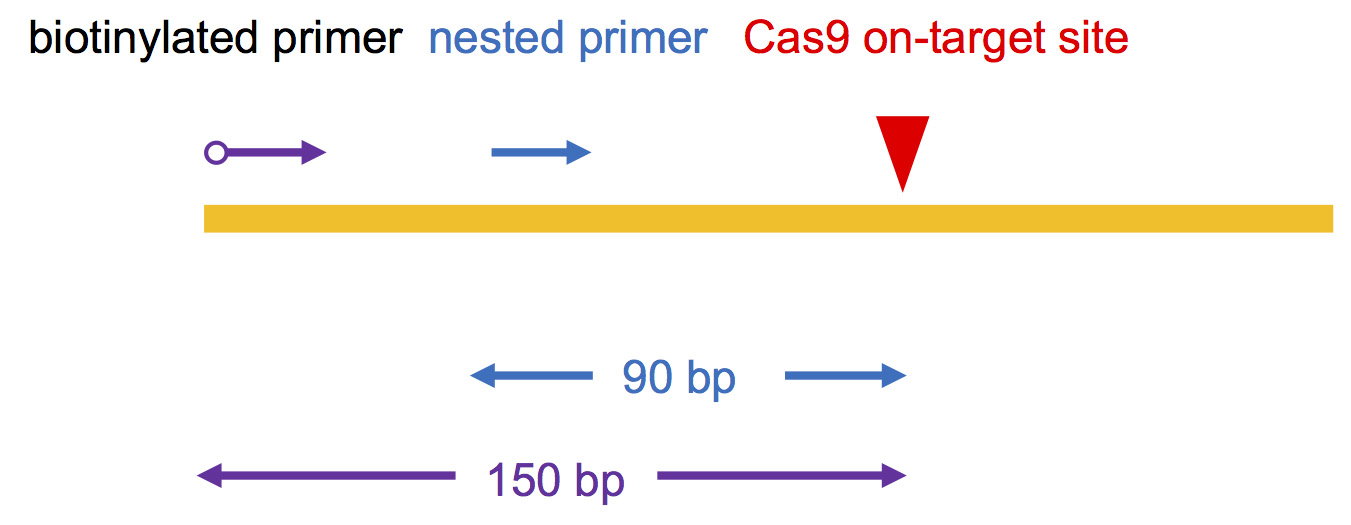
Figure 1. Primer design for iHTGTS. The red arrow indicates the Cas9 on-target cutting site. The blue arrow indicates the nested primer and purple indicates the biotinylated primer. The distance to the cutting site is shown below.
Table 1. The sequences of primers and bridge adapters. The Phor indicates the phosphorylation. MID indicates the index base for data demultiplex. REDPRIMER indicates nested primer, which varies between different loci.
- Extract Genomic DNA (gDNA)
- Forty-eight hours after transfecting HEK293T cells with the Cas9 plasmids, collect 107 transfected HEK293T cells (we have also tried K562, HCT116 and U2OS, all work well) in a 1.5 ml tube and add 500 µl cell lysis buffer. Incubate the tube in Thermomixer at 56 °C, 500 x g for 10-18 h.
Note: Protease K solution should be added into lysis buffer just before use (10 ng/ml final concentration). - Add 500 µl isopropyl and mix thoroughly till you can see a white flocculent DNA pellet.
- Using a pipet to transfer the pellet into another 1.5 ml tube with 1 ml 70% ethanol. Centrifuge at 13,000 x g for 5 min.
- Discard the supernatant. Centrifuge again and deplete residual 70% ethanol. Add 500 µl TE and incubate the tube in Thermomixer at 56 °C, 500 x g for at least 2 h.
Note: Adjust the volume of added TE to make sure the DNA concentration not less than 200 ng/µl. - Quantify the DNA using NanoDrop. The A260/A280 should be higher than 1.8.
- Recommend 20 µg gDNA for iHTGTS library construction.
- Forty-eight hours after transfecting HEK293T cells with the Cas9 plasmids, collect 107 transfected HEK293T cells (we have also tried K562, HCT116 and U2OS, all work well) in a 1.5 ml tube and add 500 µl cell lysis buffer. Incubate the tube in Thermomixer at 56 °C, 500 x g for 10-18 h.
- Fragment gDNA by sonication
- Add 20 µg gDNA into a PCR tube. Set Covaris with the following parameters: PIP = 50 watts, DF = 30%, CPB = 200, Time = 60 s.
- After sonication, take 200 ng DNA for 1% agarose page. The range of the DNA smear should be at 0.2-2 kb with a peak at 0.75 kb. (Figure 2)
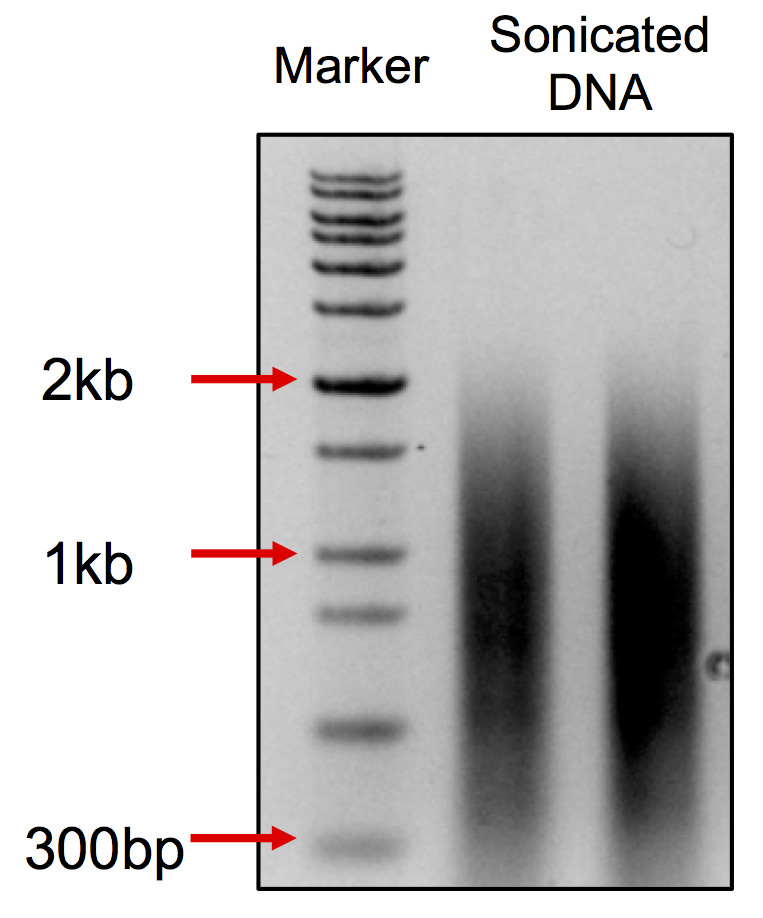
Figure 2. DNA smear pattern after sonication. The smear ranges from 300 bp to 2 kb with the peak at about 750 bp.
- LAM-PCR
- Set up the reaction in 4 x 50 µl PCR tubes as following (DNA template for each PCR reaction can be 1-10 µg):

- Set up the PCR program:
95 °C for 2 min
[95 °C for 30 s, 58 °C for 30 s, 72 °C for 1.5 min] (80 cycles)
72 °C for 2 min
10 °C forever - Deplete surplus biotin primer using AMPure XP beads
- Add 50 µl AMPure XP beads into each PCR tube, mix gently and incubate the tubes at RT for 5 min.
Note: This step aims to remove fragments less than 300 bp; the volume of the beads can be upscaled according to different batches.
Note: All the following Steps (D3b-D3f) are operated at RT.- Put the tubes on an 8-well magnet stand for 5 min.
- Remove the supernatant, add 200 µl 70% ethanol. After standing for 30 s, remove the supernatant.
- Repeat the Step D3c.
- Add 50 µl dH2O, mix the beads completely using a pipette and incubate at RT for 2 min.
- Put the tubes on the magnet for 2 min and pool the supernatants (about 200 µl) into a new 1.5 ml tube.
- Add 50 µl AMPure XP beads into each PCR tube, mix gently and incubate the tubes at RT for 5 min.
- Set up the reaction in 4 x 50 µl PCR tubes as following (DNA template for each PCR reaction can be 1-10 µg):
- Streptavidin beads binding
- Add 50 µl 5 M NaCl, 2.5 µl 0.5 M EDTA into the PCR product from the last step. Add 30 µl streptavidin beads and rotate for 4 h at RT. (The streptavidin beads should be washed twice with 1x B&W buffer before use)
- Put the beads against the 1.5 ml tube magnet stand and remove the supernatant. Wash the beads three times each using 400 µl 1x B&W buffer.
- Wash the beads with 400 µl dH2O and then resuspend the beads in 42.4 µl dH2O.
- On-beads ligation for bridge adapter

Set the reaction in a 1.5 ml tube in a rotator and ligate overnight at RT. - Nested PCR
- Add 80 µl 2x B&W buffer and 160 µl 1x B&W buffer. Put the beads against the 1.5 ml tube magnet stand and remove the supernatant. Wash the beads using 400 µl 1x B&W buffer three times and 400 µl dH2O once. Resuspend the beads in 80 µl dH2O.
- Set up the PCR reaction in 2x PCR tubes as following:
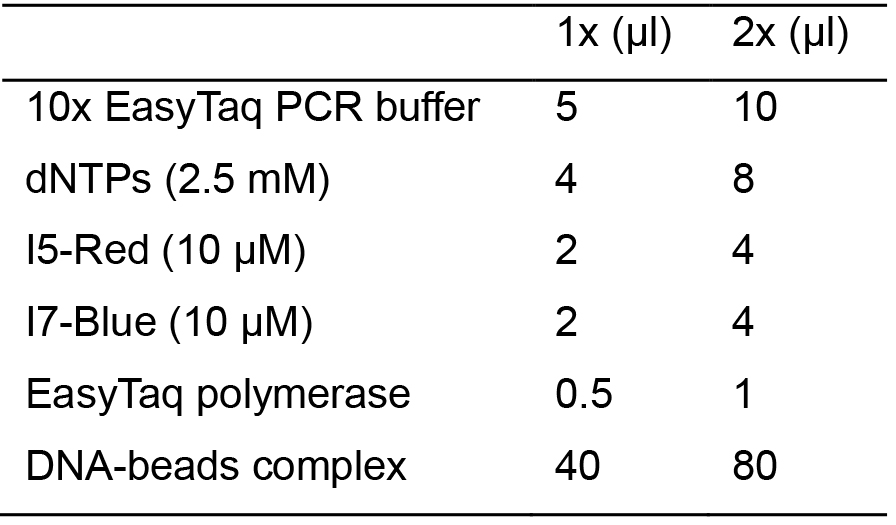
- Set up the PCR program:
95 °C for 5 min
[95 °C for 1 min, 58 °C for 30 s, 72 °C for 1 min] (15 cycles)
72 °C for 10 min
10 °C forever - Recycle the PCR products using AMpure XP beads as described in Step D3. Elute the PCR products with 35 µl dH2O for each PCR tube. Gather the PCR products together and measure the concentration.
Note: This step aims to remove DNA fragments less than 500 bp, upscale the volume of the beads according to different batches.
- Add 8 µl 10x enzyme buffer, 10 U blocking enzyme, incubate at 37 °C for 1 h or longer.
- Purify the DNA with GeneJET column, elute with 70 µl dH2O, and check the concentration.
- Add 80 µl 2x B&W buffer and 160 µl 1x B&W buffer. Put the beads against the 1.5 ml tube magnet stand and remove the supernatant. Wash the beads using 400 µl 1x B&W buffer three times and 400 µl dH2O once. Resuspend the beads in 80 µl dH2O.
- Tagged PCR
- Set up the PCR reaction in 2 x PCR tubes as following:
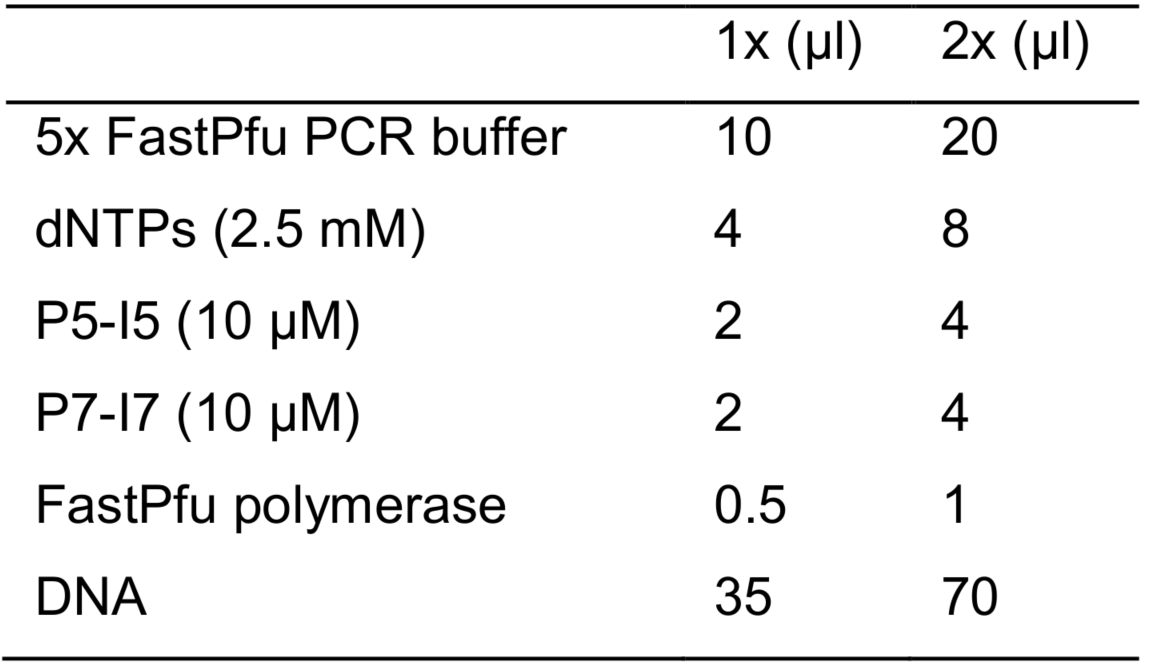
- Set up the PCR program:2.Set up the PCR program:
95 °C for 3 min
[95 °C for 20 s, 60 °C for 30 s, 72 °C for 1 min] (10-15 cycles)
72 °C for 5 min
10 °C forever
Note: The PCR cycle number is dependent on the DNA concentration from the last step.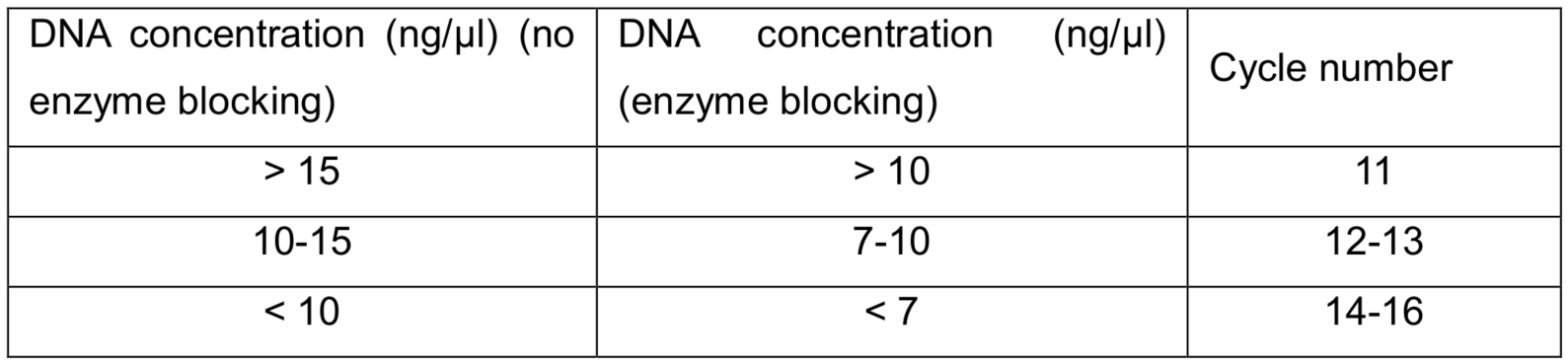
- Set up the PCR reaction in 2 x PCR tubes as following:
- Purified PCR products
Pool the DNA together, run all the DNA on 1% agarose gel in TAE buffer, cut products between 500-900 bp (Figure 3), purify through a Gel extraction column, elute with 30 µl dH2O twice. Now the PCR product is ready for Hiseq sequencing.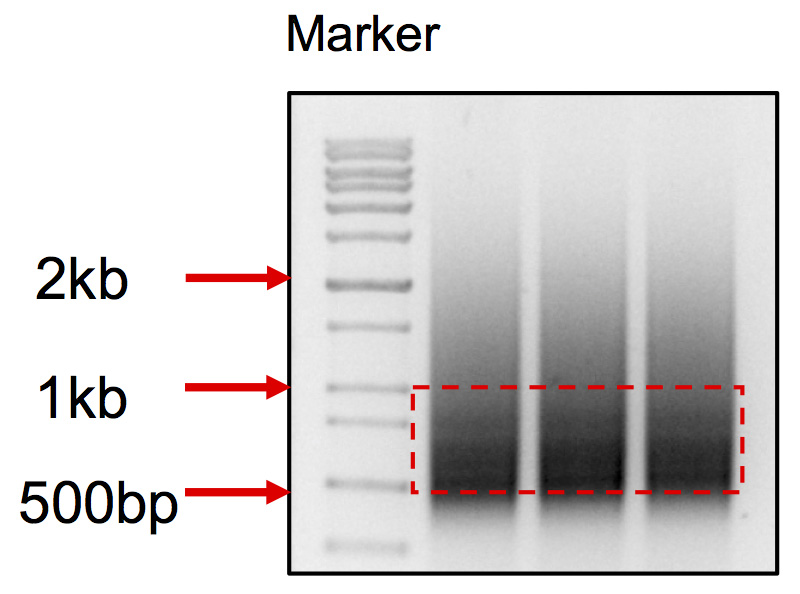
Figure 3. DNA smear pattern for iHTGTS library. Cut the DNA within the range from 500 bp to 1 kb.
Data analysis
The detailed operation and mechanism for data analysis is the same with LAM-HTGTS, which can be found in the step 48 to step 51 for Hu et al. (2016).
Recipes
Note: The reagents used in this protocol are almost the same with those in Hu et al. (2016). The following recipes are adapted from it.
- TAE
40 mM Tris-HCl
20 mM Acetic Acid
1 mM EDTA - Proteinase K
Dissolve 0.1 g of proteinase K powder in 5 ml of H2O to make a 20 mg/ml stock
Divide the solution into 0.5 ml aliquots and store them at -20 °C for up to 3 months - 5 M NaCl
Dissolve 292.5 g of NaCl in H2O, and adjust the total volume to 1 L
Autoclave the solution and store it at room temperature (RT; 20-25 °C) for up to 1 year - 0.5 M EDTA (pH 8.0)
Dissolve 186.12 g of EDTA-Na2·2H2O in H2O, adjust the pH to 8.0 using 2.5 N NaOH and then adjust the total volume to 1 L
Autoclave the solution and store it at RT for up to 1 year - 1 M Tris-HCl (pH 7.4)
Dissolve 121.14 g of Tris base in H2O, adjust the pH to 7.4 using HCl, and then bring the total volume to 1 L
Autoclave the solution and store it at RT for up to 1 year - Cell lysis buffer
200 mM NaCl
10 mM Tris-HCl (pH 7.4)
2 mM EDTA (pH 8.0)
0.2% (wt/vol) SDS
Store it at RT for up to 6 months - TE buffer
10 mM Tris-HCl (pH 7.4)
0.5 mM EDTA (pH 8.0)
Store it at RT for up to 6 months - 50% (wt/vol) PEG 8000
Dissolve 5 g of PEG 8000 in H2O at 56 °C, and then adjust the total volume to 10 ml
Filter the solution through a 0.22 μm syringe filter, prepare 1 ml aliquots and store them at -20 °C for up to 1 year - 2x B&W buffer
2 M NaCl
10 mM Tris-HCl (pH 7.4)
1 mM EDTA (pH 8.0)
Dilute it with H2O to make 1x B&W buffer. Store it at RT for up to 1 year - Annealing buffer
25 mM NaCl
10 mM Tris-HCl (pH 7.4)
0.5 mM EDTA (pH 8.0)
Store it at RT for up to 1 year - 50 mM bridge adapter
- Dissolve the two DNA oligos (Table 1) in annealing buffer to a final concentration of 400 μM
- Mix equal volumes of the two dissolved oligos in a new 1.5 ml microtube, put the tube in 1 L of boiling water with a foam floating tube rack, boil it for 5 min and then cool it down slowly in water to ~30 °C on the bench (adapter concentration is 200 μM). Alternatively, the oligos can be annealed on a PCR thermoblock
- Dilute fourfold (concentration is 50 μM) with H2O, prepare 100 μl aliquots and store them at -20 °C for up to 2 months
- Thaw the adapter on ice before use
Acknowledgments
Thanks for Thousand Talents Plan Youth Program to J. H. and PKU-TSU Center for Life Sciences.
This protocol is adapted from Hu et al. (2016).
Competing interests
The authors declare they have no conflict of interest or competing interests.
References
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A. and Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121): 819-823.
- Frock, R. L., Hu, J., Meyers, R. M., Ho, Y. J., Kii, E. and Alt, F. W. (2015). Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol 33(2): 179-186.
- Hu, J., Meyers, R. M., Dong, J., Panchakshari, R. A., Alt, F. W. and Frock, R. L. (2016). Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat Protoc 11(5): 853-871.
- Jinek, M., East, A., Cheng, A., Lin, S., Ma, E. and Doudna, J. (2013). RNA-programmed genome editing in human cells. Elife 2: e00471.
- Kim, D., Bae, S., Park, J., Kim, E., Kim, S., Yu, H. R., Hwang, J., Kim, J. I. and Kim, J. S. (2015). Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods 12(3): 237-243, 231 p following 243.
- Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., Norville, J. E. and Church, G. M. (2013). RNA-guided human genome engineering via Cas9. Science 339(6121): 823-826.
- Tsai, S. Q., Nguyen, N. T., Malagon-Lopez, J., Topkar, V. V., Aryee, M. J. and Joung, J. K. (2017). CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods 14(6): 607-614.
- Tsai, S. Q., Zheng, Z., Nguyen, N. T., Liebers, M., Topkar, V. V., Thapar, V., Wyvekens, N., Khayter, C., Iafrate, A. J., Le, L. P., Aryee, M. J. and Joung, J. K. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33(2): 187-197.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Yin, J., Liu, M., Liu, Y. and Hu, J. (2019). Improved HTGTS for CRISPR/Cas9 Off-target Detection. Bio-protocol 9(9): e3229. DOI: 10.21769/BioProtoc.3229.
Category
Molecular Biology > DNA > DNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.