- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Triple Fluorescence Anisotropy Reporter Imaging in Living Cells
Published: Vol 9, Iss 9, May 5, 2019 DOI: 10.21769/BioProtoc.3226 Views: 6260
Reviewed by: Zinan ZhouBrahma MuluguPooja Verma
Original research article
The authors used this protocol in:

Jul 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
FRET-based genetically encoded biosensors incorporate two fluorescent proteins into their design to enable ratiometric biosensing of signaling activities in live cells. While emission ratios are generally useful for quantitative studies, they leave little room in the optical spectrum for additional sensors and optogenetic tools. Homotransfer-based reporters, such as the FLuorescence Anisotropy REporters (FLAREs), incorporate two fluorescent proteins of the same color into their design. Conversion to a single color opens the visible spectrum for the use of complementary sensors. Here, we present a protocol for measuring three independent intracellular signals in living cells. We describe the configuration and calibration of a widefield microscope for multicolor FLARE imaging. Three FLARE sensors for intracellular calcium, MAPK activity, and PKA phosphorylation are co-transfected into HEK293 cells, and triple FRET imaging is performed. Compared to heterotransfer FRET biosensors, the polarization-based multiplex imaging can track multiple signaling activities concurrently in a targeted cell population.FRET-based genetically encoded biosensors incorporate two fluorescent proteins into their design to enable ratiometric biosensing of signaling activities in live cells. While emission ratios are generally useful for quantitative studies, they leave little room in the optical spectrum for additional sensors and optogenetic tools. Homotransfer-based reporters, such as the FLuorescence Anisotropy REporters (FLAREs), incorporate two fluorescent proteins of the same color into their design. Conversion to a single color opens the visible spectrum for the use of complementary sensors. Here, we present a protocol for measuring three independent intracellular signals in living cells. We describe the configuration and calibration of a widefield microscope for multicolor FLARE imaging. Three FLARE sensors for intracellular calcium, MAPK activity, and PKA phosphorylation are co-transfected into HEK293 cells, and triple FRET imaging is performed. Compared to heterotransfer FRET biosensors, the polarization-based multiplex imaging can track multiple signaling activities concurrently in a targeted cell population.
Keywords: HomotransferBackground
The timing and location of cell signaling activities are frequently regulated. Genetically-encoded biosensors can reveal the dynamic nature of proteins and second messengers that transduce intracellular signals in living cells and organisms. There are several strategies available for engineering such reporters. Those that are based on FRET offer certain advantages, including design flexibility and a ratiometric readout that facilitates quantitative studies (Mehta and Zhang, 2011). Conventionally, two differently colored fluorescent proteins are incorporated into FRET biosensors (e.g., cyan/yellow or green/red). While effective, the two-color strategy leaves little room in the visible spectrum for multiplexing with other sensors or optical modulators. Recently, we have converted several FRET-based reporters to FLuorescence Anisotropy REporters, or FLAREs, that employ FRET pairs of the same color (Markwardt et al., 2018; Ross et al., 2018; Seckinger et al., 2018), thereby enabling measurement with several biosensors in the same specimen. Homotransfer FRET is measured by fluorescence polarization microscopy. This protocol describes the configuration of a widefield microscope for multiplexed FLARE imaging, sample preparation, experimentation and image analysis. Also, this protocol may be adapted for experiments with FLARE sensors co-imaged with other non-homotransfer biosensors or optogenetic tools.
Materials and Reagents
- Pipette tips (TipOne, USA Scientific, catalog numbers: 1111-3800, 1111-0806, 1111-2821)
- 35-mm glass bottom imaging dishes (#1.5 coverglass, 14-mm round microwell opening) (MatTek, catalog number: P35G-1.5-14-C)
- 1.5 ml microcentrifuge tubes (Eppendorf, catalog number: 022364111)
- T-25 cell culture flasks (Sigma-Aldrich, Corning, catalog number: CLS430639)
- HEK293 cells (ATCC, catalog number: CRL3216, passage number according to ATCC recommendations)
- FLARE mVenus-cp172Venus EKAR plasmid (Ross et al., 2018)
- FLARE mCherry-mCherry AKAR plasmid (Ross et al., 2018)
- FLARE mCerulean3-mCerulean3 Cameleon plasmid (Ross et al., 2018)
- DPBS (Calcium-free, magnesium-free) (Gibco, catalog number: 14190-144)
- Fetal bovine serum (FBS) (qualified, US origin) (Gibco, catalog number: 26140-079)
- Trypsin-EDTA (0.05%, Gibco, catalog number: 25300)
- Penicillin-streptomycin (10,000 U/ml) (GE Healthcare, Hyclone, catalog number: SV30010)
- Bovine serum albumin (BSA) (fraction V) (Sigma, catalog number: A9418)
- Distilled water (Gibco, catalog number: 15230-170)
- Forskolin (50 mM solution in DMSO, 1,000x) (Sigma, catalog number: F3917)
- 3-Isobutyl-1-methylxanthine (IBMX) (100 mM solution in DMSO, 1,000x) (Sigma, catalog number: I7018)
- Epidermal Growth Factor (EGF) (100 µg/ml in 10 mM acetic acid, 0.1% BSA) (Sigma, catalog number: E9644)
- Thapsigargin (1 mM solution in DMSO, 1,000x) (Sigma, catalog number: T9033)
- DMSO (Sigma, catalog number: D8418)
- Fluorescein (Aldrich, catalog number: F2456)
- 95% ethanol
- Poly D-lysine hydrobromide (MW 70,000-150,000) (Sigma, catalog number: P0899)
- PolyJet transfection reagent (SignaGen, catalog number: SL100688)
- Serum-free DMEM (Gibco, catalog number: 11965)
- NaCl (Sigma-Aldrich, catalog number: S9888)
- KCl (Sigma-Aldrich, catalog number: P5405)
- Na2HPO4 (Sigma-Aldrich, catalog number: S5136)
- KH2PO4 (Sigma-Aldrich, catalog number: S5655)
- CaCl2 (Sigma-Aldrich, catalog number: C5670)
- MgSO4·7H2O (Sigma-Aldrich, catalog number: 63138)
- HEPES (Sigma-Aldrich, catalog number: H3375)
- D-glucose (Sigma-Aldrich, catalog number: G7021)
- 10 μM fluorescein solution (see Recipes)
- HBSS (Hanks’ Balanced Salt Solution) Imaging Buffer (see Recipes)
- STOCK 1
- STOCK 2
- STOCK 3
- STOCK 4
Equipment
- Pipettes
- Water jacketed 37 °C incubator (Thermo Electron Corporation, Forma Series II, HEPA Class 100, model: 3130)
- Class II biological safety cabinet (The Baker Company, SterilGard III Advance)
- Water bath (Fisher Scientific, Isotemp 205)
- Centrifuge (Sorvall, Legend Mach 1.6 R)
- Microscope (Figure 1)
- Inverted microscope (Zeiss, model: Axio Observer.Z1/7) with temperature control (Pecon, Heating insert P S1, Heating unit XL S2)
- CCD Camera (Zeiss, model: Axiocam 506)
- Objective lens 20x/0.75 NA (Zeiss, catalog number: 440649-0000-000)
- Inverted microscope (Zeiss, model: Axio Observer.Z1/7) with temperature control (Pecon, Heating insert P S1, Heating unit XL S2)
- Zeiss Colibri.2
A Zeiss Colibri.2 is used for illumination. We configured the unit with the following:- 420 nm LED fitted with 438/24 excitation filter (Semrock, FF02-438/24-25)
- 505 LED (Zeiss) with 509/22 BrightLine Bandpass Filter (Semrock, FF01-509/22-25)
- 555 LED with 578/21 BrightLine Bandpass Filter (Semrock, FF01-578/21-25)
- Beam combiners included a 458 nm dichroic beamsplitter (Semrock, FF458-Di02-25x36) and a 526 nm dichroic beamsplitter (Semrock, FF526-Di01-25x36)
- Fluorescence filter cubes
- For CFP and mCherry, we use a filter cube with a 459/526/596 dichroic beamsplitter (Semrock, FF459/526/596-Di01-25x36) and a 475/543/702 bandpass filter (Semrock FF01-475/543/702-25).
- For YFP, we use a filter cube equipped with a 514 long pass dichroic (Chroma, # ZT514rdc-UF1) and an ET545/40m emission filter.
- Polarizers
- Excitation polarization is controlled using a 1-inch wire grid polarizer (Meadowlark Optics, VFR-100-NIR) oriented perpendicularly to the optical bench.
- Parallel (P) and perpendicular (S) emission polarizations are split using an OptoSplit II (Cairn) image splitter with polarization optics.
Software
- Zen 2.3 (blue edition, Zeiss)
- Excel (Microsoft)
- FIJI (Schindelin et al., 2012) with the StackReg plugin (Thévenaz et al., 1998)
Procedure
- Configuration of the microscope for polarization
The fluorescence polarization microscopy setup should be set up so that a polarized excitation source illuminates the sample and the emitted light is split into two channels, one that is parallel to the excitation polarization and one that is perpendicular. This setup is achieved by placing a polarizer in the excitation path, and then setting up an image splitter with polarization optics to collect the two different polarization channels simultaneously.- Remove existing polarization components (e.g., Nomarski DIC optics) from the microscope.
- Place a linear polarizer in the excitation path. It should be oriented perpendicularly to the optical bench.
- Align the image splitter with polarization optics per the manufacturer's recommendations (Figure 1). It should be set up so that one half of the image will collect light polarized parallel to the excitation polarizer, and the other half should collect light with polarization perpendicular to the excitation polarizer.
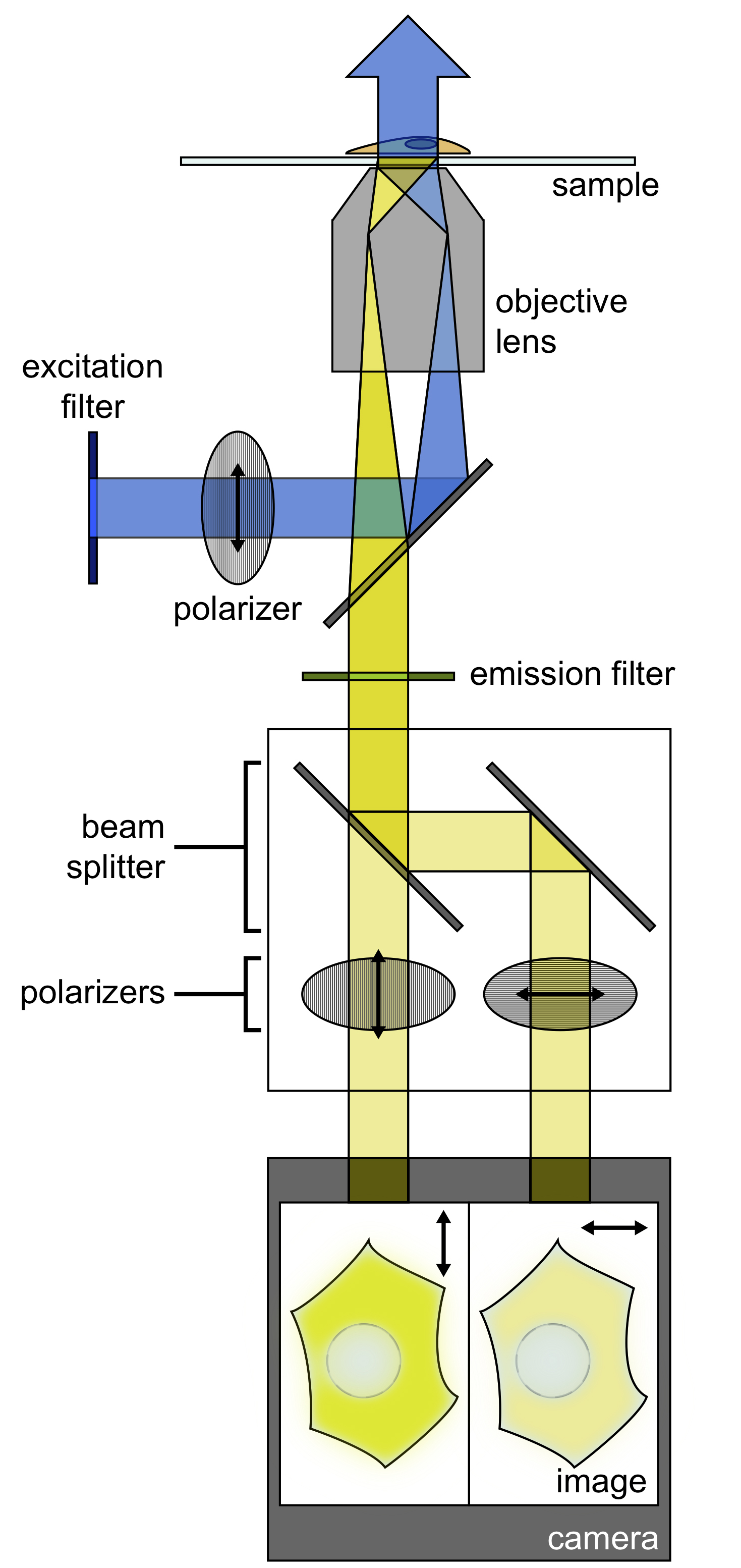
Figure 1. Schematic of fluorescence polarization microscopy imaging setup. A polarizer filters the light from an illumination source, and this light is deflected to an objective, where it illuminates the sample. The emitted light from the sample is then filtered by an emission filter. A beam splitter then separates the emitted light into P and S polarizations. Images of both polarizations are projected simultaneously on a single camera.
- Measurement of the corrective (g) factor
To correct for polarization biases within the instrument, it is important to measure a correction factor, called g. This factor is measured by using a small organic fluorescent dye. Due to its small size, the dye will rotate faster than its fluorescence lifetime, and therefore, its emitted light when excited with a polarized light source should be isotropic. Therefore, any anisotropy that we measure from it can be attributed to the polarization biases of the optical setup.- Prepare 10 μM fluorescein solution (Recipe 1), and aliquot 2 ml into a 35 mm MatTek dish.
- Image the solution using ‘YFP’ capture conditions (505 nm illumination, EYFP cube), capturing images that are parallel (P) and perpendicular (S) to the excitation polarization plane.
- Collect a background image using the same camera exposure conditions without illumination.
- Calculate corrective factor g:
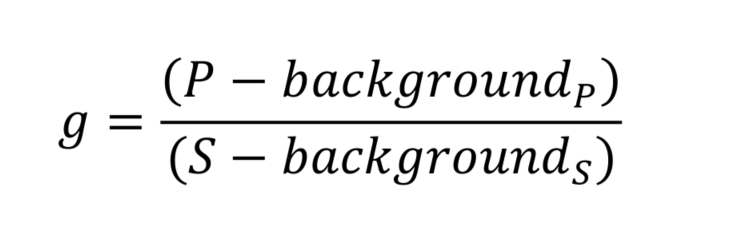
- Cell line maintenance
Maintain HEK293-T cells in T-25 cell culture flasks in a 37 °C incubator at 5% CO2. Passage cells every two to three days when they reach about 80% confluence, according to the following protocol:- Gently aspirate media from the flask, leaving the cells unperturbed.
- Wash cells twice with DPBS.
- Trypsinize with 0.05% trypsin at 37 °C for 1 min.
- Neutralize the trypsin with culture media.
- Split the HEK293-T cells at a ratio of 1:20 into a new flask with fresh culture medium. Place in a 37 °C incubator with 5% CO2.
- Seeding cells onto imaging dishes
Cells should be seeded onto imaging dishes one to three days before imaging is to be performed.- Coat MatTek imaging dishes with 100 μg/ml Poly D-lysine for 1 h. Wash twice with DPBS, and dry overnight at room temperature.
- One to three days before imaging, seed a suitable number of cells onto the coated MatTek dishes with fresh medium. Cells can be seeded on a day in which the cells are being passed after the trypsin is neutralized, and the number of cells seeded depends on the number of days before imaging. Seed enough cells so that confluency is ~80% the day before imaging. Keep these imaging dishes in a 37 °C incubator at 5% CO2.
- Transfection
Transfection should be performed the day before imaging. If you seed the cells onto the imaging dishes on the same day, wait at least four hours before transfection, and confirm that cells are well adhered before starting transfection. Confluency should be approximately 80% at the time of transfection.- Prepare plasmid DNA for biosensors of interest. To monitor Erk activity, Protein Kinase A activity, and calcium, use FLARE mVenus-cp172Venus EKAR, FLARE mCherry-mCherry AKAR, and FLARE mCerulean3-mCerulean3 Cameleon. For these sensors, the N-terminal fluorescent protein (FP) is cloned between BamHI and SphI restriction sites, the molecular switch is cloned between SphI and SacI sites, and the C-terminal FP is cloned between SphI and EcoRI, in a pcDNA3.1+ mammalian expression vector. A stop codon is present at the end of the C-terminal FP. A header sequence, with a poly-His tag and a Kozak consensus sequence is added at the N-terminal position between HindIII and EcoRI. Because the SphI and SacI sites are not unique in the pcDNA3.1 plasmid, cloning steps should be performed in the bacterial expression vector pRSET-B backbone, and then the final sensors should be subcloned into the pcDNA3.1+ mammalian expression vector.
- Combine 0.33 μg of each plasmid in 50 μl of serum-free DMEM in a 1.5 ml microcentrifuge tube (A).
- In a second 1.5 ml microcentrifuge tube (B), separately add 3 μl PolyJet transfection reagent to 50 μl of serum-free DMEM.
- Add the contents of from reagent mixture B dropwise into microcentrifuge tube A taking care to avoid contact with the container walls.
- Incubate for 15 min at room temperature.
- Add the combined PolyJet-DNA mixture dropwise into the center of the imaging dish.
- Gently swirl the imaging dishes to mix.
- Incubate the imaging dishes overnight in a 37 °C incubator and 5% CO2.
- Imaging
- Before imaging, prepare 1,000x stock solutions of drugs, including Forskolin, IBMX, EGF, and thapsigargin. Aliquot 2 μl into 1.5 ml microcentrifuge tubes, and keep these on ice.
- On the day of imaging, pipet out old culture media and wash three times with imaging buffer (HBSS imaging buffer containing 2.0 g/L of D-glucose, pH-adjusted to 7.4, and filter sterilized). After the washes, add 2 ml of HBSS. Make sure that all phenol-red from the culture media has been washed away. During these wash steps, be careful not to disturb the cells, as HEK293-T cells are only weakly adherent, and perform the wash steps gently.
- Place the imaging dish on the microscope stage and secure it.
- Focus the image on the cells, and determine the appropriate exposure time and excitation power for each color, so that none of the channels are over-exposed. Because of the image splitter with polarizers, the image should have two halves, one half reflecting the P and one the S polarization, with the P fluorescence approximately 2-3 fold brighter than the S.
- Image a time course, imaging each color channel every 30 s. Acquire a baseline, which consists of the first ten time points.
- After the baseline, pause the acquisition to add Forskolin/IBMX, to stimulate PKA. With a P1000 pipette, take up a few hundred microliters of imaging media from the imaging dish, and add to the microcentrifuge tube with the drug aliquot. Pipette up and down to mix. Carefully add this volume back to the imaging dish, on the side of the imaging dish, as to not disturb the cells, with the pipette tip submerged in the media. Pipette gently up and down to mix. Resume the acquisition immediately after drug addition.
- Repeat drug additions at the desired times for the other stimulations.
Data analysis
Image Processing
- Channel alignment
The image splitter divides the camera chip such that half of the image collects the S-polarized light and the other half collects the P-polarized light. However, it is critical to separate these two channels and align them so that we can accurately calculate the fluorescence anisotropy at each point (Figure 2).- Install FIJI (http://fiji.sc/#) (Schindelin et al., 2012).
- Install StackReg plugin (http://bigwww.epfl.ch/thevenaz/stackreg/).
- Open an image file in FIJI (Figure 2A)
- Draw a rectangular ROI around the half of the dish that is P polarized, and duplicate to make a separate stack with just the P polarization image. (Image → Duplicate, make sure “Duplicate Stack” is checked). Add this ROI to the ROI manager (Analyze → Tools → ROI Manager).
- Drag this ROI to encapsulate the S polarization half of the original image, and duplicate to make an S polarization image stack (Image → Duplicate, make sure “Duplicate Stack” is checked).
- Merge the S and P polarization stacks to make a composite stack, with the two polarizations in different channels. (Image → Color → Merge Channels; select the P polarization stack as C1 and the S polarization stack as C2, make sure “Create Composite” is checked) (Figure 2B).
- Run the StackReg plugin. Choose either Translation or Rigid Body for the transformation. If the alignments require rotation as well as translation, then choose Rigid Body. If no rotation is needed, then Translation should suffice (Figure 2C).
- Press OK, and wait for alignment to finish.
- Verify alignment by visualization.
- Repeat this process for each of the color channels.
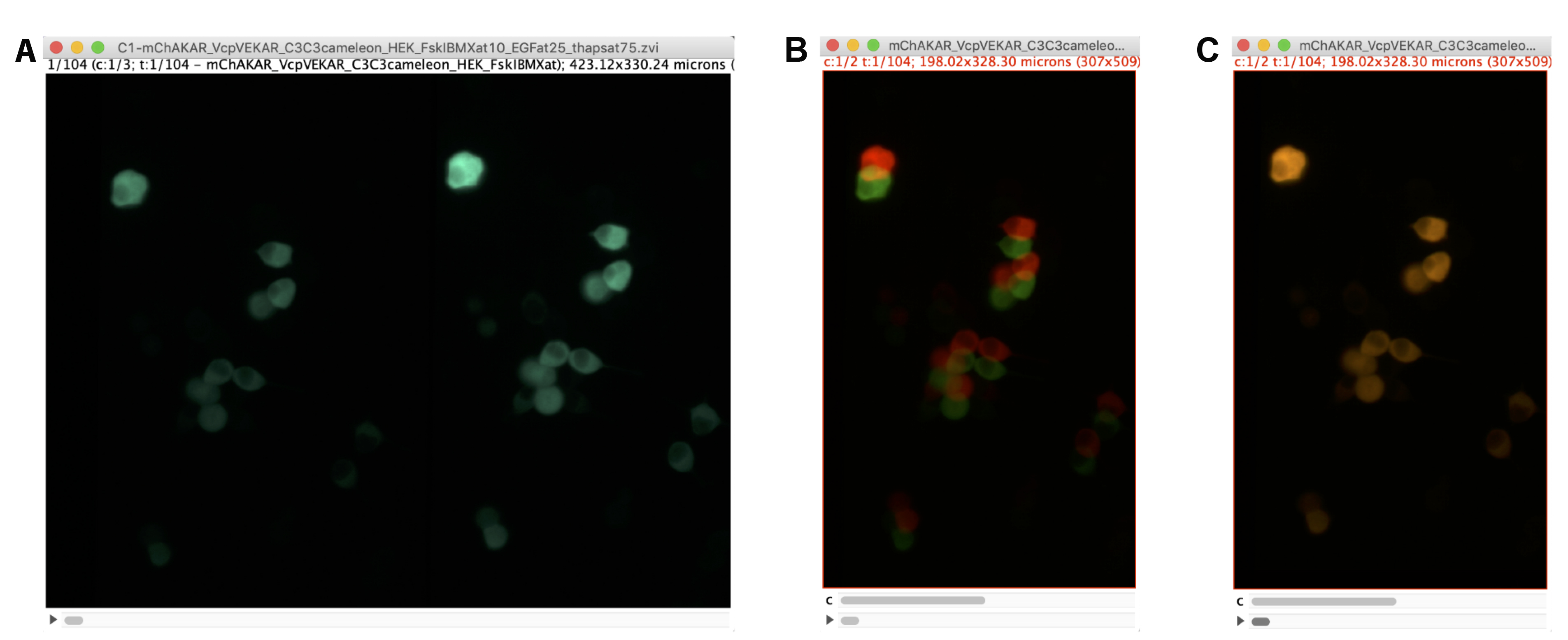
Figure 2. Alignment of Polarization Images. A. Example of the cyan channel image in an imaging experiment with HEK293-T cells expressing FLARE mCer3-mCer3 Cameleon, FLARE mVenus-cp172Venus EKAR, and FLARE mCherry-mCherry AKAR in FIJI software. Because of the image splitter with polarization optics, the left side of the image represents the S-polarized image (perpendicular to excitation source), while the right side represents the P-polarized image (parallel to excitation source). B. Screenshot of the two polarization images as a stack before channel alignment. An ROI for each side of the split image in (A) was drawn and duplicated. The two images were merged such that the two channels reflect different polarizations. The red lookup table reflects the P-polarized image, and the green reflects the S-polarized image. C. Image of the same stack after channel alignment using the StackReg plugin in FIJI.
- Data analysis
Once the S- and P- polarization images are aligned, it is possible to calculate the mean anisotropy for each of the color channels, and then plot the mean anisotropy over time. Changes in the anisotropy in the cyan, yellow, and red channels will reflect changes in calcium concentration, Erk activity, and PKA activity respectively.- In the aligned stack, select regions of interest using a selection tool, drawing around the area of the cell with the freehand selections tool.
- Add ROIs to ROI manager (Analyze → Tools → ROI Manager).
- Draw an ROI that is exclusively in the background. It is sometimes helpful to adjust the contrast to confirm that this ROI has only background pixels and does not contain cells (Figure 3A).
- Go to Analyze → Set Measurements, and select “Mean gray value.”
- In the ROI Manager, go to More → Multi Measure, to collect the mean intensity value of all the ROIs. Make sure “One row per slice” is selected. Example data extracted is shown in Figure 3B. Copy and paste this data into Microsoft Excel.
- Background subtract the mean intensities of each ROI at each time point by subtracting the intensity of the background region from the intensity of the cell, for each cell.
- Anisotropies (r) can be calculated using background subtracted P and S values for each channel, with the corrective factor (g), measured earlier.
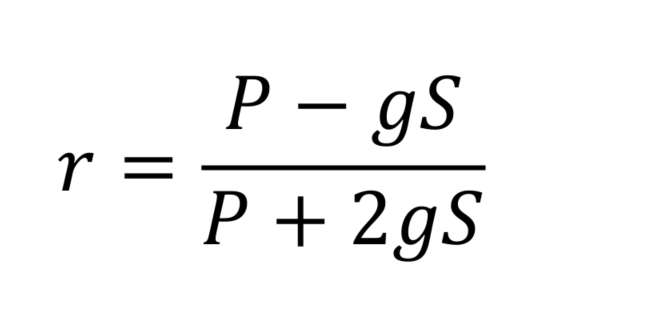
- Subtract the anisotropy from the time point just before the first drug addition (t = 0 min) from all the anisotropy measurements, for each cell to calculate the anisotropy change.
- Repeat this process for each of the color channels. Figure 4 shows representative anisotropy time course data for such an experiment.
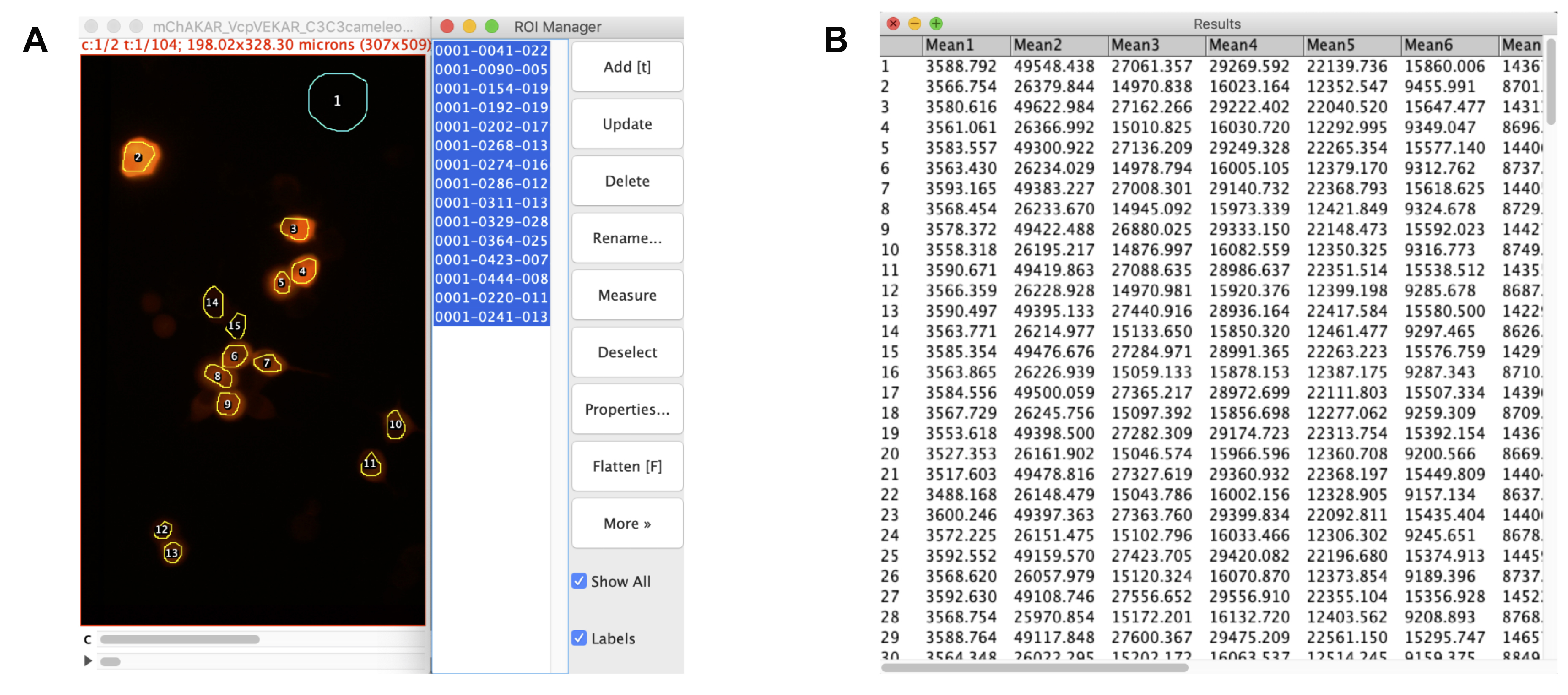
Figure 3. Drawing regions and extracting data. A. Aligned stack for cyan channel of experiment mCer3-mCer3 FLARE Cameleon, Venus-cpVenus FLARE EKAR, and mCherry-mCherry FLARE AKAR, with ROIs of interest drawn and logged with FIJI’s ROI Manager tool. The first ROI is of the background, while the rest are of the cells. B. Raw data extracted from the ROI manager using Multi Measure. The values reflect the mean intensity values for each ROI. All odd-numbered rows reflect the time-course for the P-polarized light, while the even-numbered rows reflect the time-course for the S-polarized light. The first column represents the background ROI, while the remaining columns display the mean intensity values for each cell. These data can be copied and pasted into Microsoft Excel to calculate anisotropy values for each cell at each time point.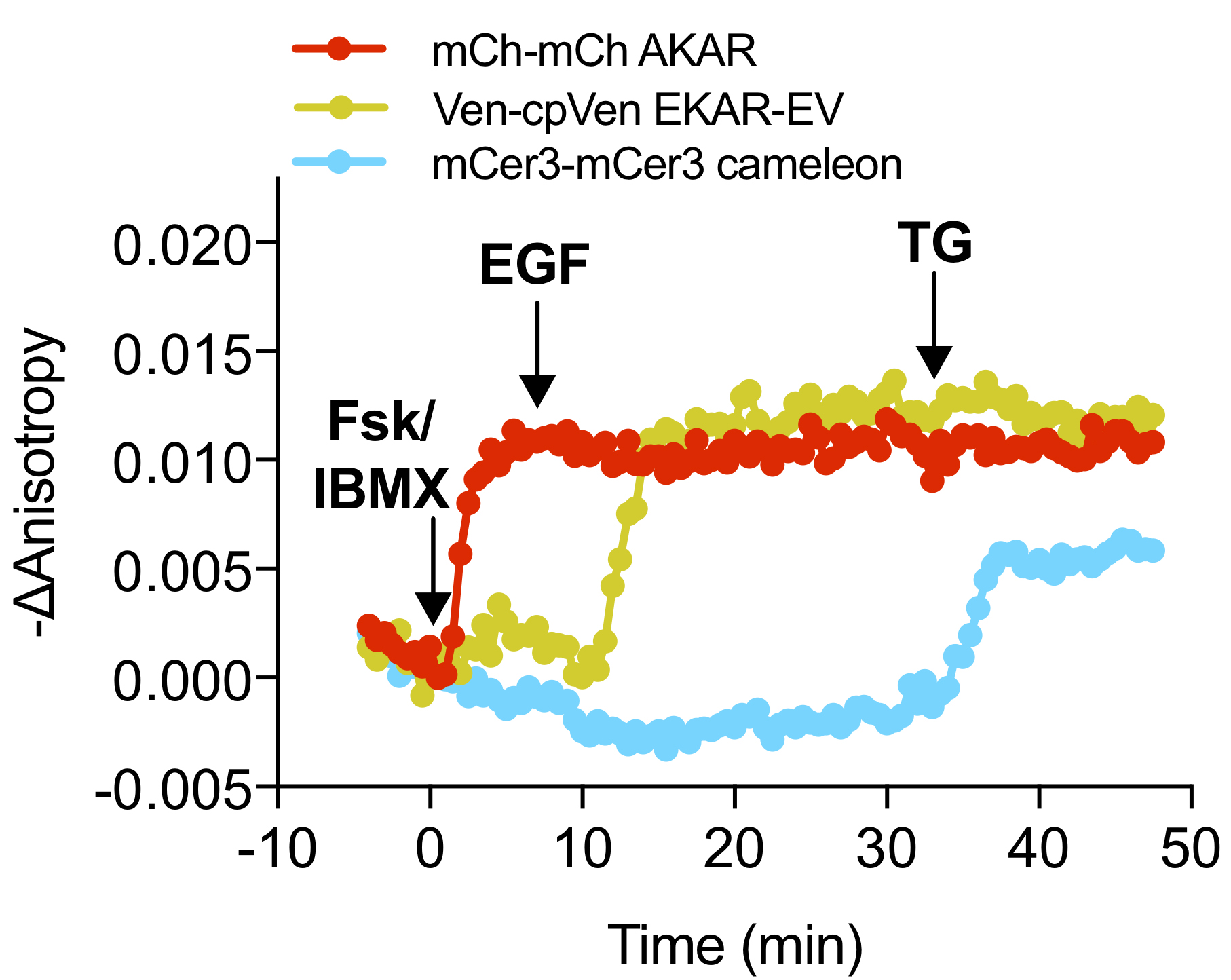
Figure 4. Example Anisotropy Time Course. Representative time course of mCer3-mCer3 FLARE Cameleon, Venus-cp172Venus FLARE EKAR, and mCherry-mCherry FLARE AKAR in HEK293 cells showing the change in anisotropy in each channel from the time point t = 0, which is defined as the time point just before the first drug addition (Ross et al., 2018). The negative change in anisotropy is plotted so that an increase in signaling activity corresponds to an increase in the graph. Robust changes in anisotropy were observed in the red, yellow, and cyan channels upon stimulation of PKA activity, Erk activity, and calcium, respectively.
- Pseudocolor image generation
If you would like to create a movie in which the anisotropy is shown in pseudocolor, we have provided the following procedure and an ImageJ macro. Because anisotropy is a ratiometric measurement, there is a problem that background pixels, which have either a value of zero or slightly above zero, will often have infinite or undefined values when the ratio is taken. To solve this problem, the script creates a pseudocolor image in which the pixel intensities are based on the fluorescent image itself, while the color information reflects the measured anisotropy. In this way, only pixels within the cell are highlighted, while pixels in the background are dark.- Separate the aligned, composite image into separate stacks (Image → Color → Split Channels).
- Background subtract each frame for each stack (Process → Subtract Background, choose an appropriate rolling ball radius, and sure “Light Background” is not checked).
- Use the Image Expression parser to create an anisotropy image (Process → Image Expression Parser, Press the plus in the lower left hand corner to add another channel. Choose the P polarization stack as “A” and the S polarization stack as “B.” In the expression box, type (A - g x B)/(A + 2 x g x B), substituting the measured value of g into the expression. Press Parse).
- Choose the desired lookup table for the anisotropy image (Image → Lookup Table).
- The anisotropy calculation results in a very noisy background due to the nature of taking a ratio after background subtraction. However, we are interested in visualizing the anisotropies within the cells, and not within the background. Therefore, it is desirable to modulate the intensity of the anisotropy image based on the fluorescence intensity. To make such a pseudocolor image of the anistropy, install and the makepseduo.ijm macro (code included below). Select the anisotropy image as the ratio image and the P polarization stack as the intensity modulator. You will then be prompted to adjust the contrast of both the anisotropy image and the P polarization stack until the contrast is in the desired range, and once you have done so press OK. The resulting image will have its hue given by the lookup table for the anisotropy image and its brightness modulated by the intensity of the fluorescence image.
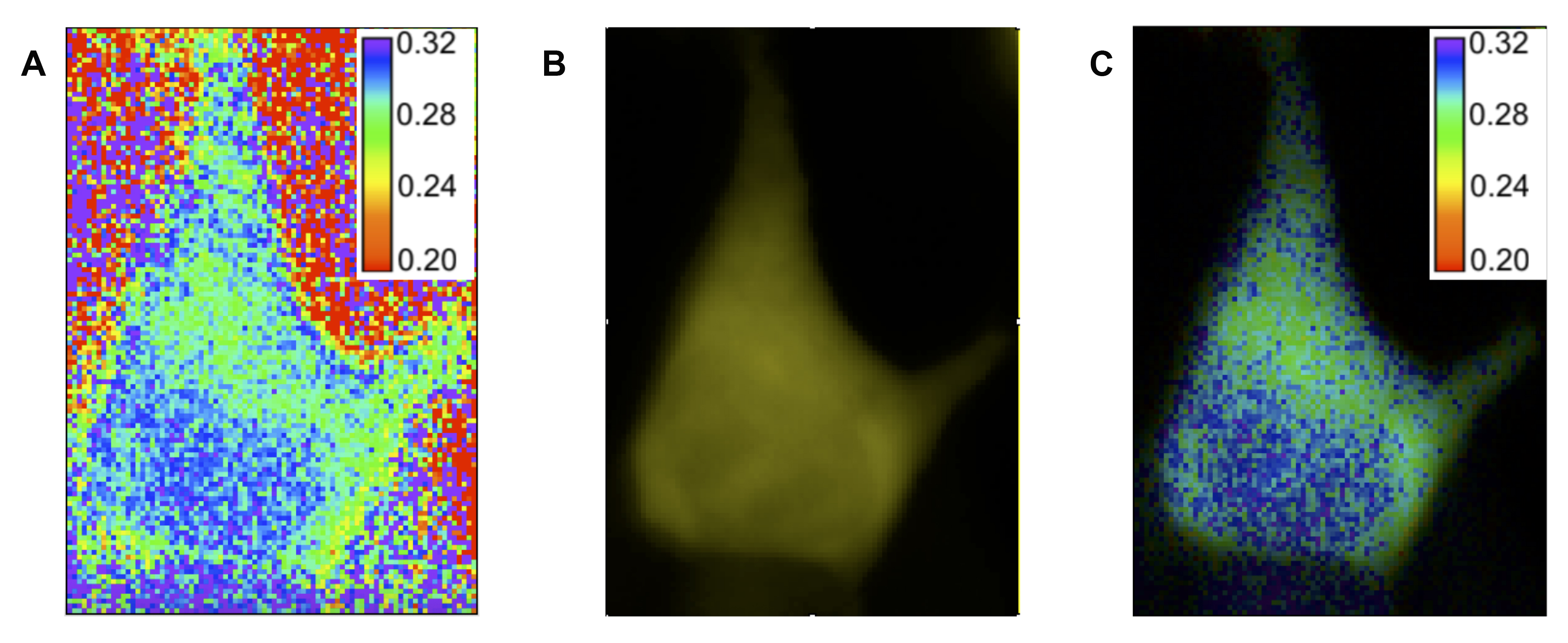
Figure 5. Making pseudocolor image. A. Ratiometric image of anisotropy values created using the Image Expression Parser tool in FIJI. The cell shown is a HEK293 cell expressing Venus-cpVenus FLARE AKAR. B. P-polarized fluorescent image of the same cell. C. Pseudocolor image created using the makepseudo.ijm plugin (below). Hue information is taken from the ratiometric image, while intensity information is taken from the fluorescent image.
Pseudocolor Image Macro (makepseudo.ijm):
Dialog.create("Making Pseudocolor");
selectImage(nImages);
Dialog.addString("Ratio image", getTitle());
if (nImages>1) {
selectImage(nImages-1);
}
else {
waitForUser("Warning","Only 1 image is open.");
};
Dialog.addString("Intensity modulator", getTitle());
Dialog.show;
rat = Dialog.getString();
ind = Dialog.getString();
psd = "pseudo";
waitForUser("Adjust the contrast of the images and press OK.");
setBatchMode(true);
selectWindow(rat);
ratsize=nSlices;
txt="title=" + rat + "_dup duplicate range=1-" + ratsize;
run("Duplicate...", txt);
run("RGB Color");
rat_d=rat+"_dup";
rename(rat_d);
for (i=1; i<=ratsize ;i++) {
if (i==1) {
selectWindow(rat_d);
setSlice(i);
run("Select All");
run("Copy");
run("Internal Clipboard");
rename(psd);
run("HSB Stack");
selectWindow(ind);
setSlice(i);
run("Select All");
run("Copy");
selectWindow(psd);
setSlice(3);
run("Paste");
run("RGB Color");
}
else {
selectWindow(rat_d);
setSlice(i);
run("Select All");
run("Copy");
run("Internal Clipboard");
rename("temp");
selectWindow(ind);
setSlice(i);
run("Select All");
run("Copy");
selectWindow("temp");
run("HSB Stack");
setSlice(3);
run("Paste");
run("RGB Color");
run("Select All");
run("Copy");
selectWindow(psd);
run("Add Slice");
run("Paste");
selectWindow("temp");
close();
}
}
selectWindow(rat_d);
close();
setBatchMode("exit and display");
selectWindow(psd);
Recipes
- 10 μM fluorescein solution
Make a 100 mM stock by dissolving 332 mg of fluorescein in 10 ml of 95% ethanol
Then dilute 1 μl of this stock solution by 1:10,000 in HBSS to make a 10 μM working solution - HBSS Imaging buffer (50 ml)
5 ml STOCK 1
0.5 ml STOCK 2
0.5 ml STOCK 3
42.5 ml ddH2O
0.5 ml STOCK 4
1 ml 1 M HEPES, pH 7.4
250 µl 2.2 M D-glucose- STOCK 1
4.0 g NaCl
0.2 g KCl
ddH2O q.s. to 50 ml - STOCK 2
0.179 g Na2HPO4 (anhydrous)
0.30 g KH2PO4
ddH2O q.s. to 50 ml - STOCK 3
0.36 g CaCl2
ddH2O q.s. to 50 ml - STOCK 4
0.615 g MgSO4·7H2O
ddH2O q.s. to 50 ml
- STOCK 1
Acknowledgments
This work was supported by National Institute of Health (R01DK077140, R01HL122827, R01MH111527 to M. A. Rizzo; and NIH R35 CA197622, R01DK073368, R01MH111516 to J. Zhang). We would like to thank Dr. Sohum Mehta for his help creating the graphic, and Dr. Gary Mo for writing the ImageJ macro for making ratiometric pseudocolor images.
Competing interests
The authors declare no conflicts of interest.
References
- Mehta, S. and Zhang, J. (2011). Reporting from the field: genetically encoded fluorescent reporters uncover signaling dynamics in living biological systems. Annu Rev Biochem 80: 375-401.
- Ross, B. L., Tenner, B., Markwardt, M. L., Zviman, A., Shi, G., Kerr, J. P., Snell, N. E., McFarland, J. J., Mauban, J. R., Ward, C. W., Rizzo, M. A. and Zhang, J. (2018). Single-color, ratiometric biosensors for detecting signaling activities in live cells. Elife 7: e35458.
- Markwardt, M. L., Snell, N. E., Guo, M., Wu, Y., Christensen, R., Liu, H., Shroff, H. and Rizzo, M. A. (2018). A genetically encoded biosensor strategy for quantifying non-muscle myosin II phosphorylation dynamics in living cells and organisms. Cell Rep 24(4): 1060-1070 e1064.
- Seckinger, K. M., Rao, V. P., Snell, N. E., Mancini, A. E., Markwardt, M. L. and Rizzo, M. A. (2018). Nitric oxide activates β-cell glucokinase by promoting formation of the "glucose-activated" state. Biochemistry 57(34): 5136-5144.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Thévenaz, P., Ruttimann, U. E. and Unser, M. (1998). A pyramid approach to subpixel registration based on intensity. IEEE Trans Image Process 7(1): 27-41.
Article Information
Copyright
![]() Ross et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Ross et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Ross, B., Wong, S. H., Snell, N. E., Zhang, J. and Rizzo, M. A. (2019). Triple Fluorescence Anisotropy Reporter Imaging in Living Cells. Bio-protocol 9(9): e3226. DOI: 10.21769/BioProtoc.3226.
- Ross, B. L., Tenner, B., Markwardt, M. L., Zviman, A., Shi, G., Kerr, J. P., Snell, N. E., McFarland, J. J., Mauban, J. R., Ward, C. W., Rizzo, M. A. and Zhang, J. (2018). Single-color, ratiometric biosensors for detecting signaling activities in live cells. Elife 7: e35458.
Category
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.