- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Visualization of Plant Cell Wall Epitopes Using Immunogold Labeling for Electron Microscopy
Published: Vol 9, Iss 7, Apr 5, 2019 DOI: 10.21769/BioProtoc.3205 Views: 7227
Reviewed by: Amey RedkarGongjun ShiSmita Nair
Original research article
The authors used this protocol in:

Nov 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Plant cell walls consist of different polysaccharides and structural proteins, which form a rigid layer located outside of the plasma membrane. The wall is also a very dynamic cell composite, which is characterized by complex polysaccharide interactions and various modifications during cell development. The visualization of cell wall components in situ is very challenging due to the small size of cell wall composites (nanometer scale), large diversity of the wall polysaccharides and their complex interactions. This protocol describes immunogold labeling of different cell wall epitopes for high-resolution transmission electron microscopy (TEM). It provides a detailed procedure for collection and preparation of plant material, ultra-thin sectioning, specimen labeling and contrasting. An immunolabeling procedure workflow was optimized to obtain high efficiency of carbohydrates labeling for high-resolution TEM. This method was applied to study plant cell wall characteristics in various plant tissues but could also be applied for other cell components in plant and animal tissues.
Keywords: Cell wallsBackground
Plant biomass is mainly composed of cell walls, which are widely used as an energy source in our daily life (Loqué et al., 2015). At the microscopic scale, cell walls consist of cellulose microfibrils embedded in complex matrix polysaccharides (hemicelluloses, pectins) and structural proteins. Cellulose microfibrils (CMFs) are the largest wall polymers with a radius of 3-5 nm and many micrometers long (Cosgrove, 2005). The orientation of CMFs determines the direction of growth and cell anisotropy (Baskin, 2005), but the CMFs also interact with other wall components all together modifying the wall properties (reviewed in Majda, 2018; Majda and Robert, 2018). The study of cell wall composition has a long history, going back to when different chemicals were applied to bind to the wall composites; however, many of them had a wide range of targets (Wallace and Anderson, 2012; Voiniciuc et al., 2018) and could be observed only via light microscope resolution. In contrast, the immunogold labeling is characterized by a high specificity of antibodies and high-resolution imaging, which can precisely localize the wall epitopes across the wall matrix (e.g., Majda et al., 2017). Despite electron microscopy being a relatively old method, it is not broadly used for plant cell walls. The reason could be that it is time-consuming, requires long training and extensive preparation time. In this protocol, I will walk you through all the steps concerning sample preparation, specificity for all the reagents and troubleshooting.
Materials and Reagents
- Adhesion slides, Polysine, 25 x 75 x 1.0 mm (VWR, catalog number: 631-0107)
- Aluminum foil
- Centrifuge tubes 15 ml (PluriSelect, catalog number: 05-00002-01)
- Centrifuge tubes 50 ml (sterile) (PluriSelect, catalog number: 05-00001-01)
- Compressed air in the can (e.g., air duster PRF 4-44)
- Disposable pH indicator paper (universal indicator paper) (Johnson, catalog number: 101.3C)
- Disposable plastic Pasteur pipettes (BRAND, catalog number: 747750)
- Double-edged razor blades (Personna, catalog number: 171930)
- Embedding capsules (8 mm flat, polypropylene capsules) (TAAB, catalog number: C095)
- Filter papers (circles, 150 mm Ø), (Whatman, catalog number: 1001150)
- Glass vials with plastic snap-cap (ca. 41 x 24 mm) (Karl Hecht, catalog number: 2783/3), or clear glass vials with snap-cap (closed top, PE transparent, 18 mm, 500 ml 20 x 40 mm) (VWR, catalog number: 548-0555)
- Grids for transmission electron microscopy, e.g., grid size 100 mesh x 250 μm pitch, nickel or copper (TAAB, maxtaform HF4, catalog number: GM021N; Sigma-Aldrich, catalog number: G1528; Agar Scientific, catalog number: G2500C)
- Light-duty 3-Ply tissue wipers (VWR, catalog number: 82003824-CS)
- Metal needle or dissection needle (VWR, catalog number: 10806-330)
- Microcentrifuge tubes 1.5 ml (Sigma-Aldrich, catalog number: Z606340)
- Microscope slides, 25 x 75 x 1.0 mm (VWR, catalog number: 48300-025 or Thermo Scientific, catalog number: 10144633CF)
- Paper tags
- Parafilm (Sigma-Aldrich, catalog number: P7543)
- Pencil
- Pipette tips
- Plastic Petri dishes (Fisherbrand, catalog number: S33580A)
- Protective gloves (Honeywell, catalog number: Dermatril 740)
- Round silicone rubber (TAAB, catalog number: G082)
- Single edge razor blades with aluminum spine (VWR, catalog number: 233-0156)
- Slide labels (Agar Scientific) or one side adhesive paper
- Square Petri dish (120 mm) (Corning, catalog number: BP124-05)
- Transparent tape (e.g., scotch)
- Waste containers
- Agar, plant agar (Duchefa Biochemie, catalog number: 9002-18-0)
- Bovine Serum Albumin (BSA), lyophilized powder ≥ 96% (agarose gel electrophoresis) (Sigma-Aldrich, catalog number: 9048-46-8)
- Disodium phosphate (Na2HPO4) (store in ambient temperature: room temperature, approx. 21 °C) (Sigma-Aldrich, catalog number: 7558-79-4)
- Distilled water
- Ethanol laboratory reagent, absolute, ≥ 99.5% (flammable, stored in designated place) (Sigma-Aldrich, catalog number: 64-17-5)
- Formaldehyde (FA) (10 ml ampule of 16% methanol-free FA) (health hazards, store closed in ambient temperature, or open in cold room/refrigerator 4 °C) (Thermo Scientific, catalog number: 28908) or paraformaldehyde (PFA) for histology (CH2O)n, (store in cold room/refrigerator 4 °C) (J.T. Baker, catalog number: S898-07)
- Formvar solution (1% formvar in dichloroethane, for microscopy) (Sigma-Aldrich, catalog number: 63148-64-1)
- Glutaraldehyde (GA) (grade I, 1 ml ampule of 25% GA in H2O, specially purified for use as an electron microscopy fixative, linear formula: OHC(CH2)3CHO (health hazards, store closed in ambient temperature, or open in cold room/refrigerator 4 °C) (Sigma-Aldrich, catalog number: 111-30-8)
- Hydrogen chloride (HCl) (Sigma-Aldrich, catalog number: 7647-01-0)
- LR white resin medium grade–catalyzed (health hazards, store in cold room/refrigerator 4 °C) (TAAB, catalog number: L012)
- Monopotassium phosphate (KH2PO4) (Sigma-Aldrich, catalog number: 7778-77-0)
- Monosodium phosphate (NaH2PO4) (store in ambient temperature) (Sigma-Aldrich, catalog number: 7558-80-7)
- Murashige and Skoog basal medium (MS) (Sigma-Aldrich, catalog number: M5519)
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: 7447-40-7)
- Primary antibodies (PlantProbes: www.plantprobes.net or the University of Georgia: www.ccrc.uga.edu)
- Secondary antibodies, e.g., EM Goat anti-Rat IgG (H+L): 10 nm Gold (BBI Solutions, catalog number: 014990) or EM Goat anti-Mouse IgG (H+L) 10 nm Gold (BBI Solutions, catalog number: EM.GMHL10)
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: 7647-14-5)
- Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: 1310-73-2)
- Sucrose (Sigma-Aldrich, catalog number: 57-50-1)
- Toluidine blue for microscopy (Sigma-Aldrich, catalog number: 6586-04-5)
- TopVision low melting point agarose (LMP agarose) (store in ambient temperature) (Thermo Scientific, catalog number: R0801)
- Tween 20 (Signa-Aldrich, catalog number: P1379)
- Uranyl acetate (UA) (health hazardous, store in ambient temperature) (VWR, catalog number: 541-09-3)
- Half strength MS basal medium (see Recipes)
- Paraformaldehyde-glutaraldehyde (4% PFA and 0.05% GA) fixative solution (see Recipes)
- Phosphate buffer (PB), 0.1 M solution (pH = 7.2) (see Recipes)
- Low melting point (LMP) agarose 1% solution (see Recipes)
- Different ethanol concentrations 10%-95% (see Recipes)
- Different LRW resin concentrations 10%-75% (see Recipes)
- Toluidine blue solution (see Recipes)
- Blocking Reagent (BR) 1% (see Recipes)
- Antibodies solutions (see Recipes)
- Phosphate Buffered Saline (PBS), 0.1 M solution (pH = 7.2) (see Recipes)
- Uranyl acetate solution (5%) (see Recipes)
Equipment
- Analytical balance
- Autoclave (YPO, model: D66161)
- Centrifuge (Marshall Scientific, Eppendorf, model: 5417C)
- Conical flask
- Desiccator (Thermo Fisher, model: 5311-0250)
- Diagonal cutting pliers e.g., stanley diagonal cutting pliers, 5 (Robosource, catalog number: 15-11)
- Diamond knife, e.g., ultra 45° (Diatome)
- Flat beaker or crystallizer (with a wide diameter)
- Forceps
- Freezer (-20 °C)
- Fume hood
- Glass baker (50 ml)
- Glass knife strips (Agar Scientific, catalog number: AGG336)
- Glass knifemakers for histology knives (LKB, catalog number: LKB 7801B; or Agar Scientific, catalog number: AGL4158)
- Grid storage box (LKB/Leica catalog number: G133, or TAAB Gilder G062)
- Lab oven/incubator (60 °C)
- Light microscope
- Magnetic hotplate stirrer with magnetic stir bar
- Metal 1.5 ml Eppendorf rack
- Microtome (Reichart Ultracut)
- Microwave oven
- pH meter
- Pipettes
- Protective clothes and mask
- Refrigerator (4 °C)
- Rotary shaker or orbital shaker (IKA KS 130 Basic)
- Slide drying rack or slide staining jar (e.g., DWK Life Sciences Wheaton)
- Small bench clamp workshop
- Thin painting brush
- Transmission Electron Microscope (JEOL, model: JEM-1230)
- Tweezers: negative-action style: thin curved tips (Dumont, catalog number: 0203-N7-PO) or thin tips (Dumont, catalog number: 0302-N0-PO-1)
- Tweezers: straight with Geneva pattern, thin tips (Dumont, catalog number: 0103-0-PO)
- Vortex mixer (Vortex-Genie 2)
- Warming plate, or slide drying hotplate (Agar Scientific, model: AGL4384) or spirit lamp burner
Procedure
- Plant material fixation
In this section, I describe the procedure for plant material preparation and fixation. This protocol was developed for Arabidopsis thaliana leaves, but it could also be applied to other organisms and tissues such as roots, shoots and woody tissues in tree species.- Sterilize the seeds before sowing
- Place a small number of seeds into 1.5 ml Eppendorf tubes (approx. 5% of the Eppendorf tube volume).
- Add 1 ml of 70% ethanol with Tween 20 for 2 min.
- Replace 70% ethanol and Tween 20 with 1 ml of 95% ethanol for 1 min.
- Remove the ethanol and wait for the seeds to dry.
- Grow Arabidopsis seedlings for 2 weeks on vertical agar plates (Recipe 1) in the chamber with long day condition (16 h) (temperature 20 °C and 18 °C at day and night, respectively).
Note: To synchronize the growth, vernalize the seeds by keeping the plates in a cold room/refrigerator at 4 °C for 2-3 days (in darkness). - Harvest plant material and place it directly in the vials filled with 3-5 μl of cold paraformaldehyde-glutaraldehyde (PFA-GA) fixation solution (Recipe 2).
Note: Remember to harvest the same leaf number from each plant (counting from the bottom to the top: cotyledons, leaf 1, leaf 2, leaf 3, leaf 4, meristem). To allow the fixative to penetrate well, cut small squares (~2 mm2 max). in the middle part of the leaf. Harvest at least 10 leaves from different plants. Conduct all the fixation steps under the fume hood with protective gloves and clothes (for handling restrictions see Safety Data Sheet SDS provided by the retailer). Mark the vials by writing the names with a permanent marker and sticking a transparent tape on these labels. - Vacuum samples in a desiccator at ambient temperature until plant pieces will sink (approx. 4 h).
Note: It might happen that some of the samples are floating after vacuuming, which indicates that samples more likely contain oxygen. Try to collect samples, which are at the bottom of the vials. - Place the vials on a rotary or orbital shaker and leave the samples mixing for a couple of hours.
- Keep the samples in a cold room/refrigerator (4 °C) overnight.
- Discard PFA-GA fixative.
- Wash the samples with phosphate buffer (PB) (Recipe 3) (twice for 30 min each).
Note: Discard PFA-GA fixative and first PB washing by using disposable plastic Pasteur pipettes in the assigned waste bottle. Wash by adding at least 5 ml of PB or distilled water (the more PB and distilled water, the better the washing). Conduct all washing steps slowly mixing on rotary or orbital shaker under a fume hood.
- Sterilize the seeds before sowing
- Embedding plant material in low melting point (LMP) agarose
Embedding small plant pieces in LMP agarose is the best way to orient the sample for sectioning (e.g., cross or longitudinal). It marks the direction of shoot/root tip (Figure 1A) or the localization of abaxial and adaxial leaf sides (Figure 1B). Embedding also facilitates the handling and protection of samples.
Figure 1. Orienting the plant pieces in low melting point (LMP) agarose. A-B. Samples embedded in cuboid-shaped agarose blocks presenting one cut edge to mark the orientation of the sample (e.g., top left in Figure 1A). A. Top part of the root is marked by a cut edge. B. The adaxial side of the leaf is in the front when the cut edge is on the top left.- Wash the samples with distilled water (twice for 30 min each).
- Pour a thin (3-5 mm thick) layer of agarose (Recipe 4) on a square plastic Petri dish and wait a few minutes until agarose cools down (but should not solidify).
- Place the plant material on agarose by using tweezers (many pieces on the same plate but keep distance between the samples approx. 2-3 cm2).
Note: Handle the plant specimens with care not to damage the tissues. - Add more agarose (± 3 mm thick layer) to cover the plant specimens (wait until agarose solidifies) (Figure 2A).
- Cut off small cubes of agarose with the embedded specimens (one specimen in one cube) (Figure 2B).
Note: The size of agarose cubes should not be bigger than the diameter of embedding capsules (8 mm Ø). - Cut one of the corners in the agarose cube to orient the sample (Figures 1, 2B and 2C).
- Move the blocks to vials filled with distilled water, then discard the water before proceeding to the next step.

Figure 2. Embedding the plant material in agarose. A. Square Petri dish filled with a layer of 3 mm thick agarose. B. Small cuboid-shaped agarose blocks with cut one of the edges (all the leaf pieces are seen from the adaxial side). C. Magnified agar cube with the abaxial side of the leaf in the front. In this case, big plant pieces were used for visualization, but they should be ± 5 times smaller.
- Dehydrating specimens embedded in agarose blocks
Here I describe a common method to remove water from samples and to enable better penetration of resin. Time intervals can be adjusted; however, longer intervals give better quality embedding. Ideally, the dehydration process would be carried over the span of two days, but all dehydration steps can also be carried out within the same day (depends on the sample size).- Dehydrate the samples in a graded ethanol series as below (Recipe 5):
10% ethanol (twice for 30 min each)
20% ethanol (twice for 30 min each)
30% ethanol (twice for 30 min each)
50% ethanol (twice for 30 min each)
Note: The amount of ethanol added varies according to the size and number of the samples embedded in agarose. Make sure that agarose blocks are completely covered by ethanol. - Leave the samples in 50% ethanol in ambient temperature (on the bench) overnight.
Note: Close the vials with plastic snap-cap to prevent ethanol evaporation. - Continue to dehydrate the samples in a graded ethanol series as below:
70% ethanol (twice for 30 min each)
80% ethanol (twice for 30 min each)
90% ethanol (twice for 30 min each)
95% ethanol (three times for 30 min each)
99.5% ethanol (three times for 30 min each)
Note: Carry all dehydration steps on rotary or orbiter shaker. The vials should be closed with plastic snap-caps. Some intermediate ethanol dilutions can be omitted (e.g., 20% and 80% ethanol), but it might affect the quality of embedding. The volume of alcohol in the last step should be the same for all vials.
- Dehydrate the samples in a graded ethanol series as below (Recipe 5):
- Resin embedding
In this step, alcohol is replaced with viscous and low soluble LR white (LRW) resin. All these steps must be carried gradually with vigorous mixing of the samples on an orbital shaker. Perform all these steps in cold room/refrigerator (4 °C) to prevent resin polymerization.
Figure 3. Embedding the plant material in resin. A. Embedding capsule and paper tags. B. Capsule after resin polymerization. C. Capsule cut along to remove the resin block. D. Resin block alone. E. Trimmed resin block into a trapezoid shape.- Slowly add a few drops of resin to the vials with a known volume of 99.5% ethanol until the resin content will reach 10% (v/v) of the overall volume.
Note: All steps, including the exchange of the resin, must be carried out using protective gloves and clothes under the fume hood. Keep the vials closed with the plastic snap-caps mixing in the orbital shaker in cold room/refrigerator (4 °C). Always discard the resin to designated waste. - Leave the vials in the orbital shaker to mix samples overnight.
- Exchange resin 10% in alcohol with a new resin in a graded resin series as below (Recipe 6):
10% resin (approx. 5 h)
25% resin (approx. 5 h)
50% resin overnight
75% resin (approx. 5 h)
100% resin overnight
100% (fresh) resin overnight
Note: Some intermediate resin dilutions can be omitted (e.g., 25% and 75%), but it might affect the quality of embedding.
- Slowly add a few drops of resin to the vials with a known volume of 99.5% ethanol until the resin content will reach 10% (v/v) of the overall volume.
- Closing samples in the embedding capsules and resin polymerization
This is the last step of the embedding procedure, in which you must pay attention to properly orientate the samples (Figure 1). The agarose blocks should lie flat in the middle of the bottom part of the capsule (Figures 4A and 4B), which need to be oriented in a parallel direction towards the knife edge (see Procedure G, Figure 6). Handle the capsules with care to prevent the slipping of the samples close to the corners, which could cause some issues in trimming the sample and sectioning.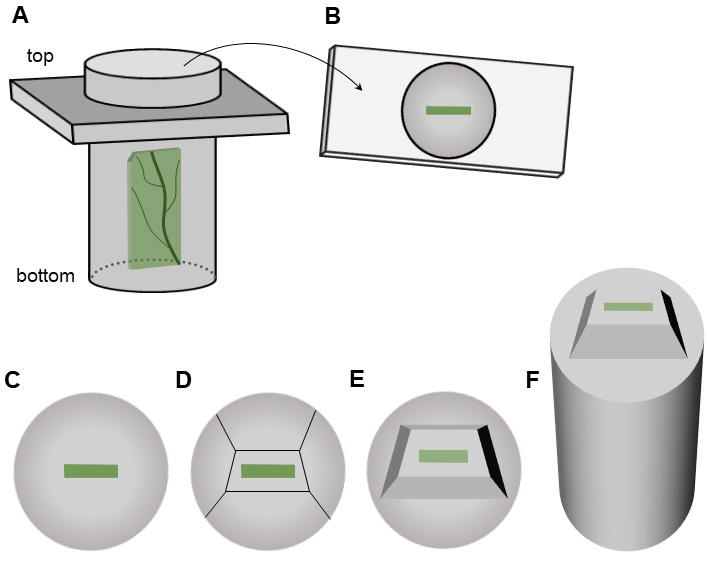
Figure 4. Trimming the plant specimen prior sectioning. A-B. A capsule with a piece of the leaf embedded in resin (A). The sample should be in the middle of the bottom part of the capsule (B). C-E. Resin with specimen alone seen from the bottom: resin before trimming (C), the direction of trimming (D), trimmed specimen (E). F. Trimmed resin block seen from the side.- Place the capsule (Figure 3A) on a rack and fill half of the capsule with resin.
- Put the sample in the capsule, orient the sample well and fill-up the capsule with fresh resin.
- Cut small paper tags, label them with a pencil and place them in the top of the capsules.
Note: The capsule should be filled with resin so that, by closing it, no space for air bubbles is left. Use a pencil to tag your samples instead of a pen or a marker, which can be washed out by the resin. - Place the capsules in the incubator/oven (60 °C) for 24 h (or until it becomes solid) to polymerize the resin.
Note: Remember to check the temperature of the oven (60 °C). A temperature that is too low can affect the polymerization (resin will not polymerize equally), while a temperature that is too high or an excessively long incubation can lead to cracking or breaking of the resin. - Samples are ready when the resin becomes stiff (Figure 3B).
Note: To know if the resin solidified, indent a metal needle to the resin surface.
- Preparing the grids for transmission electron microscope (TEM)
The samples for the TEM are mounted on a small grid, which is then inserted inside the TEM. The grid must be coated with formvar, which is an adhesive layer holding the sections. While preparing the grids, make sure that microscope slides, tweezers and laboratory glassware are clean (use tissue wipers and compressed air to clean the dust on the surfaces).- Add approx. 50 ml of formvar solution to a small beaker (Figure 5A).
- Grab the microscope slide in a vertical position (holding a dull side) and vigorously dip approx. half of the slide in the formvar solution (no more than approx. 5 s in the solution) (Figure 5B).
Note: The formvar will form a thin film on the side. - Dry the slide with a film in a vertical position on a drying rack for 3-10 min.
Note: Do not touch any part of the slide, which was in contact with the formvar solution. - Prepare glassware with a wide diameter (≥ 10 cm) and fill it up with distilled water. Place the grids on a round silicone rubber or filter paper (Figure 5C).
- Scratch the slide edges with a sharp razor blade and blow moist air on the slide.
- Dip the slide vertically (90° or ≥ 45°) in the distilled water bath.
Note: You will see the film detaching from the glass and then floating on the water surface. - Using fine tweezers, put the grids onto a floating film with the dull side in contact with formvar (put as many grids as possible) (Figures 5D and 5E).
Note: Each grid has two sides: a dull side and a shiny side (Figure 7A). Sections will be mounted on the formvar-coated dull side (this side should be in contact with buffers, antibodies, distilled water, contrasting stain) (Figure 7B), and the shiny side will be in contact with filter paper during the drying of the samples (it is up to you which side you want to coat with formvar and put the sections on but remember to be consistent). - Prepare a new slide covered by one side adhesive paper or slide label (Figure 5F).
- Grab that slide in vertical position and touch one of the edges of the floating film (Figure 5G).
- Dip the slide into the water, which will cause adhesion of the film with grids to the slide (Figures 5H and 5I).
- Leave the slide with grids covered with formvar until dry.
Note: Store the slides in a closed box or plastic Petri dish in cold room/refrigerator (4 °C).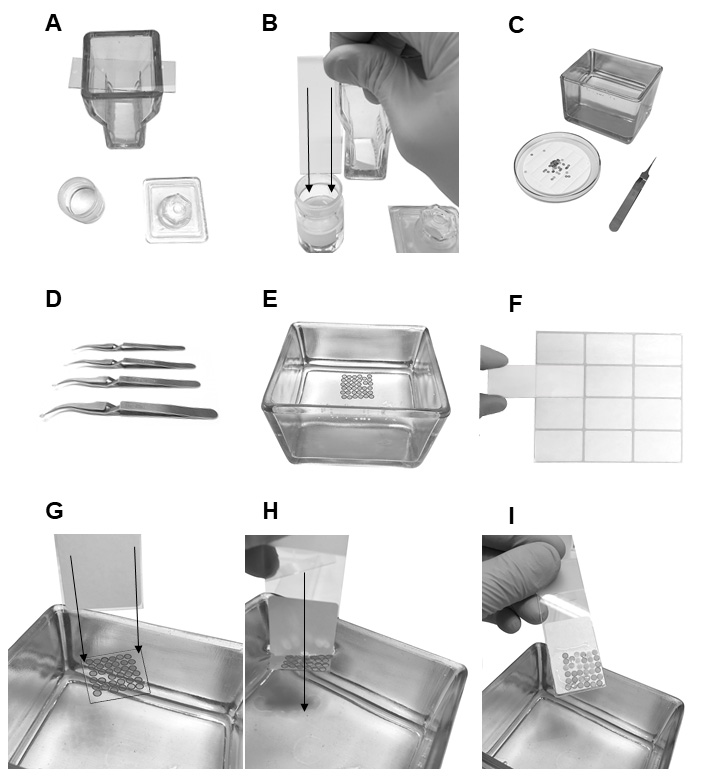
Figure 5. Preparing the grids for TEM. A. Baker filled with formvar solution and a slide on the slide staining jar. B. Dipping of the microscope slide in formvar solution. C. Glassware filled up with distilled water, Petri dish with round silicone rubber and grids on it, tweezers. D. Tweezers holding the outer ring of the grids. E. Grids on a floating formvar film with the dull side of the film. F. Sticking a slide label to the slide. G. Slide in vertically before touching the edges of the floating film. H. Dipping the slide in water causing the adhesion of film with grids to the slide. I. Slide with formvar and grids on it.
- Ultrathin sectioning for electron microscopy
The most important thing in the preparation of the sections is to properly orient the sample. Remember to regularly control the sections quality. Make sure that the cells/tissue/organ is oriented in parallel to the knife (Figure 6) and if they are not, then correct the angle between the sample and the knife edge.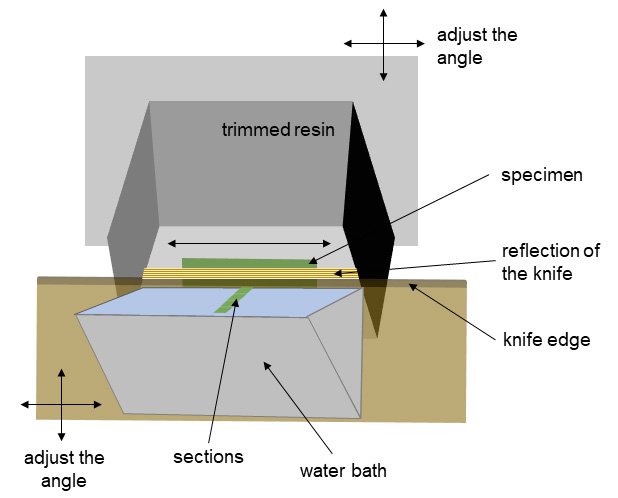
Figure 6. Ultrathin sectioning for electron microscopy. Begin with orienting the specimen in parallel to the knife edge. When resin is close to the knife, a reflection of the knife will be seen on the surface of the resin. This reflection should be parallel to the sample (if properly oriented). With sectioning progression, make sure that the cells/tissue/organ are oriented in parallel to the knife (remember to correct the angle between the sample and the knife edge).- Immobilize the capsules using a small bench clamp workshop.
- Remove the capsules using stiff single edge razor blades and diagonal cutting pliers (Figures 3C and 3D).
- Trim the bottom of the sample with a stiff single edge razor blade and then use thin double-edged razor blades for more precise cutting (Figures 3E and 4C-4F).
Note: The bottom of the sample, which will be in parallel to the knife edge (see Procedure E), should be trimmed in a trapezoid shape with the major (longer) base on the bottom and minor (shorter) base on the top (Figures 3E, 4D-4F and 6). The size of the trapezoid should not exceed the length of the diamond knife (≤ 2 mm). - Mount the resin block on an ultramicrotome holder with the bottom of the sample oriented towards the knife.
- Orient the resin block with the sample in the way that the trimmed bottom of the sample (Figure 4A) will be parallel to the glass knife edge (Figure 6).
Note: When the sample is close to the knife you will be able to see the reflection of the knife on the surface of the resin (this reflection should be parallel). - Cut a few sections using a glass knife.
Note: The sections can be more than 1 µm thick. The purpose of this point is to see how the sections look like and to orient the specimen properly. - Place a drop of distilled water on a regular or a polylysine-coated slide and put a few sections on that drop.
Note: Use a wet painting brush (the thinner, the better) to collect the sections. - Dry sections on the slide (see Equipment 30).
- Put a drop of toluidine blue stain (Recipe 7) onto the sections for a minute and wash under tap water.
- Observe the sections under the light microscope.
Note: If the sample is oriented in the proper way, you can begin ultra-thin sectioning. If it is not oriented properly (cells are cut askew), adjust the sample position and repeat sectioning (Steps G5-G10). - Mount a diamond knife onto the ultramicrotome and fill the bath with distilled water.
- Orient the resin block with the sample in an analogical way as above (see Step G5).
- Begin ultrathin sectioning (70 nm thin sections) until you see the ribbon of sections floating on the water.
- Using the forceps dip the grid under the water (the dull side with formvar on the top) and collect some sections.
- Leave the grids with section on the filter paper or on the round silicon rubber until dry (Figure 7A, numbers 7-9), store in the grid storage box.
Note: Remember to place the shiny side on the bottom, and dull side with the section on the top.
- Immunogold labeling
Here I describe how to perform the labeling of cell wall epitopes. In this process, primary antibodies bind to specific cell wall epitopes, which are then recognized by secondary antibodies coupled with gold particles for visualization in TEM. The immunolocalization procedure relies on moving the grids over a series of small droplets containing different solutions, antibodies and stain for contrasting, all are placed on the parafilm (Figures 7 and 8). Remember to include controls such as wild type, untreated plants as well as secondary antibody alone, which should not give any labeling.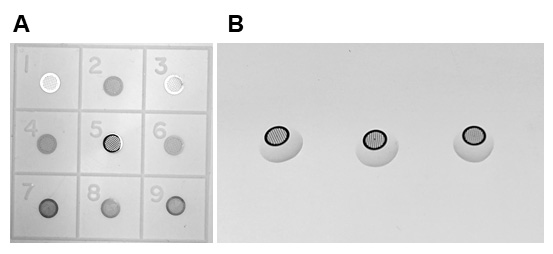
Figure 7. Grids for electron microscopy. A. Grids with two sides: a dull side and a shiny side. Grids with the shiny side correspond to numbers 1, 3, 5. Grids with the dull side correspond to numbers 2, 4, 6, 7-9. Sections are mounted on the formvar-coated dull side (numbers 7-9). B. The dull side is in contact with droplets. Usually, the shiny side must be in contact with the filter paper while drying the samples (not shown).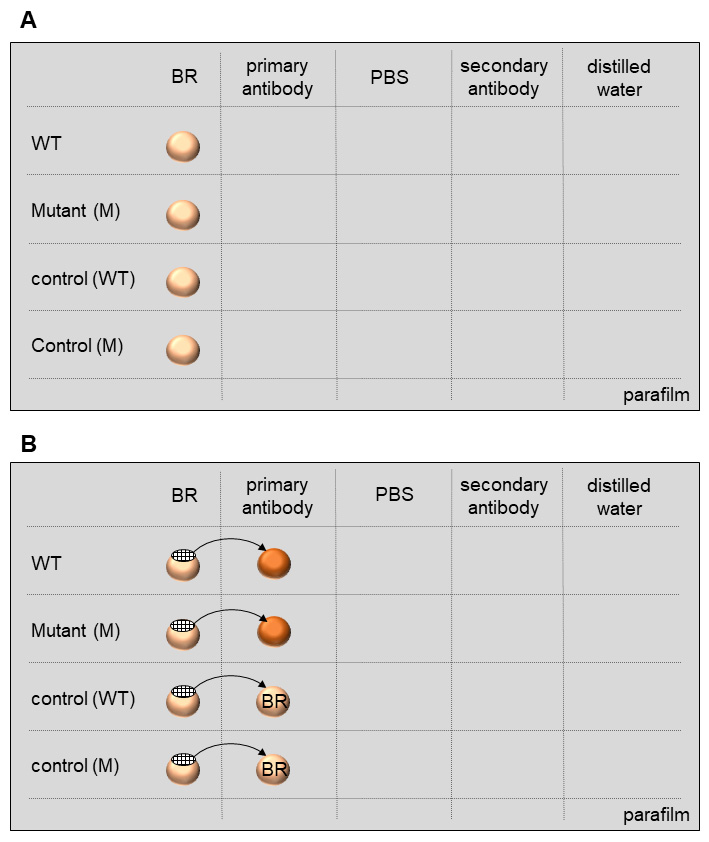
Figure 8. Immunolocalization procedure. The labeling relies on moving the grids over a series of small droplets containing different solutions (blocking reagent [BR], primary antibodies, PBS buffer, secondary antibody, distilled water) all placed on the parafilm.- Place a piece of parafilm on a clean table.
Note: Use 70% ethanol to clean the table. The side of parafilm covered by paper is clean and should be on top (where droplets of different solutions will be placed). Avoid touching the parafilm with hands to not contaminate it. Add a few drops of water below the parafilm and stick the corners of the parafilm with a tape to stabilize it and keep flat. - Place a few droplets of BR (depending on the number of grids) in a row on the parafilm (for BR see Recipe 8) (Figure 8A).
- Place the grids on droplets of BR and leave it for 15 min at ambient temperature (the dull side of the grid containing the section should be in contact with the droplet) (Figures 7B and 8B).
Note: Always transfer the grids with clean tweezers. It is recommended to use a set of tweezers (negative-action style). To avoid cross-contamination between antibodies, before touching the grids, always clean tweezers with 70% ethanol, rinse in distilled water and dry with a clean tissue wiper before contact with a grid. Always handle the grids with care. The tweezers should be in contact only with the external ring of the grid. - Put the droplets of primary antibody solutions (Recipe 9) in the second row (use BR instead of primary antibodies for the controls, which will be treated only with secondary antibodies).
- Take the grid out of BR droplet, touch the edge of the grid with a clean filter paper (to absorb the reagent) and transfer the grid onto a droplet with primary antibody.
Note: From the circles of the filter paper, cut small triangles, use the sharp tip to touch the grid edge. - Incubate with primary antibody for 45-60 min at ambient temperature.
Note: During the incubation in primary and secondary antibodies make sure that the solution will not dry out. To prevent drying, cover the samples with e.g., a large glass Petri dish. - Put droplets of PBS (Recipe 10) in the four next rows.
- Take the grid out of the primary antibody droplet, touch the edge of the grid with a clean filter paper (to absorb the reagent), and transfer the grid onto a droplet with PBS.
Note: Rinse the grids four times in PBS. Leave the grids for 5 min on each droplet. Before transferring to the next PBS droplet, always absorb previous PBS with a filter paper. - Put the droplets of the secondary antibody solution (Recipe 10) into the next row.
- Transfer the grids in an analogical way to above (Steps H5 and H8).
- Incubate with the corresponding secondary antibody for 60 min at ambient temperature.
- Put the droplets of distilled water into the four next rows.
- Transfer the grids in an analogical way to above (Steps H5 and H8).
Note: Rinse the grids four times in distilled water. Leave the grids for 5 min on each droplet. Before transferring to the next water droplet, always absorb previous water with a filter paper. - Take out the grid from the distilled water droplet, touch the edge of the grid with a clean filter paper (to absorb the reagent) and transfer the grid on a filter paper or on a silicon rubber (the dull side with the section should be on top).
- Leave the grids for at least 60 min or until dry (cover with a large Petri plate to prevent contamination).
- Put a droplet of uranyl acetate (UA) (Recipe 11) on a new piece of the parafilm.
Note: Briefly spin at maximum speed before use. - Transfer the grid onto a droplet of uranyl acetate (UA) and leave it for 10 min at ambient temperature in darkness (UA is light sensitive).
- Take out the grid from UA droplet, touch the edge of the grid with a clean filter paper (to absorb the reagent) and transfer the grid onto the distilled water droplet.
- Holding the grids with negative-action style tweezers rinse each grid under tap water for 30 s, then place the grids onto the filter paper or silicon rubber, wait until dry (the dull side with the section should be on the top) (Figure 7A, points 7-9) and store in a grid storage box.
Note: All the steps involving the use of UA must be carried with special attention by using protective gloves and clothes under the fume hood. All UA waste must be placed in assigned waste bins. Avoid too long incubation in UA as it can give too high contrast.
- Place a piece of parafilm on a clean table.
Recipes
- Half strength MS basal medium
- Add 2.2 g of MS
- Add 10 g of sucrose (1% v/v)
- Add distilled water to a final volume of 1 L
- Mix the solution on a magnetic stirrer with a magnetic stir bar
- Adjust pH to 5.6 by decreasing pH with hydrogen chloride (HCl) or increasing pH with sodium hydroxide (NaOH)
- Add 7 g of plant agar (0.7% v/v)
- Autoclave the medium
- Paraformaldehyde-glutaraldehyde (4% PFA and 0.05% GA) fixative solution
- Add 4 g of paraformaldehyde (PFA) powder to a conical flask with 100 ml of 0.1 M phosphate buffer (4% of PFA for total concentration)
- Add a magnetic stir bar, cover with aluminum foil to prevent evaporation and place the flask on a magnetic hotplate stirrer (90-100 °C)
- Add a few drops of sodium hydroxide solution (NaOH) to speed up dissolving the powder (use 0.1 M concentration of NaOH or lower) and then leave it mixing until the powder dissolves completely (it can take a couple of hours)
- Adjust pH to 7.2 by decreasing pH with hydrogen chloride (HCl) or increasing pH with sodium hydroxide (NaOH)
- Add 200 μl of 25% glutaraldehyde (GA) (0.05% of GA for total concentration)
- Store the fixative solution in a designated (ventilated) refrigerator (for a short storage) or freeze the fixative in -20 °C (for long storage)
- Phosphate buffer (PB), 0.1 M solution (pH = 7.2)
- Prepare a stock solution of 0.1 M disodium phosphate (Na2HPO4) and another solution of 0.1 M monosodium phosphate (NaH2PO4) by dissolving these salts in distilled water
- Adjust phosphate buffer pH to 7.2 by mixing 68.4 ml of disodium phosphate solution (Na2HPO4) with 31.6 ml of monosodium phosphate solution (NaH2PO4) (Sambrook et al., 1982)
- Low melting point (LMP) agarose 1% solution
- Dissolve 1 g of LMP agarose in 100 ml of distilled water
- Warm it up in a microwave until boiling
- Wait to cool it down before contact with the plant material
- Different ethanol concentrations 10%-95%
- Mix ethanol absolute ≥ 99.5% with distilled water to obtain different concentrations
- Prepare around 100 ml of each concentration and keep it in closed bottles
- Different LRW resin concentrations 10%-75%
Mix LWR resin with ethanol absolute ≥ 99.5% to obtain different concentrations - Toluidine blue solution
- Dissolve 0.5 g of toluidine blue powder in 50 ml of 70% ethanol.
- Prepare 1% sodium hydroxide (NaOH) solution by dissolving 0.5 g of NaOH in 50 ml of distilled water
- Mix toluidine blue solution with 1% NaOH in ratio 1:5
- Adjust pH of NaOH to 2.5 by decreasing pH with hydrogen chloride (HCl) or increasing pH with sodium hydroxide (NaOH)
- Blocking Reagent (BR) 1%
Dissolve 1 g of Bovine Serum Albumin (BSA) in 100 ml of PBS (for 1% of BR for total concentration) - Antibodies solutions
Primary antibodies are diluted in BR (1:10) and secondary antibodies are diluted in BR (1:50) - Phosphate Buffered Saline (PBS), 0.1 M solution (pH = 7.2)
8 g sodium chloride (NaCl)
0.2 g potassium chloride (KCl)
1.15 g disodium phosphate (Na2PO4)
0.2 g monopotassium phosphate (KH2PO4)
Refill with distilled water up to 1 L
Adjust pH to 7.2 by decreasing it with hydrogen chloride (HCl) or increasing it with sodium hydroxide (NaOH)
Note: Autoclave phosphate buffer solution before storing in ambient temperature. - Uranyl acetate solution (5%)
- Add 0.5 g of UA to 5 ml of double distilled water in a 15 ml tube
- Adjust the pH to 3.5 by decreasing it with hydrogen chloride (HCl) or increasing it with sodium hydroxide (NaOH)
- Fill the tube with double distilled water up to 10 ml and energetically shake the content
- Filter the solution through a filter paper or spin the solution in the centrifuge at maximum speed for 10 min and transfer the liquid part to a new tube
- Keep UA solution in the dark
Acknowledgments
I would like to greatly acknowledge Stéphanie Robert and Richard S. Smith for comments on the manuscript and support of the work. I acknowledge Nicola Trozzi for proofreading the manuscript. I would like to thank Lenore Johansson, Kjell Olofsson and Janusz Kubrakiewicz for sharing their experience. This work was performed at Umeå Core Facility Electron Microscopy at Umeå University. The work was supported by the Swedish Research Council Vetenskapsrådet (grant nos. VR2012-2343 and VR2016-00768), Vinnova (Verket för Innovationssystem) and ERA-CAPS. This protocol was adapted from the published study (Majda et al., 2017).
Competing interests
I declare no conflicts of interest or competing interests.
References
- Baskin, T. I. (2005). Anisotropic expansion of the plant cell wall. Annu Rev Cell Dev Biol(21): 203-222.
- Cosgrove, D. J. (2005). Growth of the plant cell wall. Nat Rev Mol Cell Biol 6(11): 850-861.
- Loqué, D., Scheller, H. V. and Pauly, M. (2015). Engineering of plant cell walls for enhanced biofuel production. Curr Opin Plant Biol 25: 151-161.
- Majda, M. (2018). Role of the cell wall in cell shape acquisition. Swedish University of Agricultural Sciences, Umeå, Sweden. Acta Universitatis agriculturae Sueciae. 10: 1652-6880. ISBN 978-9-17-760160-9.
- Majda, M. and Robert, S. (2018). The role of auxin in cell wall expansion. Int J Mol Sci 19(4): E951.
- Majda, M., Grones, P., Sintorn, I. M., Vain, T., Milani, P., Krupinski, P., Zagorska-Marek, B., Viotti, C., Jonsson, H., Mellerowicz, E. J., Hamant, O. and Robert, S. (2017). Mechanochemical polarization of contiguous cell walls shapes plant pavement cells. Dev Cell 43(3): 290-304 e294.
- Sambrook, J., Fritsch, E. F. and Maniatis, T. (1982). Molecular cloning: A laboratory manual. 2nd edition. Cold Spring Harbor Laboratory.
- Voiniciuc, C., Pauly, M. and Usadel, B. (2018). Monitoring polysaccharide dynamics in the plant cell wall. Plant Physiol 176(4): 2590-2600.
- Wallace, I. S. and Anderson, C. T. (2012). Small molecule probes for plant cell wall polysaccharide imaging. Front Plant Sci 3: 89.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Majda, M. (2019). Visualization of Plant Cell Wall Epitopes Using Immunogold Labeling for Electron Microscopy. Bio-protocol 9(7): e3205. DOI: 10.21769/BioProtoc.3205.
Category
Plant Science > Plant cell biology > Cell wall
Plant Science > Plant developmental biology > Morphogenesis
Cell Biology > Cell imaging > Electron microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.