- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Multiple Modification of Chromosomal Loci Using URA5.3 Selection Marker in the Unicellular Red Alga Cyanidioschyzon merolae


Published: Vol 9, Iss 7, Apr 5, 2019 DOI: 10.21769/BioProtoc.3204 Views: 5914
Reviewed by: Dennis J NürnbergGeneviève BallJuan Facundo Rodriguez Ayala
Original research article
The authors used this protocol in:

Nov 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The unicellular red alga Cyanidioschyzon merolae has been used as a eukaryotic photosynthetic model for various basic and applied studies. Although the nuclear genome of C. merolae can be modified by homologous recombination with exogenously introduced DNA, it has been difficult to modify multiple chromosome loci within the same strain because of the limited number of available positive selection markers. Recently, we reported a modified URA5.3 gene cassette (URA5.3T), which can be used repeatedly for nuclear genome transformation using the pMKT plasmid vectors for epitope tagging (3x FLAG- or 3x Myc-) of nuclear-encoded proteins. In addition, these plasmid vectors can also be used to knock out multiple genes one by one. This report describes the construction of DNA fragments for transformation and the detailed transformation procedure.
Keywords: Cyanidioschyzon merolaeBackground
The standard method of transformation in C. merolae uses the uracil-auxotrophic strains M4 (Minoda et al., 2004) or T1 (Taki et al., 2015), which harbor a frameshift or a complete deletion mutation in the gene URA5.3 (CMK046C). The gene URA5.3 encodes a protein composed of an orotate phosphor ribosyl transferase (OPRTase) domain and an orotidine-5'-phosphate decarboxylase (OMPdecase) domain, which synthesizes uridine-5'-monophosphate from orotate. The transformant is selected by introducing URA5.3 as a selection marker into the uracil-auxotrophic strain. A recent study has reported the chloramphenicol resistance CAT gene as a second selectable marker gene for the nuclear transformation experiment in C. merolae (Fujiwara et al., 2017). However, it is difficult to modify more than three genome loci. Thus, we developed the URA5.3 selection marker gene recycling system using a modified URA5.3 gene, named URA5.3T (Takemura et al., 2018). The URA5.3T includes the URA5.3 terminator sequence (462 bp) at the 5’ end of the URA5.3 promoter-terminator cassette to make homologous repeats, the epitope tag region which encodes the recognition sequence for three different proteases (TEV, Factor Xa and thrombin), and a 3x FLAG or 3x Myc tag-encoding sequence (Figure 1). The URA5.3 promoter-terminator cassette is eliminated by frequently occurring DNA excision through the homologous repeat sequences located on both sides of URA5.3T. In this method, the URA5.3 eliminated transformant is selected using 5-fluoroorotic acid (5-FOA), which is converted to cytotoxic 5-fluorouracil via URA5.3 activity. The marker-eliminated transformant can be used for the next round transformation experiment using the URA5.3T marker gene (Figure 1). In this report, we present the procedure for construction of the transformation template DNA, which includes the recycling marker URA5.3T, for the purpose of gene tagging or gene knock out.
Figure 1. Outline from transformation to URA5.3 marker elimination using URA5.3T. The DNA includes the homologous regions for the genome-specific locus (grey box) and the URA5.3T. The URA5.3T includes the epitope tag region, a homologous repeat sequence (red hatched box) and the URA5.3 promoter-terminator cassette. The epitope tag region contains the recognition sequence for three different proteases (TEV, Factor Xa and thrombin) and a 3x FLAG or 3x Myc tag-encoding sequence.
Materials and Reagents
- Pipette tips (NIPPON Genetics Co., Ltd., catalog numbers: FG-102, FG-301, FG-401)
- Toothpicks
- Cuvettes (As One Corporation., catalog number: 1-2855-02)
- 1.5 ml Microtube (BM Equipment Co., Ltd., catalog number: NT-175)
- 50 ml Centrifuge tube (Corning Incorporated, catalog number: 352196)
- 24-well tissue culture test plate (BM Equipment Co., Ltd., catalog number: 92424)
- Sterilized plastic plate (Φ 90 mm) (As One Corporation, catalog number: GD90-15)
- AnaeroPouch (Mitsubishi Gas, catalog number: 2-3764-02)
- AnaeroPack (Mitsubishi Gas, catalog number: 2-3765-01)
- C. merolae T1 strain (Taki et al., 2015)
- Escherichia coli DH5α (Takara Bio, catalog number: 9057)
- Wild type C. merolae DNA
- Salmon sperm DNA (FUJIFILM Wako Pure Chemical Industries Corporation, catalog number: 047-17322)
- Plasmid vector pMKT (Takemura et al., 2018; available upon request to authors)
- KOD-Plus-Neo DNA polymerase (Toyobo, catalog number: KOD-401)
- GoTaq DNA polymerase (Promega, catalog number: M7123)
- StuI (NEB, catalog number: R0187S)
- DpnI (Takara Bio, catalog number: 1235A)
- Specific primers [set for amplifying the Upstream and Downstream fragments with the homologous sequence for the StuI-digested pMKT (see Figure 2A), ordered at Integrated DNA Technologies (IDT) Inc.]
- Confirmation primer sets (annealing to the outside sequence of the homologous regions, see Figure 3, ordered at Integrated DNA Technologies (IDT) Inc.)
- Primer sets for colony PCR (ordered at Integrated DNA Technologies (IDT) Inc.):
Forward primer for up: 5’-CGAGGTCGACGGTATCGATAAGC-3’
Reverse primer for up: 5’-GATCTAGTAACATAGATGACACCGC-3’
Forward primer for down: 5’-GCTGCTAGGGATTGTGGCGCGA-3’
Reverse primer for down: 5’-CCGCTCTAGAACTAGTGGATCCC-3' - In-fusion HD cloning kit (Takara Bio, catalog number: 639648)
- Phenol, Crystals (FUJIFILM Wako Pure Chemical Corporation, catalog number: 169-22445)
- Chloroform (FUJIFILM Wako Pure Chemical Corporation, catalog number: 035-02616)
- Isoamyl alcohol (3-Methyl-1-butanol) (FUJIFILM Wako Pure Chemical Corporation, catalog number: 135-12015)
- Trizma® base Primary Standard and Buffer, crystalline (Sigma-Aldrich, catalog number: T1503-500G)
- HCl (FUJIFILM Wako Pure Chemical Corporation, catalog number: 081-03475)
- Sodium dodecyl sulfate (SDS) (FUJIFILM Wako Pure Chemical Corporation, catalog number: 194-13985)
- NaOH (FUJIFILM Wako Pure Chemical Corporation, catalog number:199-4895)
- 8-quinolinol (FUJIFILM Wako Pure Chemical Corporation, catalog number: 178-00752)
- Agarose ME (Iwai Chemical Company, catalog number: 50013R)
- Wizard SV gel and PCR clean-up system (Promega, catalog number: A9281)
- Polyethylene glycol 4000 (FUJIFILM Wako Pure Chemical Corporation, catalog number: 5322-68-3)
- Cornstarch (Kawamitsu-Bussan, catalog number: 4901 486 02701 6)
- Top starch solution (Takemura et al., 2019)
- Gellan gum (FUJIFILM Wako Pure Chemical Corporation, catalog number: 073-03071)
- Uracil (Sigma-Aldrich, catalog number: U0750-5G)
- 5-Fluoroorotic acid monohydrate (5-FOA) (FUJIFILM Wako Pure Chemical Corporation, catalog number: 066-03663)
- Sodium acetate (FUJIFILM Wako Pure Chemical Corporation, catalog number: 190-01071)
- Ethanol (FUJIFILM Wako Pure Chemical Corporation, catalog number: 055-00457)
- (NH4)2SO4 (FUJIFILM Wako Pure Chemical Corporation, catalog number: 019-03435)
- MgSO4∙7H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 138-00415)
- H2SO4 (FUJIFILM Wako Pure Chemical Corporation, catalog number: 192-04696)
- H3BO3 (FUJIFILM Wako Pure Chemical Corporation, catalog number: 021-02195)
- MnCl2∙4H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 133-00725)
- ZnCl2 (FUJIFILM Wako Pure Chemical Corporation, catalog number: 268-01022)
- Na2MoO4∙2H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 198-02471)
- CoCl2∙6H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 038-03681)
- CuCl2∙2H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 039-04135)
- KH2PO4 (FUJIFILM Wako Pure Chemical Corporation, catalog number: 169-04245)
- CaCl2∙2H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 038-19735)
- FeCl3∙6H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 090-02802)
- Na2EDTA (FUJIFILM Wako Pure Chemical Corporation, catalog number: 345-01865)
- DNA solution (see Recipes)
- PEG solution (see Recipes)
- MA2 medium (Kobayashi et al., 2010, see Recipes)
- MA2 solution I
- A6 minor salts
- MA2 solution II
- MA2 solution III
- MA2 solution IV
- MA2 solid gellan gum plate (see Recipes)
- DNA extraction buffer (see Recipes)
- Neutralized phenol (see Recipes)
Equipment
- Pipettes (Gilson, Inc., catalog numbers: F123600, F123601, F123602)
- Refrigerated centrifuge (Koki Holdings Co., Ltd., model: CF16RXII)
- Microcentrifuge (TOMY Seiko Co., Ltd., model: MX150)
- 500 ml flask (IWAI, catalog number: 4442FK500)
- Heat Block (Chiyoda Science Co., Ltd., model: MiniT-100)
- Bio incubator (TOMY Seiko Co., Ltd., model: CLE-303)
- NanoDrop (Thermo Fisher Scientific Inc., model: ND-LITE)
- Thermal Cycler (Bio-Rad Laboratories, Inc., model: T100 Thermal Cycler)
- Electrophoresis apparatus (Atto Corporation, model: WSE-1710)
- FAS gel imager (Nippon Genetics Co., Ltd., model: FAS-IV)
- Spectrophotometer (Beckman coulter, Inc., model: DU730, A23616)
Procedure
- Construction of the introducing DNA fragment
Overview of the construction: PCR-amplified DNA fragments (Upstream and Downstream of homologous regions for the genome-specific locus) and StuI-digested pMKT (including two fragments Digested vector L and S) are connected using the In-fusion system to generate the pMKT-UPandDOWN. The resultant plasmid is used as the template DNA for amplification of the introducing DNA fragment by PCR (Figure 2).
PCR amplification of DNA fragments (Figure 2A):- Specific primer sets are designed as given below to amplify the DNA fragments (500-1,500 bp each) for homologous recombination to the genome-specific locus. Upstream and Downstream DNA fragments do not need to share the same length. For tagging, the upstream DNA fragment should include an open reading frame devoid of the termination codon, which is fused in frame to the epitope tag. Each colored region (20 bp) is the homologous sequence of the end of relevant StuI-digested pMKT (The colors correspond to Figure 2A) and each repeated ‘Ns’ (20-25 bp) indicates the annealing sequence to amplify the Upstream and Downstream DNA fragments. The Tm of the annealing sequence (‘Ns’) of each primer should be designed to be above 63 °C.
Upstream Forward (UF):
5’-GACACGTGAATTTAAATAGGNNNNNNNNNNNNNNNNNNNN-3’
Upstream Reverse (UR):
5’-TGAAAATAAAGATTTTCAGGNNNNNNNNNNNNNNNNNNNN-3’
Downstream Forward (DF):
5’-AGGGATTGTGGCGCGACAGGNNNNNNNNNNNNNNNNNNNN-3’
Downstream Reverse (DR):
5’-GACACGTGAATTTAAATAGGNNNNNNNNNNNNNNNNNNNN-3’ - PCR reactions are carried out using KOD-Plus-Neo DNA polymerase under the following reaction conditions: 94 °C for 2 min, followed by 30 cycles of 10 s at 98 °C, 30 s/1 kb at 68 °C, and a final extension of 2 min/1 kb at 68 °C with specific primers sets (UF/UR for Upstream and DF/DR for Downstream) and 50 ng wild type C. merolae DNA as a template. PCR-amplified DNA fragments are purified using the Wizard SV Gel and PCR Clean-Up System following the manufacturer’s protocol, and the concentration of DNA is measured.
Note: We routinely obtain reproducible amplification results by the above condition. The PCR results should be checked by gel electrophoresis to confirm the amplification.
- Specific primer sets are designed as given below to amplify the DNA fragments (500-1,500 bp each) for homologous recombination to the genome-specific locus. Upstream and Downstream DNA fragments do not need to share the same length. For tagging, the upstream DNA fragment should include an open reading frame devoid of the termination codon, which is fused in frame to the epitope tag. Each colored region (20 bp) is the homologous sequence of the end of relevant StuI-digested pMKT (The colors correspond to Figure 2A) and each repeated ‘Ns’ (20-25 bp) indicates the annealing sequence to amplify the Upstream and Downstream DNA fragments. The Tm of the annealing sequence (‘Ns’) of each primer should be designed to be above 63 °C.
- pMKT vector digestion by StuI to obtain two DNA fragments, here named Digested vector L (Long, approximately 3.6 kbp) and S (Short, approximately 3.0 kbp, Figure 2B)
- Incubate 1 μg of pMKT DNA in 50 μl 1x CutSmart Buffer (i.e., provided buffer of StuI) containing 10 units of StuI at 37 °C for 1 h.
- Add 100 μl of dH2O and 150 μl of Phenol:Chloroform:Isoamyl alcohol = 25:24:1 solution and mix vigorously. Centrifuge at 12,000 x g for 10 min at room temperature. Take 150 μl of supernatant and add 15 μl of 3 M sodium acetate and 375 μl of 100% EtOH. Mix vigorously and centrifuge at 12,000 x g for 30 min at 4 °C. Discard the supernatant, add 75% EtOH to the precipitate, and centrifuge at 12,000 x g for 5 min at 4 °C. Discard the supernatant, completely remove the remaining drops by pipette after flush centrifugation, and air dry the pellet briefly on the bench at room temperature. Then add 20 μl of dH2O and dissolve the precipitated DNA. Measure the DNA concentration.
Note: When using phenol-chloroform-isoamyl alcohol, personal protective equipment needs to be used according to local health and safety regulations. Always work under a fume hood.
- In-fusion reaction to generate the template plasmid (Figure 2C)
- Gently mix 5 μl of DNA mixture [StuI-digested pMKT (Digested vector L and S):upstream:downstream ≈ 100 fmol:50 fmol:50 fmol] and 5 μl of 2x In-fusion reaction mix. Incubate the reaction mixture at 50 °C for 15 min and then cool it down on ice.
- Transform Escherichia coli DH5α with the reaction mixture by using a chemical transformation method as common use. Find the expected plasmid construct by colony PCR. PCR reactions are carried out using GoTaq DNA polymerase under the following reaction conditions: 95 °C for 3 min, followed by 30 cycles of 30 s at 95 °C, 30 s at 55 °C, 1 min/1 kb at 72 °C, and a final extension of 2 min at 72 °C with primers sets (Forward primer for up: 5’-CGAGGTCGACGGTATCGATAAGC-3’/Reverse primer for up: 5’-GATCTAGTAACATAGATGACACCGC-3’ for the upstream sequence, and Forward primer for down: 5’-GCTGCTAGGGATTGTGGCGCGA-3’/Reverse primer for down: 5’-CCGCTCTAGAACTAGTGGATCCC-3’ for the downstream sequence). Subsequently, the positive candidates are examined by DNA sequence analysis with primer sets for colony PCR.
- Extract and purify the plasmid DNA (pMKT-UPandDOWN).
- Amplification of DNA for transformation by PCR to prepare the linearized DNA fragment and completely remove the template circular plasmid DNA (Figure 2D)
Note: Inclusion of the template plasmid DNA frequently may result in random integration of the plasmid into the genome. Thus, the template plasmid DNA should be carefully removed.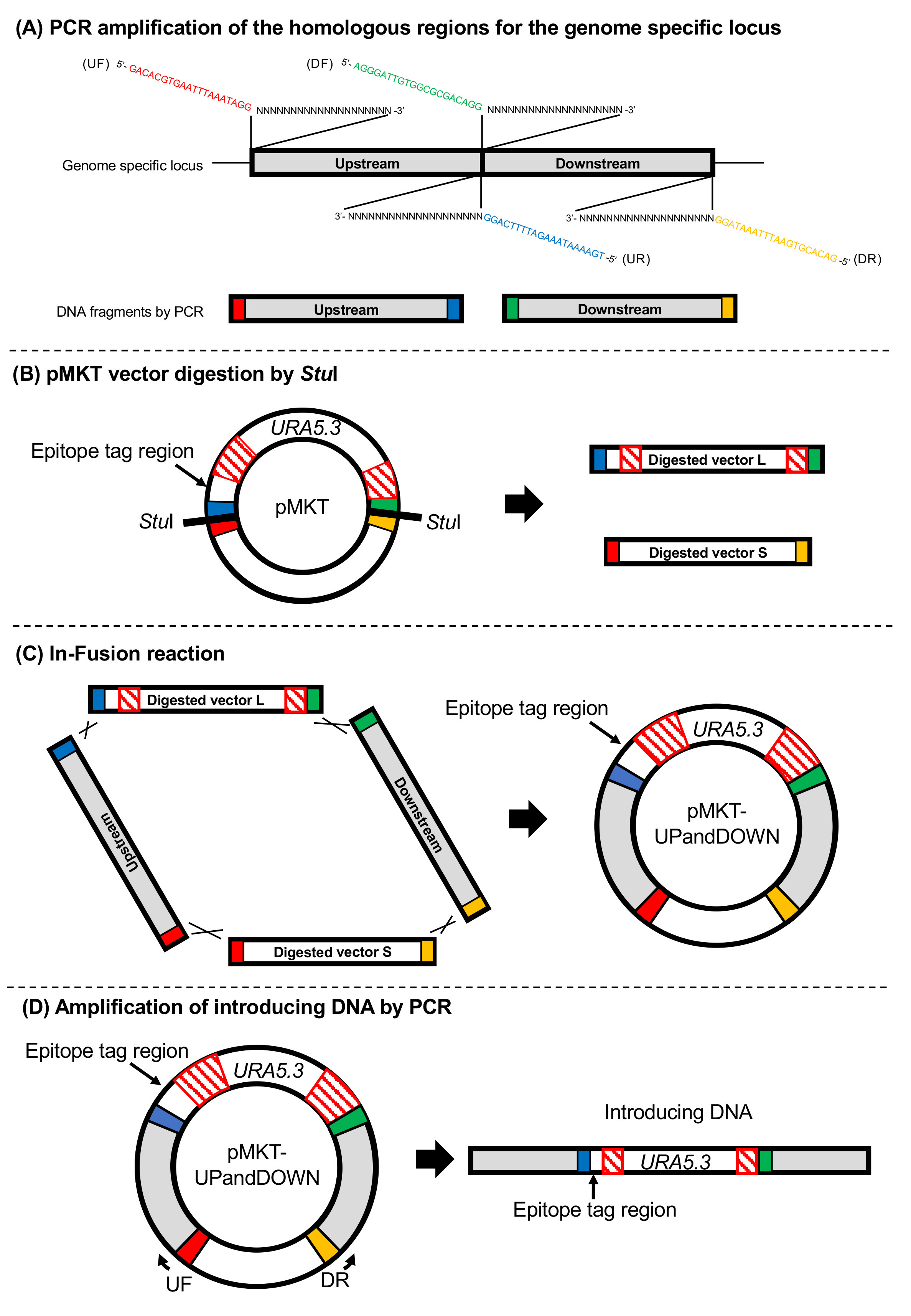
Figure 2. Overview of the template vector (pMKT-UPandDOWN) and DNA constructs for transformation. A. DNA regions (Upstream and Downstream) corresponding to the target gene are amplified by PCR with the specific primer sets having the 20 bp homologous sequence for the pMKT (shown as red, blue, green and yellow). B. Digestion of the pMKT vector with StuI enzyme generates 2 DNA fragments named Digested vectors L and S after their lengths (L-long; S-short). The red hatched areas indicate the homologous repeat sequence (i.e., URA5.3 terminator region). C. Four DNA fragments (Digested vector L, Digested vector S, Upstream and Downstream) are connected in one reaction tube using the In-fusion system, to make a plasmid pMKT-UPandDOWN). D. The DNA fragment for transformation is amplified by PCR with the primer set UF and DR using pMKT-UPandDOWN as the template.- PCR is carried out as described in Step A2 with the specific primer sets (Upstream Forward: UF and Downstream Reverse: DR) and pMKT-UPandDOWN as the template.
- To digest the plasmid DNA, treat the amplified PCR mix with 10 units of DpnI at 37 °C for 1 h.
- Purify the specific band using the Wizard SV Gel and PCR Clean-Up System after 1.2% agarose electrophoresis (applying a voltage of 100V for 30 min). Prepare 2-5 μg of DNA fragments for transformation in 40 μl of dH2O.
- Transformation of C. merolae cells
Transform the C. merolae T1 cells following the polyethylene glycol-mediated protocol as described by Ohnuma et al. (2008) and Fujiwara and Ohnuma (2018).- When OD750 reaches ~10, dilute the cell culture in 50 ml MA2 medium to yield an OD750 of 0.4 and cultivate in the glass cylinders (3 cm in diameter and 35 cm in length) with cotton plug bubbled with 2% CO2 in air under constant light (20 μmol photons m-2 s-1) at 40 °C for 20-24 h.
- Harvest the cells (OD750 < 0.7) by centrifugation (1,200 x g, for 5 min at 40 °C), and discard the supernatant. Resuspend the pellet with 200-300 μl of MA2 medium.
- Combine 2-5 μg of the DNA fragment in 40 μl of dH2O and 60 μl of DNA solution (see Recipes).
- Add 25 μl of the resuspended cells to the tube and mix gently.
- Add 125 μl of the PEG solution (see Recipes) immediately and mix the liquid by gently pipetting 4-5 times using the 200 μl pipette.
- Transfer the mixture to 30-40 ml of MA2 medium and incubate under the same condition as described in Step E1 for 20-24 h.
- Harvest the cultured cells by centrifugation (1,200 x g, for 5 min at 40 °C) and spread the cells onto the MA2 solid gellan gum plate without uracil and 5-FOA by the top starch method to increase the plating efficiency (Takemura et al., 2019). Incubate the plate at 40 °C supplemented with 5% CO2 in AnaeroPack with AnaeroPouch.
- After 2-4 weeks, colonies will appear on the surface of the MA2 solid gellan gum plate.
- Pick up well-separated colonies (usually 20-24 candidates) using sterilized toothpicks, and inoculate 2 ml of MA2 liquid medium prepared in a 24-well tissue culture test plate.
- After incubation for 1-2 weeks in the same incubator without agitation, harvest the 20-24 candidate colonies and extract the DNA as described below (Kobayashi et al., 2010). To select the positive transformants, perform the PCR with the DNA using the ‘‘Confirmation primer sets’’ to anneal to the outside sequence of the homologous regions used to introduce the DNA fragment (Figure 3). PCR reactions are carried out using KOD-Plus-Neo DNA polymerase under the following reaction conditions: 94 °C for 2 min, followed by 30 cycles of 10 s at 98 °C, 30 s/1 kb at 68 °C, and a final extension of 2 min/1 kb at 68 °C.
- DNA extraction from C. merolae cells
- Harvest C. merolae cells by centrifugation at 3,000 x g for 5 min at 4 °C, and discard the medium.
- Resuspend the cell pellets with 400 μl of DNA extraction buffer [50 mM Tris-HCl (pH = 8.0), 5 mM EDTA, 0.5% Sodium dodecyl sulfate].
- Add 400 μl of neutralized phenol.
- Incubate at 65 °C for 10 min with shaking every 2 min by hand vigorously.
Note: A vortex should not be used. - Centrifuge at 12,000 x g for 5 min at room temperature. Transfer 375 μl of the above aqueous phase into a fresh tube, and add 375 μl of PCI (neutralized phenol:chloroform:isoamyl alcohol = 25:24:1). Invert the tube by hand vigorously.
- Centrifuge at 12,000 x g for 5 min at room temperature. Transfer 350 μl of the above aqueous phase into a fresh tube, and add 350 μl of PCI. Invert the tube by hand vigorously.
- Centrifuge at 12,000 x g for 5 min at room temperature. Transfer 300 μl of the above aqueous phase into a fresh tube, and add 30 μl of the 3 M sodium acetate and 750 μl of 100% ethanol. Invert the tube by hand vigorously.
- Centrifuge at 12,000 x g for 30 min at 4 °C. Discard the supernatant and add 200 μl of 75% ethanol.
- Centrifuge at 12,000 x g for 5 min at 4 °C. Discard the supernatant and let the liquid evaporate completely.
- Dissolve the precipitate of DNA in distilled water.
Note: When using phenol-chloroform-isoamyl alcohol, personal protective equipment needs to be used according to local health and safety regulations. Always work under a fume hood.
- Elimination of URA5.3 gene marker from the genome
- The transformant cell using URA5.3T as a gene selectable marker is cultured in 50 ml of MA2 medium until the OD750 reaches 0.5-5.0.
- Centrifuge 50 ml of the culture at 1,200 x g for 5 min at 40 °C.
- Discard the supernatant, add 1.5 ml of MA2 medium and resuspend the precipitated cells.
- Add 1 ml of the top starch solution (Takemura et al., 2019) to 1 ml of the cell suspension, mix gently, and spread onto the MA2 solid gellan gum plate containing 0.5 mg/ml uracil and 0.8 mg/ml 5-FOA.
- Add 1 ml of MA2 medium to the remaining cell suspension. Take 1 ml of the diluted cell suspension, and spread onto the MA2 solid gellan gum plate as in Step F4.
- After 2-4 weeks, the colonies of the URA5.3 eliminated cells appear on the MA2 solid gellan gum plate.
- Pick up well-separated colonies (usually 20-24 candidates) using sterilized toothpicks, and inoculate each in 2 ml of MA2 liquid medium prepared in 24-well tissue culture test plate.
- After incubation for 1-2 weeks in the same incubator, extract DNA from each green colored inoculate as described in Step E10. To confirm URA5.3 elimination, perform the PCR with the DNA using the “Confirmation primer sets” to anneal to the outside sequence of the homologous regions used to introduce the DNA fragment as described in Step E10 (Figure 3).
- After confirmation of URA5.3 elimination by PCR, the strain is ready for the next transformation experiment using URA5.3T as a gene selectable marker.
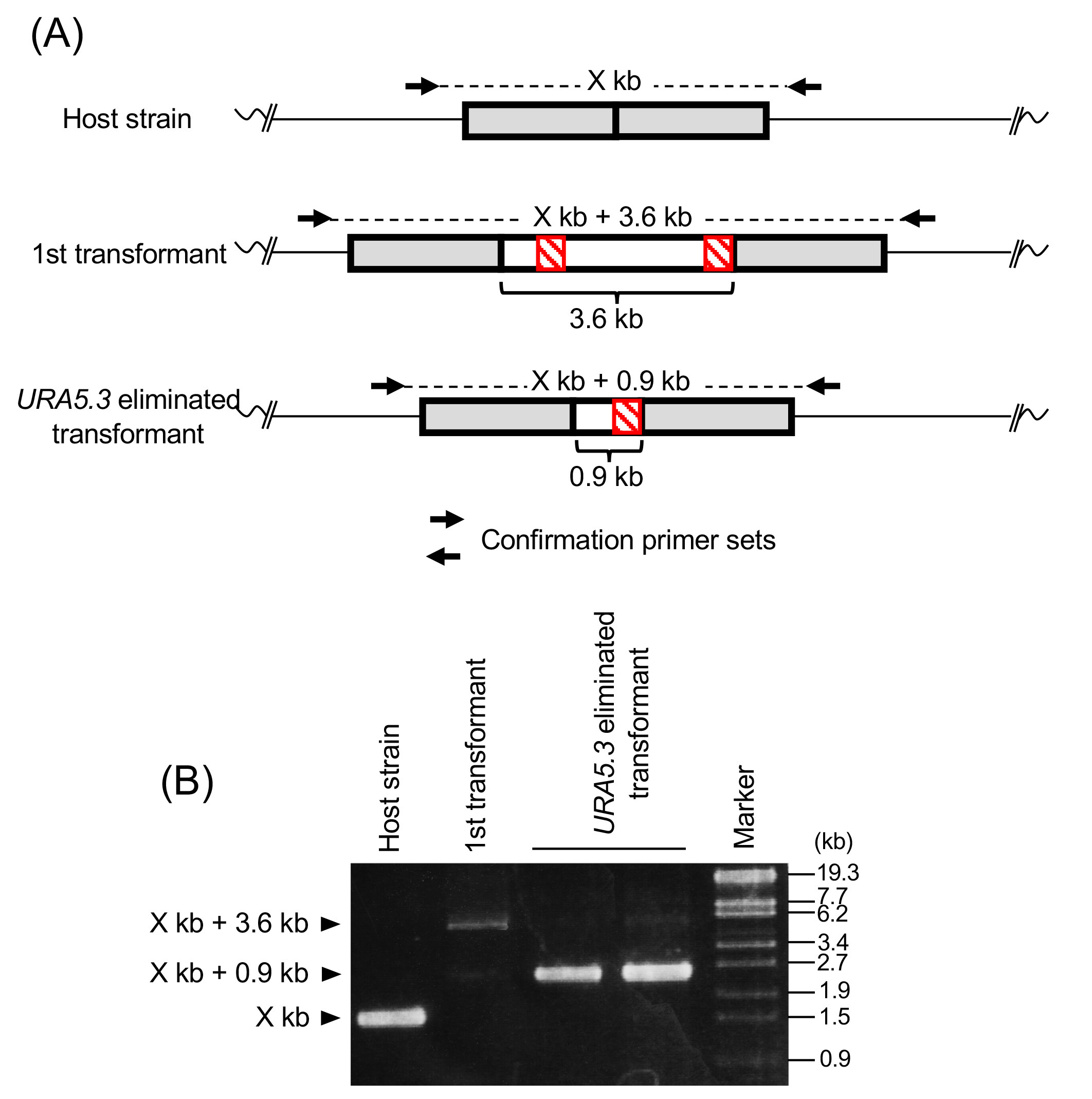
Figure 3. Schematic representation of confirmation of the transformation and URA5.3 elimination by PCR. A. Schematic representation of the modified gene loci of the host strain, 1st transformant and URA5.3 eliminated transformant. The arrows indicate the specific primer sets for confirmation of the transformation (named “Confirmation primer sets”). These primers should be designed to anneal to the outside sequence of the homologous regions. B. Example of gel electrophoresis image of PCR to confirm the expected recombination events using confirmation primer sets.
Recipes
- DNA solution
- Mix 34 μl of dH2O and 6 μl of 10x MA2 solution I in a 1.5 ml microtube
- Add 20 μl of 10 mg/ml salmon sperm DNA to the above microtube
- Incubate the microtube at 40 °C until use
- PEG solution
- Take 0.6 g of polyethylene glycol 4000 in a 1.5 ml microtube
- Add 470 μl of 10x MA2 solution I to the microtube
- Incubate the microtube at 65 °C for 10 min shaking every 2 min by hand
- MA2 medium
- Mix 100 ml of MA2 solution I, 10 ml of MA2 solution II, 1 ml of MA2 solution III and 885 ml of dH2O to a glass bottle and sterilize by autoclaving at 121 °C for 20 min
- Add 4 ml of MA2 solution IV to the mixture
MA2 solution I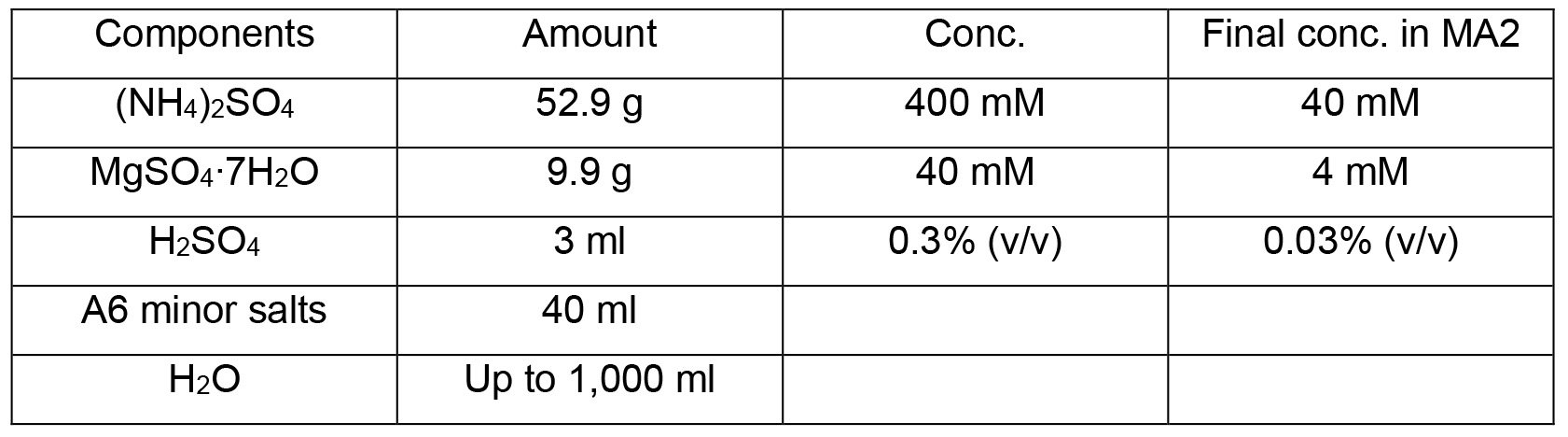
A6 minor salts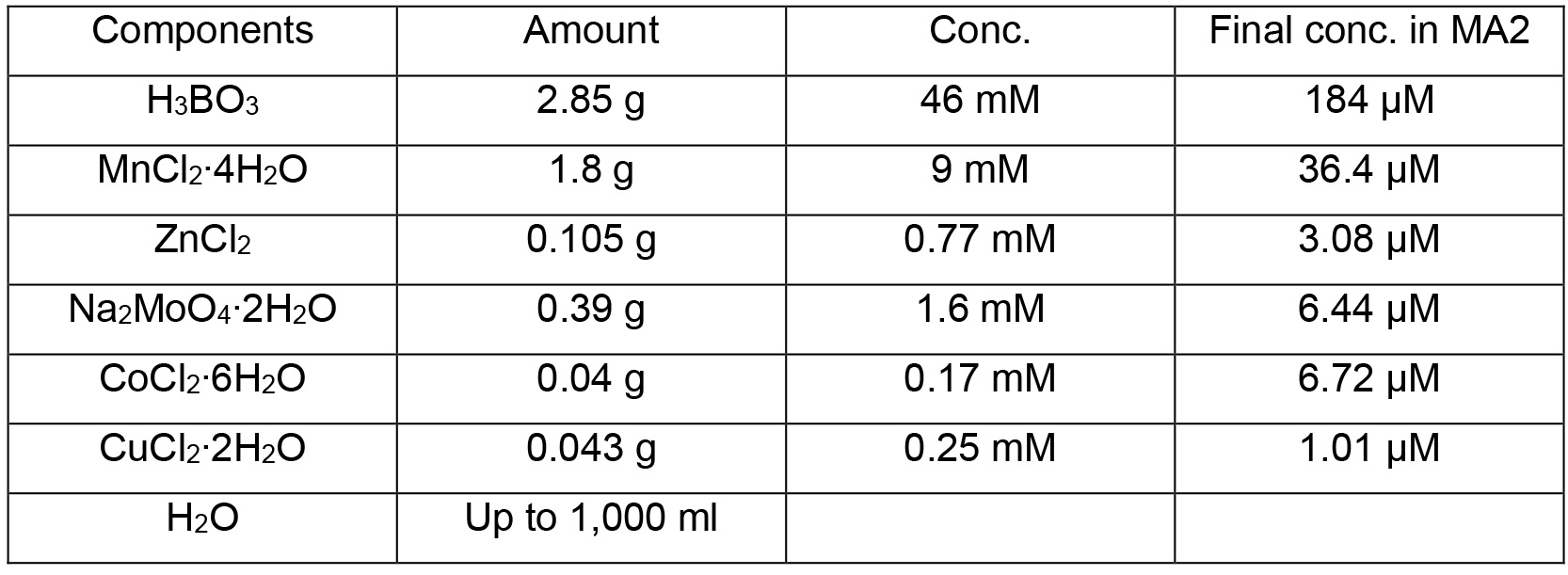
MA2 solution II
MA2 solution III
MA2 solution IV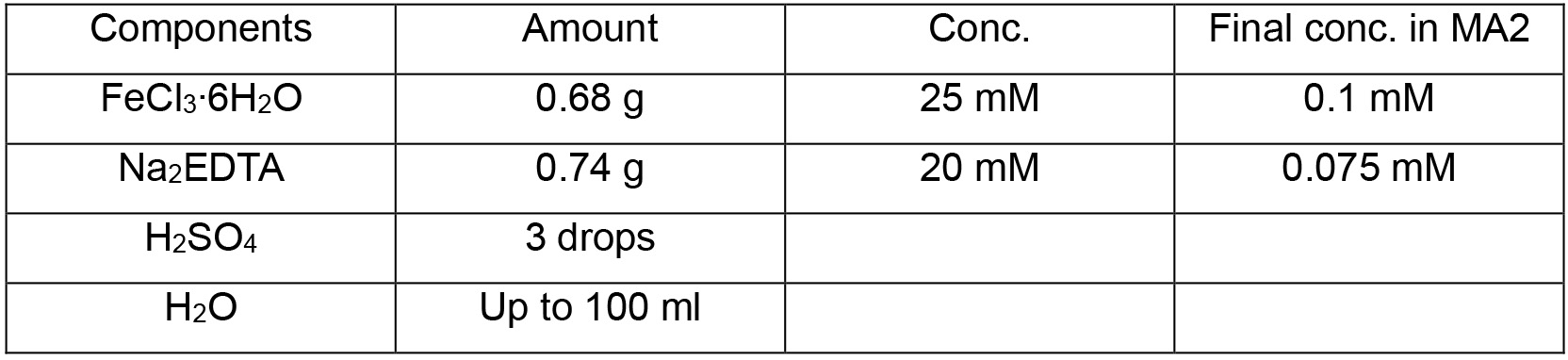
Note: This MA2 solution IV should be sterilized by filtration and stored at 4 °C in the dark.
- MA2 solid gellan gum plate (for making 9-10 plates)
- Mix 30 ml of MA2 solution I, 3 ml of MA2 solution II, 300 μl of MA2 solution III, and 70 ml of dH2O in a 500 ml flask. Mix 200 ml of dH2O and 1.5 g gellan gum in another 500 ml flask. Sterilize these flasks by autoclaving
- After autoclaving, mix each solution with 1.2 ml of MA2 solution IV in one flask. In case of 5-FOA selection, add directly 150 mg of uracil (final concentration: 0.5 mg/ml) and 240 mg of 5-FOA (final concentration: 0.8 mg/ml) in powder to the flask
- Pour the mixed solution onto the sterilized plastic plates
- Dry the plates at room temperature for 15 min
- If plates are to be stored, they should be kept in the dark at 4 °C
- DNA extraction buffer
- Dissolve 12.1 g of Trizma® base to 100 ml of dH2O in a glass bottle and adjust pH to 8.0 with HCl to make 1 M Tris-HCl (pH = 8.0) solution. Sterilize by autoclaving
- Dissolve 10.0 g of sodium dodecyl sulfate to 100 ml of dH2O in a glass bottle to make 10% SDS solution
- Dissolve 18.6 g of EDTA∙2Na to 100 ml of dH2O in a glass bottle and adjust pH to 8.0 with NaOH to make 0.5 M EDTA solution. Sterilize by autoclaving
- Mix 5 ml of 1 M Tris-HCl (pH = 8.0) solution, 5 ml of 10% SDS solution, 1 ml of 0.5 M EDTA solution and 89 ml of dH2O in a glass bottle
- Store at room temperature
- Neutralized phenol
- Dissolve 100 g of phenol in a 200 ml glass bottle by heating at 65 °C
- After dissolving phenol, add 0.1 g of 8-quinolinol to phenol
- Add the mixture of 50 ml of 1 M Tris-HCl (pH = 8.0) solution and 50 ml of dH2O to the phenol and mix well for 5 min at room temperature
- After mixing, incubate the phenol mixture for 10 min at room temperature
- Remove the 80 ml of the upper phase (liquid layer)
- Add the mixture of 50 ml of 1 M Tris-HCl (pH = 8.0) solution and 50 ml of dH2O to the phenol and mix well for 5 min at room temperature
- After mixing, incubate the phenol mixture for 10 min at room temperature
- Remove the 100 ml of the upper phase (liquid layer)
- Store at 4 °C in the dark
Acknowledgments
This protocol was adapted from (Takemura et al., 2018 and 2019). The authors thank the Biomaterials Analysis Division, Tokyo Institute for Technology for DNA sequence analysis. This study was supported by MEXT/JSPS KAKENHI (Grant numbers: 15K14539 to K.T., 17K07438 to S.I., 17K07439 to Y.K.) and by Advanced Low Carbon Technology Research and Development Program (ALCA) of Japan Science and Technology Agency (JST) to K.T.
Competing interests
No competing interests declared.
References
- Fujiwara, T., Ohnuma, M., Kuroiwa, T., Ohbayashi, R., Hirooka, S. and Miyagishima, S. Y. (2017). Development of a double nuclear gene-targeting method by two-step transformation based on a newly established chloramphenicol-selection system in the red alga Cyanidioschyzon merolae. Front Plant Sci 8: 343.
- Kobayashi, Y., Ohnuma, M., Kuroiwa, T., Tanaka, K., and Hanaoka, M. (2010) The basics of cultivation and molecular genetic analysis of the unicellular red alga Cyanidioschyzon merolae. Endocytobiosis Cell Res 20: 53-61.
- Minoda, A., Sakagami, R., Yagisawa, F., Kuroiwa, T. and Tanaka, K. (2004). Improvement of culture conditions and evidence for nuclear transformation by homologous recombination in a red alga, Cyanidioschyzon merolae 10D. Plant Cell Physiol 45(6): 667-671.
- Fujiwara, T. and Ohnuma, M. (2018). Procedures for transformation and their applications in Cyanidioschyzon merolae. In: Kuroiwa, T., Miyagishima, S.Y., Matsunaga, S., Sato, N., Nozaki, H., Tanaka, K. and Misumi, O. (Eds.). Cyanidioschyzon merolae. Singapore, Springer Singapore: 87-103.
- Ohnuma, M., Yokoyama, T., Inouye, T., Sekine, Y. and Tanaka, K. (2008). Polyethylene glycol (PEG)-mediated transient gene expression in a red alga, Cyanidioschyzon merolae 10D. Plant Cell Physiol 49(1): 117-120.
- Takemura, T., Imamura, S., Kobayashi, Y. and Tanaka, K. (2018). Construction of a selectable marker recycling system and the use in epitope tagging of multiple nuclear genes in the unicellular red alga Cyanidioschyzon merolae. Plant Cell Physiol 59(11): 2308-2316.
- Takemura, T., Imamura, S., Kobayashi, Y., and Tanaka, K. (2019). Top starch plating method for the efficient cultivation of unicellular red alga Cyanidioschyzon merolae. Bio-protocol 9(4): e3172.
- Taki, K., Sone, T., Kobayashi, Y., Watanabe, S., Imamura, S. and Tanaka, K. (2015). Construction of a URA5.3 deletion strain of the unicellular red alga Cyanidioschyzon merolae: A backgroundless host strain for transformation experiments. J Gen Appl Microbiol 61(5): 211-214.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Takemura, T., Imamura, S., Kobayashi, Y. and Tanaka, K. (2019). Multiple Modification of Chromosomal Loci Using URA5.3 Selection Marker in the Unicellular Red Alga Cyanidioschyzon merolae. Bio-protocol 9(7): e3204. DOI: 10.21769/BioProtoc.3204.
Category
Microbiology > Microbial genetics > Genome editing
Plant Science > Phycology > DNA > Genome editing
Molecular Biology > DNA > DNA cloning
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.