- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantification of Queuosine Modification Levels in tRNA from Human Cells Using APB Gel and Northern Blot
Published: Vol 9, Iss 6, Mar 20, 2019 DOI: 10.21769/BioProtoc.3191 Views: 6871
Reviewed by: Gal HaimovichJoshua S TitlowAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Queuosine (Q) is a hypermodified base in the wobble anticodon position of tRNAs coding for the amino acids Tyr, His, Asn, and Asp. tRNA Q-modification is introduced by a queuine tRNA-ribosyltransferase (TGT) that replaces the guanine base at G34 at these tRNAs with the modified base. tRNA Q-modification is widely distributed among prokaryotic and eukaryotic organisms, but only bacteria synthesize Q-modified tRNA de novo. In mammals, tRNA Q-modifications strictly rely on the presence of gut microbiomes or diets to produce the queuine base. Despite decades of study, cellular roles of tRNA Q-modification are still not fully understood. Here we describe a method to quantify tRNA Q-modification levels in individual tRNAs from human cells based on the presence of a cis-diol in the Q modification. This cis-diol moiety slows modified tRNA migration through polyacrylamide gels supplemented with N-acryloyl-3-aminophenylboronic acid (APB) compared to the unmodified tRNA. This difference can be visualized by Northern blots using probes for specific tRNA.
Keywords: QueuosineBackground
Transfer RNAs (tRNA) contain nucleotide modifications that perform a wide range of roles in translation and in the generation of tRNA fragments that are functional small RNAs. Queuosine (Q, Figure 1) is a tRNA modification that occurs in the wobble anticodon position of tRNAs coding for amino acids Tyr, His, Asn, and Asp. All these tRNAs contain G34U35N36 anticodon sequences. In bacteria, G34-to-Q34 modification can be synthesized de novo through a cascade of enzyme reactions (El Yacoubi et al., 2012). In mammals, the G34 base in these tRNAs can be replaced with queuine (Figure 1) derived from either gut microbiome or diet by a two-protein complex enzyme QTRT1 (catalytic subunit) and QTRT2 encoded in the host genome. Q is widely distributed among prokaryotic and eukaryotic organisms, but its cellular roles are still not fully understood (Rakovich et al., 2011; Fergus et al., 2015; Tuorto et al., 2018; Wang et al., 2018). For example, tRNA Q-modification levels have been correlated with cancer progression and cellular metabolism.
To understand the role of Q-tRNA modification in mammalian cells, we performed comparative studies of cells having various levels of Q-modification (Wang et al., 2018). We found that tRNAs in cultured cell lines are Q-modified at 5%-60% in standard culture medium. Using dialyzed serum medium which was deprived of small molecules like queuine and continued passage of cell divisions, tRNA Q-modification levels continuously decreased until they became undetectable (0Q cells). The addition of 0.1-1 µM queuine to the media restored Q-modification to 100% levels within 12-24 h (100Q cells). To measure the level of tRNA Q-modification in individual tRNA, we used a previously developed method on the basis of the structure of queuosine (Igloi and Kossel, 1985; Zaborske et al., 2014). Cis-diol moieties, such as the 3’ ribose of a tRNA, slow migration through polyacrylamide gels supplemented with N-acryloyl-3-aminophenylboronic acid (APB). Since queuosine has an additional ribose moiety, the migration of Q-modified tRNA in APB gel is slower compared to unmodified tRNA. This differential migration of Q-modified versus unmodified tRNA can be determined by Northern blot analysis using probes specific for each tRNA. The mammalian tRNAAsp and tRNATyr are further glycosylated at the cis-diol of the modified base with mannose (man-Q) or galactose (gal-Q) which prevents the reaction between the Q-base and APB; our approach therefore only works for tRNAHis and tRNAAsn in mammalian cells. Using this method and other techniques, we showed that Q-modification protects cognate tRNAs against ribonuclease cleavage in vitro and in cells which alters the abundance of tRNA fragments (tRF) in human cells (Wang et al., 2018).
Previously, the Q-content in total tRNA in mammalian cells was evaluated by taking advantage of the irreversible nature of the N-glycosidic bond of the Q-base to the sugar-ribose backbone. Total tRNAs were isolated and the extent of Q-modified in total tRNA was measured by the exchange of 3H-guanine with the guanine base of unmodified tRNAs catalyzed by the E. coli TGT enzyme. This exchange reaction was blocked by the Q-modification of the same tRNAs. Total Q-content was estimated based on the fraction of tRNAs that did not show exchange. This approach was first used to determine the Q-modification content in mammalian tissues and bacterial cells during different stages of development and aging (Singhal et al., 1981). Although useful, this previous method could not measure Q-modification level of individual tRNA. In contrast, our APB-gel based method directly measures the Q-modification levels of individual tRNA species starting with as little as 1-2 µg of total RNA.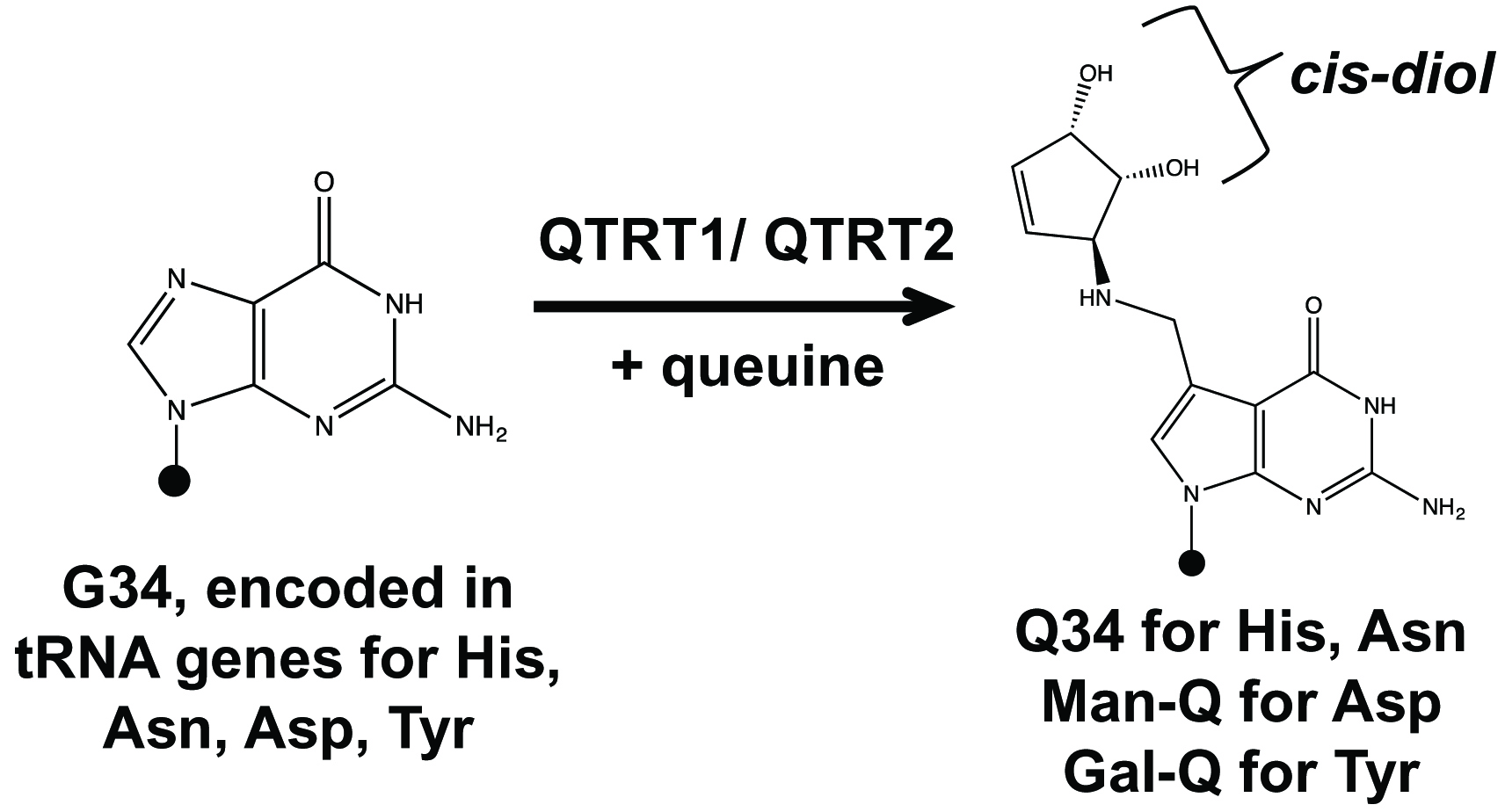
Figure 1. Queuosine (Q) tRNA modification in four tRNAs. Diagram of G34 to Q34 modifications in the wobble anticodon position of tRNAHis/Asn/Asp/Tyr. Queuine corresponds to the nucleobase of Q-nucleotide. QTRT1 and QTRT2 (formerly QTRTD1) proteins are encoded in the human genome. Black dots represent the tRNA body; the QTRT1/QTRT2 complex introduces Q modification in the place of G using queuine as substrate. Q34 in tRNAAsp and tRNATyr are further glycosylated at the cis-diol of the modified base with mannose (man-Q) or galactose (gal-Q) in mammals. Our method measures tRNAHis and tRNAAsn, but not tRNAAsp and tRNATyr because of their glycosylation.
Materials and Reagents
Note: All reagents were purchased from Thermo Fisher if not indicated otherwise and were stored at room temperature.
- Pipette tips
- Plastic foil
- Syringe
- Gel spacers 0.8 mm (Cole-Parmer, UX-28573-12)
- Hybridization tubes 2 x 200 mm (VWR, catalog number: 32645-030)
- Whatman paper (WhatmanTM, 10427812 Grade)
- Hybond-XL membrane (GE Healthcare, catalog number: 45-001-148)
- 5’ 32P-labeled oligonucleotide probes (see Procedure B)
- [γ-32P] ATP (PerkinElmer, catalog number: NEG035C005MC) (storage: -20 °C)
- T4 Polynucleotide kinase (10 U/µl) (Thermo Fisher, catalog number: EK0032) (storage: -20 °C)
- 1x T4 PNK buffer
- 3-Acrylamidophenylboronic acid (Frontier Scientific, catalog number: A10094-5g) (storage: 4 °C)
- 40% acrylamide/bis solution, 29:1 (Bio-Rad, catalog number: 161-0146)
- Acetic acid
- DMEM (Thermo Fisher, catalog number: 11965092)
- EDTA
- Ethanol, 95.5% USP (Ricca, catalog number: R2916000-1A)
- Fetal Bovine Serum dialyzed (Thermo Fisher, catalog number: 26400044)
- Queuine Hydrochloride (Toronto Research Chemicals, catalog number: Q525000)
- Sodium acetate
- Sodium chloride (NaCl)
- Sodium dodecyl sulfate (SDS)
- Sodium phosphate monobasic (NaH2PO4) (Thermo Fisher, catalog number: BP329)
- Sodium phosphate dibasic (Na2HPO4) (Thermo Fisher, catalog number: S374)
- 10x TAE buffer (Sigma-Aldrich, catalog number: 11666690001)
- Tris (Bioland Scientific, catalog number: CT01-5kg)
- Urea
- Bromophenol blue
- Xylene cyanol
- 10x TBE buffer (EMD Millipore, catalog number: 8850-OP)
- Sodium acetate (KOAc)
- Sodium chloride (KCl)
- Nuclease-free water
- 2x Gel loading buffer (see Recipes)
- 10% denaturing polyacrylamide gel (see Recipes)
- 10% APB polyacrylamide gel (see Recipes)
- Dialyzed serum medium (see Recipes)
- Gel extraction buffer (see Recipes)
- Hybridization buffer (see Recipes)
- 1 M Sodium phosphate buffer (pH 7.0) (see Recipes)
- 1 M Tris-HCl buffer (pH 9.0) (see Recipes)
- Washing solution (see Recipes)
Equipment
- Pipettes
- Microcentrifuge (Eppendorf, model: 5415D)
- Hybridization Oven (UVP, model: 95-0030-01)
- Phosphoimager (Bio-Rad, Molecular Imager Pharosfx Plus System, 170-9460)
- Phosphor screens (Bio-Rad or Fuji)
- X-ray Cassette K 35 x 43 cm (Bio-Rad)
- Polyacrylamide gel electrophoresis apparatus with glass plates 17 x 15 cm (Biometra, Model V16-2)
- Thermoblock 24 x 1.5 ml (Eppendorf, 022670522)
- Tube rotator (Cole-Parmer Ltd, model: SB3)
- Vacuum gel dryer (Bio-Rad, Model 583 Gel Dryers)
- Incubator
- 4 °C refrigerator
- -20 °C freezer
Software
- Quantity One (Bio-Rad Laboratories, Inc., www.bio-rad.com/)
Procedure
- Human cell culture
- Obtaining Q-deprived (0Q) human cells
Culture your cells in dialyzed serum medium for at least five doublings. The Q-modification level is expected to decrease by a factor of 2 after each doubling. - Generating fully Q-modified (100Q) human cells
Add queuine hydrochloride solution to the cell culture media at a final concentration of 0.1-1 µM and incubate for at least 12 h.
Note: Conditions provided here were verified for HEK293T cells, these need to be optimized separately for each cell line used. - RNA extraction (described below)
- Obtaining Q-deprived (0Q) human cells
- 32P-labeling and purification of Northern probes
Before proceeding to the Northern blot analysis, prepare 5’ 32P-labeled oligonucleotide probes that are complementary to tRNA species of interest. If needed, prepare probes for a tRNA not modified with Q (e.g., tRNAAla) as a negative control.
Warning: [γ-32P] ATP is highly radioactive. Follow all safety procedures required in handling radioactive material (e.g., ordering, storage, personal protection, shielding, preventing and detecting contamination etc.). Contact the appropriate lab/department safety inspector for more information, also about available trainings.
Oligonucleotide probe sequences for human tRNAs are:
(tRNAHis)5’TGCCGTGACTCGGATTCGAACCGAGGTTGCTGCGGCCACAACGCAGAGTACTAACCACTATACGATCACGGC;
(tRNAAsn)5’CGTCCCTGGGTGGGCTCGAACCACCAACCTTTCGGTTAACAGCCGAACGCGCTAACCGATTGCGCCACAGAGAC;
Oligonucleotides can be ordered from DNA synthesizing company (e.g., IDT, gel purified is good enough).- Labeling the oligos with [γ-32P] ATP by T4 PNK reaction
- Use 20 pmol of each oligo for labeling.
- Set up the reaction with 1x T4 PNK buffer from the supplier, 6 U/µl T4 PNK, 28 µCi/µl [γ-32P] ATP and add water to the final volume of 10 µl.
- Incubate for 30 min at 37 °C in a thermoblock.
- Inactivate T4 PNK by adding 2x gel loading buffer.
- Gel purification of the probes
- Load the samples on 10% denaturing polyacrylamide gel.
- Run the gel at 18 W until the first dye BPB is at the bottom of the gel (typically about 1 h).
- Expose the gel to a phosphorimager screen for 1 min (this time can vary depending on the radioactivity of the probes).
- Based on the signal, excise the bands corresponding to the probe.
- Incubate crushed gel slices with gel extraction buffer and rotate the mixture at 4 °C overnight on a tube rotator.
- Precipitation of the probes
- Centrifuge the samples at maximum speed for 30 min and collect the solution from the gel slices above.
- Precipitate the DNA using 3 volumes of 100% ethanol at -20 °C for 2 h.
- Centrifuge the samples at maximum speed for 30 min and remove the supernatant.
- Wash the pellet with 70% ethanol.
- Centrifuge the samples at maximum speed for 5 min and remove the supernatant.
- Resuspend the pellet in 400 µl of nuclease-free water. Use 1/4 of the prepared probe for each blot (described below).
Note: Ideally, fluorescent probes could be used in place of 32P-labeled probes. However, we have not optimized the use of fluorescent probes ourselves. The sensitivity of fluorescent probes at this time can be quite a bit lower than 32P-probes, so more RNA samples might be needed.
- Labeling the oligos with [γ-32P] ATP by T4 PNK reaction
- APB gel separation of total RNA
- Total RNA extraction
- Extract total RNA from mammalian cells (e.g., Trizol method, see Reference 6)
Note: If using RNA extraction kit, choose one that does not lose small RNAs. - Measure the RNA concentration of your samples.
- Aliquot 5 µg or more of RNA for each analyzed sample. If not available, as low as 1 µg total RNA may be used. Keep the total volume of the RNA sample low, so the whole sample can fit in one lane after adding 2x gel loading buffer (final volume: about 20 µl, Step B2c).
- Extract total RNA from mammalian cells (e.g., Trizol method, see Reference 6)
- Deacylation of RNA to remove amino acids attached to the 3’ end of tRNAs
- Mix the RNA with 100 mM final concentration of Tris-HCl (pH 9.0).
- Incubate for 30 min at 37 °C in a thermoblock.
- Combine deacylated RNA samples with an equal volume of denaturing 2x gel loading buffer. (Store the samples at -20 °C if needed for future use)
- APB gel
- Prepare 10% urea denaturing polyacrylamide gel containing 5% 3-Acrylamidophenylboronic acid (adjust the volumes of the gel solution to the glass plates and gel spacers available in the lab).
- Pre-run the gel in 1x TAE buffer for 30 min (e.g., at constant 18 W using the electrophoresis apparatus listed in Equipment).
- Wash the wells of the gel using a syringe and 1x TAE buffer.
- Load your samples onto the gel.
- Run gel electrophoresis in 1x TAE buffer at 18 W until the first dye (BPB) reaches the bottom of the gel.
- Total RNA extraction
- Northern blots
- Gel transfer
- After electrophoresis, take apart the glass plates.
- Put a Hybond-XL membrane on the gel (mark the orientation of the gel).
- Take the gel off the glass plate and create a “sandwich” as follows (from the bottom): Whatman filter paper-membrane-gel-plastic foil.
- Transfer RNA onto the membrane using vacuum gel dryer for 2 h at 80 °C.
- Pre-hybridization
- Remove the membrane from the gel using distilled water.
- Wash the membrane twice for 30 min each in hybridization buffer at room temperature.
- Hybridization
- Put the membrane into hybridization tubes of appropriate size (adjusted to the size of your membrane).
- Add 15 ml of hybridization buffer per one tube.
- Add 100 µl of the 5’ 32P-labeled oligonucleotide probe prepared beforehand (described in Procedure A).
- Incubate and rotate the tubes in a hybridization oven for 16 h at 60 °C.
- Washing the membrane
- Remove the hybridization buffer.
- Wash the membranes twice for 30 min each at 60 °C in a washing solution.
- Take the membranes out of the tubes and dry them until you don’t see any bubbles from washing solution.
- Membrane exposure
Put the membranes into a cassette, cover with plastic foil and expose to phosphor screen from 6 to 16 h. - Reading the signal
Use phosphoimager to scan and quantify the intensity of detected bands with the Imager software. An example of the result is shown in Figure 2.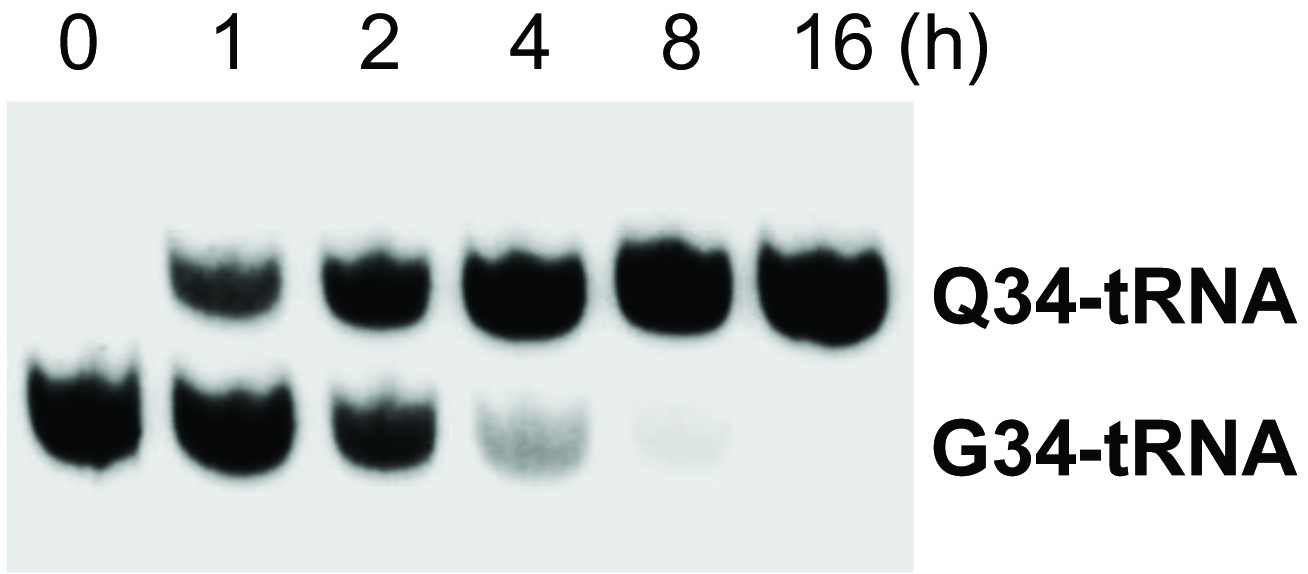
Figure 2. Q-modification kinetics. Time course of Q modification for tRNAAsn starting with the addition of 1 μM queuine to 0Q cell medium (5 µg total RNA loaded in each lane).
Data analysis
Scanned gel image was analyzed with the use of Quantity One imager software.
If procedure was performed correctly, the final image should show separate bands corresponding to the Q-modified and/or G-containing fractions of the intended tRNA species (Figure 2). If larger separation is desired, run the gel for longer time while preventing running the tRNAs out using the dye migration as a guide, or use a longer gel. While performing the transferring step, make sure to cut out the section of the gel containing the tRNA molecules of interest.
When quantifying results, assume that all signals in one lane (Q-tRNA + G-tRNA) represents total tRNA of this species loaded in this lane. Calculate the fraction of Q-modified tRNAs for each sample as [Q-band]/([Q-band] + [G-band]).
For negative controls, repeat the same procedure using a probe for a tRNA not modified with Q (e.g., tRNAAla), which should only show one band on the APB gel.
Another control is to perform peroxide oxidation of the RNA sample first which destroys the cis-diol of the Q-base and the 3’ end of the RNA, so that the probed tRNA species runs faster than the G-band (Igloi and Kossel, 1985; Zaborske et al., 2014).
Recipes
All indicated concentrations are final concentrations of each chemical.
- 2x Gel loading buffer
9 M urea
100 mM EDTA
0.2% (w/v) bromophenol blue
0.2% (w/v) xylene cyanol - 10% denaturing polyacrylamide gel
10% acrylamide/bis solution, 29:1
9 M urea
1x TBE - 10% APB polyacrylamide gel
10% acrylamide/bis solution, 29:1
9 M urea
1x TAE
5% 3-Acrylamidophenylboronic acid - Dialyzed serum medium (HEK293T and HeLa)
DMEM
10% dialyzed FBS - Gel extraction buffer
50 mM KOAc, pH 7.0
200 mM KCl - Hybridization buffer
20 mM sodium phosphate, pH 7.0
300 mM NaCl
1% sodium dodecyl sulphate (SDS) - 1 M Sodium phosphate buffer (pH 7.0)
390 mM NaH2PO4
610 mM Na2HPO4 - 1 M Tris-HCl buffer (pH 9.0)
1 M Tris
Adjust pH with HCl - Washing solution
20 mM sodium phosphate, pH 7.0
300 mM NaCl
0.1% SDS
2 mM EDTA
Acknowledgments
This method was based on the protocol developed previously (Zaborske et al., 2014; Wang et al., 2018). We thank the support from DoD/CDMRP (BC160450 to T.P.).
Competing interests
There are no conflicts of interest or competing interest.
References
- El Yacoubi, B., Bailly, M. and de Crecy-Lagard, V. (2012). Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu Rev Genet 46: 69-95.
- Fergus, C., Barnes, D., Alqasem, M. A. and Kelly, V. P. (2015). The queuine micronutrient: charting a course from microbe to man. Nutrients 7(4): 2897-2929.
- Igloi, G. L. and Kossel, H. (1985). Affinity electrophoresis for monitoring terminal phosphorylation and the presence of queuosine in RNA. Application of polyacrylamide containing a covalently bound boronic acid. Nucleic Acids Res 13(19): 6881-6898.
- Rakovich, T., Boland, C., Bernstein, I., Chikwana, V. M., Iwata-Reuyl, D. and Kelly, V. P. (2011). Queuosine deficiency in eukaryotes compromises tyrosine production through increased tetrahydrobiopterin oxidation. J Biol Chem 286(22): 19354-19363.
- Singhal, R. P., Kopper, R. A., Nishimura, S. and Shindo-Okada, N. (1981). Modification of guanine to queuine in transfer RNAs during development and aging. Biochem Biophys Res Commun 99(1): 120-126.
- TRIzolTM Reagent RNA extraction protocol: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/trizol_reagent.pdf.
- Tuorto, F., Legrand, C., Cirzi, C., Federico, G., Liebers, R., Muller, M., Ehrenhofer-Murray, A. E., Dittmar, G., Grone, H. J. and Lyko, F. (2018). Queuosine-modified tRNAs confer nutritional control of protein translation. EMBO J 37(18) pii: e99777.
- Wang, X., Matuszek, Z., Huang, Y., Parisien, M., Dai, Q., Clark, W., Schwartz, M. H. and Pan, T. (2018). Queuosine modification protects cognate tRNAs against ribonuclease cleavage. RNA 24(10): 1305-1313.
- Zaborske, J. M., DuMont, V. L., Wallace, E. W., Pan, T., Aquadro, C. F. and Drummond, D. A. (2014). A nutrient-driven tRNA modification alters translational fidelity and genome-wide protein coding across an animal genus. PLoS Biol 12(12): e1002015.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Matuszek, Z. and Pan, T. (2019). Quantification of Queuosine Modification Levels in tRNA from Human Cells Using APB Gel and Northern Blot. Bio-protocol 9(6): e3191. DOI: 10.21769/BioProtoc.3191.
Category
Microbiology > Microbial biochemistry > RNA
Molecular Biology > RNA > RNA detection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.