- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Preparation of RNA 3’ End Sequencing Libraries of Total and 4-thiouracil Labeled RNA for Simultaneous Measurement of Transcription, RNA Synthesis and Decay in S. cerevisiae

Published: Vol 9, Iss 6, Mar 20, 2019 DOI: 10.21769/BioProtoc.3189 Views: 5484
Reviewed by: Gal HaimovichAnca SavulescuIndranil Malik
Original research article
The authors used this protocol in:

Aug 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

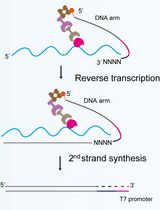
Abstract
Cellular RNA levels are determined by the rates of RNA transcription from the gene template and subsequent RNA stability. Knowledge about both transcription and RNA decay is, therefore, necessary to interpret RNA levels and gene expression, especially during cellular processes where these parameters change. Numerous experimental strategies have been developed to measure transcription and RNA decay rates. However, to our knowledge, none of those techniques can simultaneously interrogate transcription and RNA decay. The presented protocol allows this and provides a simple approach to simultaneously estimate total RNA levels, transcription and decay rates from the same RNA sample. It is based on brief metabolic labeling of RNA and subsequent concurrent sequencing of polyA+ and polyA- RNA 3’ ends. The protocol was developed in S. cerevisiae and should be broadly applicable.
Keywords: TranscriptionBackground
All cells express a multitude of RNA species and control of their RNA levels inside cells is a central function of life. Cells control levels of individual RNA species via regulation of their transcription and RNA decay rates. Hence, measuring those parameters has been a traditional focus area in molecular biology, and numerous techniques have been developed to interrogate them. Among the many methods to measure transcription, chromatin immunoprecipitation (ChIP) of RNA polymerase and its higher resolution variant ChIP-exo (Rhee and Pugh, 2011), global run-on sequencing (GRO-seq, Core et al., 2008) and its successor ‘Precision nuclear Run On sequencing (PRO-seq, Kwak et al., 2013), and nascent elongating transcript sequencing (NET-seq, Churchman and Weissman, 2011) are all readily applicable to genome-wide analysis and have, arguably, received most attention in recent years. RNA stability is usually measured by distinct methodology including pulse-chase type experiments or by following RNA decline after transcription shut-off. More recently, pulse-only application of metabolic labeling (i.e., with 4-thiouridine or 5-bromouridine) has been shown to allow for measurement of RNA stability without employing a chase (Miller et al., 2011; Sun et al., 2012, Neymotin et al., 2014; Rabani et al., 2014).
A major limitation of the above-mentioned approaches is that they rely on significantly different experimental approaches and measuring transcription and RNA decay, therefore, requires two different lines of experimentation. Hence, obtained measurements for transcription and decay rates are not directly comparable.
Recently, strategies based on brief metabolic labeling times combined with fragmentation of labeled RNA before purification (termed TT-seq or iTC-seq) were shown to provide a transcription-like measure from metabolic labeling experiments (Riising et al., 2014, Schwalb et al., 2016). However, even such an optimized procedure does not directly distinguish transcription from early post-transcriptional effects occurring during the labeling period.
To overcome these limitations, we developed a new approach, which is based on existing 4tU based protocols (Barrass et al., 2015), that allows to assay transcription together with total RNA levels and RNA decay rates (Schmid et al., 2018). Our method employs RNA 3’ end sequencing instead of conventional RNA-seq to measure RNA levels of both total and labeled RNA. Obtained data can be used to measure total RNA levels from the amount of pA+ RNA 3’ ends originating from gene 3’ ends, RNA production from the amount of pA+ RNA 3’ ends originating from gene 3’ ends in the labeled RNA fraction. Transcription can be directly estimated using the amount of pA- RNA 3’ ends within gene bodies. Finally, RNA decay rates can be derived from comparing RNA production to total RNA levels, or, alternatively, by comparing transcription and total RNA levels. The approach is simple, based on standard molecular biology techniques and does not require specialized equipment. Hence, it should be applicable to a wide range of experimental questions and model organisms. Here, we provide a detailed protocol for the RNA-seq library preparation part of the protocol.
Materials and Reagents
- 2 ml and 1.5 ml Eppendorf tubes (Hounisen, catalog numbers: 72.695.500 and 72.706)
- 15 ml Falcon tubes
- Zeba Spin Desalting Columns 7KMWCO (Thermo Scientific, catalog number: 89890)
- Oligonucleotides used
- S. pombe spike-in
ACT1_S. pombe_fwd CCATTCTTGCTTCTCTTTCTACTTTCC (Sigma-Aldrich, custom-made)
ACT1_S. pombe_rev CGCTCTCATCATACTCTTGCT (Sigma-Aldrich, custom-made) - S. cerevisiae pA+ RNAs
HHF1_fwd ACTGCCCGGTTTTTCTTCT (Sigma-Aldrich, custom-made)
HHF1_rev CCTAAACCCGCTATAATACACTCAT (Sigma-Aldrich, custom-made)
PMA1_fwd TACTGTCGTCCGTGTCTGGATCT (Sigma-Aldrich, custom-made)
PMA1_rev CCTTCATTGGCTTACCGTTCA (Sigma-Aldrich, custom-made)
TLC1_pA+_fwd TACTGTCGTCCGTGTCTGGATCT (Sigma-Aldrich, custom-made)
TLC1_pA+_rev CCTTCATTGGCTTACCGTTCA (Sigma-Aldrich, custom-made) - S. cerevisiae non-polyadenylated RNAs
SCR1_fwd CCCGGCTATAATAAATCGATCT (Sigma-Aldrich, custom-made)
SCR1_rev GCTGACGCTGGATAAAACT (Sigma-Aldrich, custom-made)
- S. pombe spike-in
- 4-thiouracil (Sigma-Aldrich, catalog number: 440736)
- Dry ice
- Phenol, pH 4.3 (Sigma-Aldrich, catalog number: P4682)
- Chloroform (Sigma-Aldrich, catalog number: 32211)
- Sodium acetate (Sigma-Aldrich, catalog number: S-8625)
- Sodium citrate (Merck, catalog number: 1.06448)
- Sodium chloride (Merck, catalog number: 1.06404)
- HEPES (Sigma-Aldrich, catalog number: H3375)
- Tris (VWR, catalog number: 103156X)
- EDTA (Merck, catalog number: 1.08418)
- Disodium hydrogen phosphate (Merck, catalog number: 1.06575)
- Sodium dihydrogen phosphate (Merck, catalog number: 1.06345)
- SDS (AppliChem, Panreac, catalog number: A1112)
- Magnesium chloride (Merck, catalog number: 1.05833)
- Glycogen (20 mg/ml) (Sigma-Aldrich, catalog number: G1508) (store at -20 °C)
- β-mercaptoethanol (Sigma-Aldrich, catalog number: M6250)
- MTSEA-biotin (Biotium, catalog number: 90064)
- Dimethylformamide (DMF) (Merck, catalog number: 1.03053)
- TURBO DNase free kit (Ambion, catalog number: AM1907M)
- SuperScript II (Invitrogen, catalog number: 18064-014)
- Platinum SYBR Green qPCR Super-Mix-UDG kit (Invitrogen, catalog number: 11733-046)
- Escherichia coli poly(A) polymerase kit (Thermo Fisher, catalog number: AM1350)
- PureLink micro RNA purification kit (Ambion, catalog number: 12183018A)
- Ribo-Zero Gold rRNA Removal kit for yeast (Illumina, catalog number: MRZY1306)
- RiboLock RNase Inhibitor (Thermo Scientific, catalog number: E00381)
- Lexogen QuantSeq 3’ mRNA-Seq Library Prep Kit REV (Lexogen, catalog number: 016.96)
- Dynabeads MyOne Streptavidin C1 (Invitrogen, catalog number: 65002)
- Ethanol 96% (VWR, catalog number: 83804)
- KOH (Merck, catalog number: P5958)
- SC amino acid mix (Merck, catalog number: Y1501; or home-made equivalent)
- Yeast nitrogen base (Merck, catalog number: Y0626)
- DMSO (Merck, catalog number: W387520)
- YPAD growth medium (Merck, catalog number: Y1375; or home-made equivalent)
- Acetic acid (Merck, catalog number: 1005706)
- NaOH (Merck, catalog number: S8045)
- HCl (Merck, catalog number: H1758)
- URA- medium (synthetic growth medium without uracil) (see Recipes)
- 4tU stock (see Recipes)
- TES (see Recipes)
- S. pombe spike-in RNA (see Recipes)
- RNA storage solution (see Recipes)
- 3 M NaOAc (see Recipes)
- MTSEA-biotin stock (see Recipes)
- 1 M HEPES-KOH, pH 7.5 (see Recipes)
- 0.5 M EDTA-NaOH, pH 8 (see Recipes)
- 1 M NaPi buffer, pH 6.8 (see Recipes)
- 10x NaMg (see Recipes)
- 1x NaMgPS (see Recipes)
- TEN1000 (see Recipes)
- 0.7 M β-mercaptoethanol (see Recipes)
- 20 mg/ml Glycogen (see Recipes)
Equipment
- DynaMag-2 magnet (Thermo Fisher Scientific, catalog number: 12321D)
- Incubator (30 °C, with agitation for 500 ml flasks)
- Centrifuge (for 50 ml tubes)
- Microcentrifuge (cooled, for Eppendorf tubes)
- Thermomixer (for Eppendorf tubes, heating up to 65 °C)
- Rotating wheel
- Real-time PCR system (Agilent, model: AriaMX, catalog number: G8830A)
- Bioanalyzer (Agilent, model: Bioanalyzer 2100)
- Qubit 2.0 Fluorometer (Life Technologies)
- NanoDrop UV spectrophotometer
- Centrifuge bucket (or flask)
- Autoclave
- Vortexer
- 4 °C refrigerator
- -20 °C freezer
- -70 °C freezer
Procedure
- Metabolic labeling of S. cerevisiae culture
- Prepare 96% ethanol for fast inactivation of cells. Prepare for each culture 1x culture volume of ethanol in a centrifuge bucket (or flask) that can accommodate 2.5x or more of culture volume. Pre-chill these flasks to around -70 °C in a box with plenty of dry ice.
- Use 100 ml culture in URA- media for a standard experiment.
- Grow cells to an optical density (OD600) of 0.4 to 0.8.
- Add 1/10,000 culture volume of 100 mM 4tU (i.e., final concentration of 10 µM) and mix thoroughly.
Note: If you process several cultures in parallel, prepare and follow an exact time schedule to be able to precisely adhere to the desired labeling times. Remember to prepare one culture that has not been treated with 4tU (negative control) and process it as the other samples. - Continue to grow for desired metabolic labeling time (usually 2 min to enrich strongly for nascent RNA).
Note: Time of labeling can be optimized by testing several time points, e.g., 1, 2, 5 and 10 min and comparing the enrichment of desired targets (see [Optional] Quality Control section). - Quickly inactivate cells by pouring culture into 1x volume of deep-chilled ethanol prepared in Step A1. Leave on normal ice until all samples ready to centrifuge.
Note: The idea behind this approach is that simply putting cells on ice or the like will allow metabolic activity for an uncontrollable amount of time, whereas pouring into deep-chilled ethanol will instantaneously stop all activity and allow for precise control over labeling time. - Spin at 1,500 x g for 3 min at 4 °C.
- Remove supernatant, resuspend in 3 ml of ice-cold ddH2O and split the suspension into 3 x 2 ml Eppendorf tubes.
- Spin cells in Eppendorf tubes at full speed for 12 s, and wash cells another 2 rounds each with 1 ml ice cold ddH2O.
- Carefully remove supernatant. Store pellet at -70 °C or continue with RNA extraction.
- Extract total RNA
Note: The 3 aliquots from above are handled separately, and all phenolizations and precipitation in this section are done in 2 ml tubes. You will need 15 tubes per sample.- Resuspend cells in 800 µl TES. Add 800 µl acidic phenol and shake vigorously for 1 h at 65 °C in a thermomixer.
- Spin for 5 min at full speed and 4 °C. Transfer top phase to a new tube and phenolize with another 700 µl acidic phenol by shaking vigorously for 5 min at 65 °C in a heated block.
- Spin for 5 min as above.
- Take top phase to a new tube and extract with 700 µl chloroform by vortexing vigorously for 20 s at room temperature.
- Spin for 1 min as above.
- Take top phase to a new tube and extract with 600 µl chloroform by vortexing vigorously for 20 s at room temperature.
- Spin for 1 min as above.
- Take 500 µl of the top phase to a new tube and precipitate RNA by adding 50 µl 3 M NaOAc and ice-cold 1.4 ml 96% ethanol.
- Spin immediately for 20 min at full speed at 4 °C.
- Wash pellet twice with 1 ml 70% ethanol each.
- Resuspend RNA in 100 µl RNA storage solution per tube.
Note: Be careful to avoid overdrying the pellets, as very dry pellets are hard to dissolve. This can be done by first removing most ethanol with a pipet, re-spin briefly, remove any remaining ethanol with a pipet P200 and then leave tubes open for ca. 1 min open at RT before resuspension. Resuspension should be fast and require only a bit of shaking and/or pipetting up and down. - Combine RNA from 3 aliquotes and measure the RNA concentration of a 1:5 dilution using, i.e., a Nanodrop.
Note: RNA concentration of the undiluted sample should be > 1 µg/µl.
- Biotinylate 4sU RNA
- Prepare coupling reaction by buffering 500 µg RNA with 4 µl 1 M HEPES-KOH pH 7.5 and 0.8 µl 0.5 M EDTA and fill with RNA storage solution to 400 µl.
Note: If 500 µg RNA is not available, the protocol is doable with 300 µg or even less. - Optional: Add spike-in RNA. We usually use 3-5 µg (1/100 w/w) S. pombe spike-in RNA (see Recipes and Data analysis).
- Add 10 µl of the 5 mg/ml MTSEA-biotin stock, vortex to mix and incubate for 30 min at 25 °C in the dark.
- During this incubation prepare Zeba columns, place Zeba columns in 15 ml Falcon tubes. Drain and wash each column 3 x with 2 ml RNA storage solution each. For each wash: apply liquid and spin 2 min at 1,000 x g.
- Apply MTSEA coupling reactions onto drained Zeba columns, let liquid soak in resin for 2 min, add then another 130 µl RNA storage solution, spin for 2 min at 1,000 x g in a fresh 15 ml Falcon and recover the flow-through which contains biotin-coupled RNA devoid of free MTSEA-biotin coupling reagent. Precipitate the flow-through (~500 µl) with 50 µl 3 M NaOAc and 1.4 ml 96% ethanol. Incubate for 30 min at 4 °C or overnight at -20 °C. Spin for 20 min at 4 °C, full speed.
- Wash 2 x 1 h at RT each with 1 ml 80% ethanol in the dark. Spin for 15 min at 4 °C, 16,000 x g between washes.
Note: As above, take care to void overdrying the pellet, i.e., simply leave tubes with pellet open at RT for 1 min and then resuspend. - Resuspend RNA in 250 µl RNA storage solution.
- Take 10 µl of input (‘Total RNA’) aliquot.
- Prepare coupling reaction by buffering 500 µg RNA with 4 µl 1 M HEPES-KOH pH 7.5 and 0.8 µl 0.5 M EDTA and fill with RNA storage solution to 400 µl.
- Isolation of the biotinylated RNA
- Add to 240 µl RNA sample: 30 µl 10x NaMg, 30 µl 1 M NaPi, 3 µl 10% SDS.
Note: This results in the RNA sample in ~300 µl with a solvent composition equivalent to 1x NaPSMg. - Prepare magnetic streptavidin-coupled beads (adjust the amount depending on number of samples):
- Use 50 µl Dynabeads MyOne Streptavidin C1 slurry per sample.
- Drain beads by applying on magnet and pipetting off the liquid.
- Wash first 1 x with 400 µl 1x NaMgPS.
- Block beads with 200 µl 1x NaMgPS + 10 µl 20 mg/ml glycogen.
- Rotate for 20 min at RT.
- Remove blocking solution from magnetic beads.
- Wash 1 x with 400 µl NaMgPS.
- Drain beads and add RNA immediately.
- Let the RNAs bind to beads for 30 min at room temperature by gentle agitation, i.e., on a rotating wheel in the dark.
- Remove supernatant from magnetic beads.
- Wash 5 x with 400 µl 1x NaMgPS each.
- Wash 1 x with 400 µl TEN1000.
- Elute biotinylated RNA by incubating 2 x with 50 µl β-mercaptoethanol. Incubate for 5 min at RT before removing eluate at each step.
- Combine eluates and precipitate RNA with 10 µl 3 M NaOAc, pH 5, 2.5 µl 20 mg/ml glycogen and 250 µl 96% ethanol.
- Incubate on dry ice for 30 min or -20 °C overnight.
- Spin for 30 min at full speed, 4 °C.
- Wash RNA pellet 2 x with 75% ethanol, to remove β-mercaptoethanol as much as possible.
- Resuspend in 20 µl RNA storage solution.
- DNase-treat 2 µl (4 µg) of the total RNA aliquot and 17.6 µl (300 ng) of the 4tU IP samples using the TURBO DNase kit from Ambion, according to the manufacturer’s instructions.
- Optional: Detect RNA quality and concentration using a Bioanalyzer and the RNA pico kit (Agilent), Qubit or similar (see Figure 1).
- Optional: use 2 µl of elution RNA for quality control (see [Optional] Quality Control section).
- Add to 240 µl RNA sample: 30 µl 10x NaMg, 30 µl 1 M NaPi, 3 µl 10% SDS.
- In vitro polyadenylation with E-PAP
- To 5 µl of DNase-treated RNA, add 4 µl 5x E-PAP buffer, 2 µl 25 mM MnCl2, 0.2 µl E-PAP (2 U/µl) from the Escherichia coli poly(A) polymerase kit (Thermo Fisher), 2 µl 10 mM ATP, 0.2 µl RiboLock and 6.6 µl RNase-free ddH2O.
- Mix and incubate for 30 min at 30 °C.
- Purify RNA from polyadenylation reactions using PureLink RNA purification kit micro (Ambion) according to the manufacturer’s instructions and elute in 22 µl RNase-free ddH2O.
- Ribo-depletion
Note: The protocol below is based on RiboGold kit from Illumina, which is no longer commercially available. And it is also applicable to other similar products (e.g., RNA-seq kit).- Carry out ribo-depletion using Ribo-Zero Gold rRNA Removal Kit for Yeast (Illumina) essentially as per the manufacturer’s instruction but using a downscaled version.
- Mix 18 µl of eluate with 2 µl reaction buffer, 2 µl of removal solution and 8 µl of RNase-free ddH2O.
- Incubate for 10 min at 65 °C and cool to room temperature for 5 min.
- Add 65 µl of rRNA magnetic beads to the reaction mix from Step F3 and first incubate for 5 min at room temperature and then 5 min at 50 °C. Transfer the unbound solution to a fresh tube containing 40 µl ddH2O and 10 µl 3 M NaOAc and 20 µg glycogen.
- Add 300 µl ethanol, precipitate RNA for 20 min at -20 °C and spin for 30 min at 4 °C.
- Wash pellet twice with 70% ethanol.
- Resuspend pellet in 10 µl of RNA storage solution.
- 3’ end RNA-seq library preparation and sequencing
- Use 5 µl of IP or 1 µl of input aliquots as starting RNA material and prepare RNA-seq libraries using QuantSeq REV kit (Lexogen) according to the manufacturer’s instructions.
- Sequence samples on Illumina HiSeq using single-read 50 nt reads or a similar machine with custom primers as defined by the QuantSeq REV specifications. Aim for around 5 million primary reads per each library to achieve necessary depth for S. cerevisiae experiments.
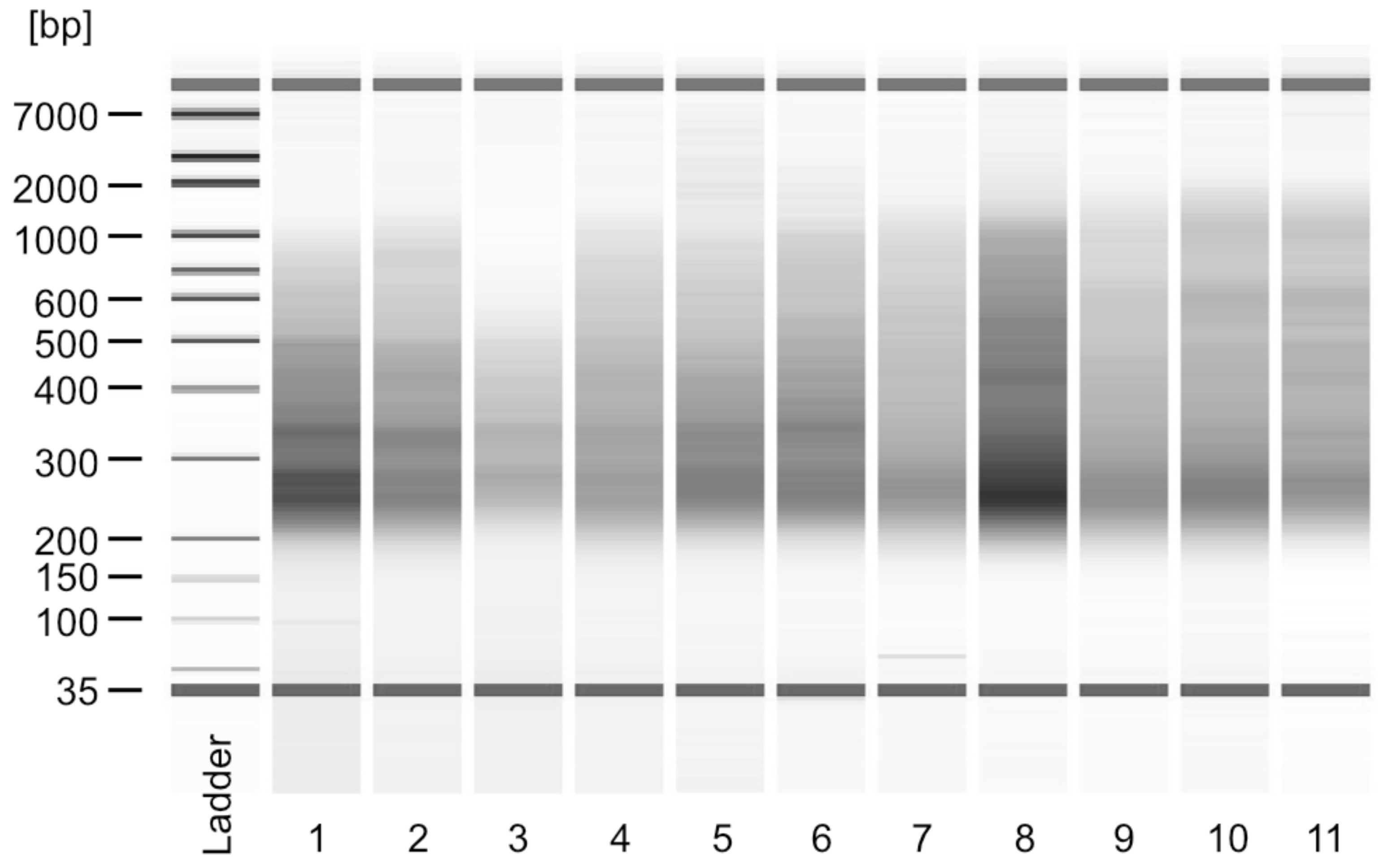
Figure 1. Example of Bioanalyzer traces of final sequencing libraries. Libraries shown are from samples that were not subjected to E-PAP treatment. Lanes 1-4 are from total RNA and lanes 5-11 from 10’ 4tU IP samples. All samples were successfully used for Illumina sequencing.
(Optional) Quality control of 4tU labeled RNAs
- Use 2 µl of input and elution sample.
- Perform reverse transcription using Superscript II or equivalent and an Oligo(dT)18 according to manufacturers’ instructions.
- Dilute RT reactions in 100 μl nuclease-free water and use 2 µl if diluted RTs per 16 µl qPCR Platinum SYBR Green qPCR Super-Mix-UDG kit reactions according to manufacturers’ instructions. Prepare reactions with 1:10 diluted RT for oligos against mRNAs and RNAPIII non-adenylated transcript SCR1.
Data analysis
RNA concentration equivalents of various targets are estimated from RT-qPCR values using the formula RNAconc = 2(-Ct); optionally including a factor for correction with measured primer efficiency. Those values are then normalized relative to the values obtained for spike-in estimate reaction against S. pombe ACT1 mRNA. The use of external spike-ins for normalization permits direct comparison of IP efficiencies across samples and labeling times and allows for removal of technical noise (i.e., IP efficiency) that may arise due to the complex nature of the experiment. Normalized values from negative control elutions (i.e., from samples without 4tU) are used to estimate the background in elution samples. Several mRNAs should be significantly enriched above background, which indicates that the experiment has worked. Note, the goal is to enrich strongly for nascent (i.e., pA-) 4tU-labeled RNAs, which is achieved with short labeling times. Enrichment of mature RNAs (pA+ mRNA, full-length snRNAs) is most efficient with increased labeling times (see Figure 2). That is, for 10 min labeling the vast majority (> 90%) of transcripts are enriched significantly above background. For 2 min labeling, the enrichment of pA+ RNAs above background is less significant and the number of transcripts detected significantly above background varies significantly between technical repeats. Usually, at least ~20% of all transcripts are found significantly enriched. Hence, we usually use 2 min labeling times to enrich for nascent RNAs (where many, but not all mature RNAs are also significantly enriched above background) and 10 min labeling times when only measuring full-length RNAs.
To control in vitro polyadenylation reactions (E-PAP treatment), ratio of non-adenylated SCR1 relative to mRNAs is compared. Those species should be enriched at least 10-fold relative to mRNAs in E-PAP treated samples.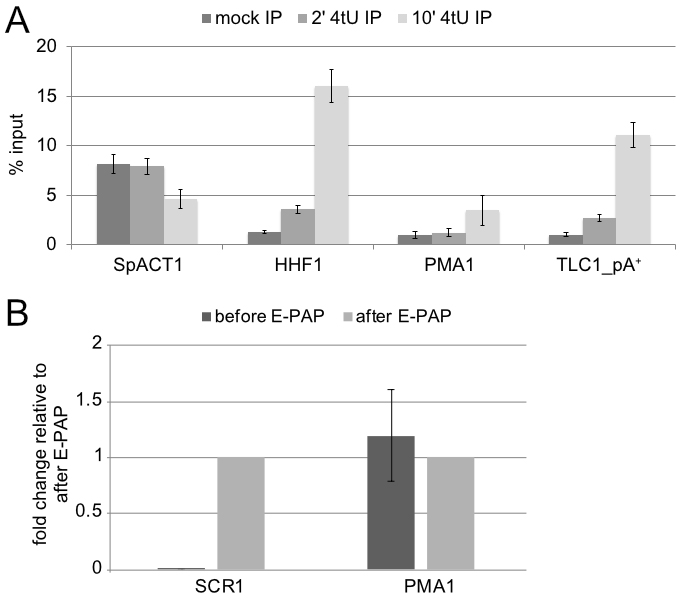
Figure 2. Recovery of RNAs after 2 and 10 min labeling and efficiency of E-PAP treatment. A. Increase in labeling efficiency in function of 4tU labeling time. Wild-type cells were treated with 4tU for 0, 2 and 10 min, respectively. The graph depicts IP efficiencies (depicted as % input) from mock, 2’ 4tU and 10’ 4tU labeling times for spike in (SpACT1, for S. pombe ACT1) and S. cerevisiae mRNAs PMA1 and HHF1, as well as for the polyadenylated precursor of telomerase RNA TLC1. B. RT-qPCR of total RNA before and after treatment with E-PAP. RNAs were reverse transcribed with Oligo(dT)18 and quantified by qPCR with primers for SCR1 (a non-polyadenylated RNAPIII product) and PMA1 (a polyadenylated mRNA). The non-polyadenylated SCR1 RNA is detected > 1,000 times more efficiently after E-PAP treatment, whereas PMA1 signal is not affected.
Recipes
- URA- medium (synthetic growth medium without uracil)
0.2% (w/v) SC amino acid mix without uracil
2% (w/v) glucose
0.67 g (w/v) yeast nitrogen base
Dissolve in ddH2O, autoclave and store at room temperature - 4tU stock
100 mM 4-thiouracil in DMSO, store at -20 °C. Warm to RT before use - TES
10 mM Tris-HCl, pH 7.5
1 mM EDTA, 1% (w/v) SDS
Prepare from RNase-free stocks and keep at RT - S. pombe spike-in RNA
- Grow standard S. pombe cells to exponential growth at 30 °C in YPAD growth medium and labeled for 15 min with 5 mM (w/v) final 4tU
- Prepare total RNA as described for S. cerevisiae above and store aliquots in RNA storage solution at -20 °C
- RNA storage solution
1 mM Na-citrate, pH 6.4
Prepare from RNase-free stocks and keep at RT - 3 M NaOAc
3 M Sodium acetate adjusted to pH to 5 with acetic acid
Prepare from RNase-free stocks and keep at RT - MTSEA-biotin stock
5 mg/ml in DMF
Store at -20 °C - 1 M HEPES-KOH, pH 7.5
1 M HEPES adjusted to pH 7.5 with KOH
Prepare from RNase-free stocks and keep at RT - 0.5 M EDTA-NaOH
0.5 M EDTA adjusted to pH 8 with NaOH
Prepare from RNase-free stocks and keep at RT - 1 M NaPi buffer, pH 6.8
Mix 1 M Na2HPO4 with 1 M NaH2PO4 to reach pH 6.8
Prepare from RNase-free stocks and keep at RT - 10x NaMg
100 mM Tris-HCl, pH 7.0
2 M NaCl
250 mM MgCl2
Prepare from RNase-free stocks and keep at RT - 1x NaMgPS
1x NaMg (1:10 from 10x)
100 mM NaPi (1:10 from 1 M)
0.1% SDS (1:100 from 10%)
Prepare fresh before use in RNase-free H2O and keep at room temperature
Note: Can form precipitate, which can be redissolved by gentle heating. - TEN1000
10 mM Tris, pH 7.5
0.5 mM EDTA
1 M NaCl
Prepare from RNase-free stocks and keep at RT - 0.7 M β-mercaptoethanol
1:20 dilution of pure β-mercaptoethanol in RNA storage solution. Prepare fresh - 20 mg/ml Glycogen
Prepare in RNase-free H2O and keep at -20 °C
Acknowledgments
We thank David Barrass and Jean Beggs for the initial 4tU labeling and IP protocol and Rikke Jespersen and Dorthe Caroline Riishøj for technical support. Work in the T.H.J. laboratory was supported by the Danish National Research Council, the Lundbeck Foundation (grant R171-2014-553) and the ERC (grant 339953).
Competing interests
The authors declare no competing interests.
References
- Barrass, J. D., Reid, J. E., Huang, Y., Hector, R. D., Sanguinetti, G., Beggs, J. D. and Granneman, S. (2015). Transcriptome-wide RNA processing kinetics revealed using extremely short 4tU labeling. Genome Biol 16: 282.
- Churchman, L. S. and Weissman, J. S. (2011). Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469(7330): 368-373.
- Core, L. J., Waterfall, J. J. and Lis, J. T. (2008). Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322(5909): 1845-1848.
- Kwak, H., Fuda, N. J., Core, L. J. and Lis, J. T. (2013). Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339(6122): 950-953.
- Miller, C., Schwalb, B., Maier, K., Schulz, D., Dumcke, S., Zacher, B., Mayer, A., Sydow, J., Marcinowski, L., Dolken, L., Martin, D. E., Tresch, A. and Cramer, P. (2011). Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol Syst Biol 7: 458.
- Neymotin, B., Athanasiadou, R. and Gresham, D. (2014). Determination of in vivo RNA kinetics using RATE-seq. RNA 20(10): 1645-1652.
- Rabani, M., Raychowdhury, R., Jovanovic, M., Rooney, M., Stumpo, D. J., Pauli, A., Hacohen, N., Schier, A. F., Blackshear, P. J., Friedman, N., Amit, I. and Regev, A. (2014). High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. Cell 159(7): 1698-1710.
- Rhee, H. S. and Pugh, B. F. (2011). Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147(6): 1408-1419.
- Riising, E. M., Comet, I., Leblanc, B., Wu, X., Johansen, J. V. and Helin, K. (2014). Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol Cell 55(3): 347-360.
- Schmid M, Tudek A, Jensen TH. (2018). Simultaneous measurement of transcriptional and post-transcriptional parameters by 3' end RNA-seq. Cell Reports 24(9): 2468-2478.
- Schwalb, B., Michel, M., Zacher, B., Fruhauf, K., Demel, C., Tresch, A., Gagneur, J. and Cramer, P. (2016). TT-seq maps the human transient transcriptome. Science 352(6290): 1225-1228.
- Sun, M., Schwalb, B., Schulz, D., Pirkl, N., Etzold, S., Lariviere, L., Maier, K. C., Seizl, M., Tresch, A. and Cramer, P. (2012). Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res 22(7): 1350-1359.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Schmid, M., Tudek, A. and Jensen, T. H. (2019). Preparation of RNA 3’ End Sequencing Libraries of Total and 4-thiouracil Labeled RNA for Simultaneous Measurement of Transcription, RNA Synthesis and Decay in S. cerevisiae. Bio-protocol 9(6): e3189. DOI: 10.21769/BioProtoc.3189.
Category
Molecular Biology > RNA > RNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.