- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Differentiation of Human Induced Pluripotent Stem Cells (iPSCs) into an Effective Model of Forebrain Neural Progenitor Cells and Mature Neurons
Published: Vol 9, Iss 5, Mar 5, 2019 DOI: 10.21769/BioProtoc.3188 Views: 18126
Reviewed by: Giusy TornilloZe YangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Induced Pluripotent Stem Cells (iPSCs) are pluripotent stem cells that can be generated from somatic cells, and provide a way to model the development of neural tissues in vitro. One particularly interesting application of iPSCs is the development of neurons analogous to those found in the human forebrain. Forebrain neurons play a central role in cognition and sensory processing, and deficits in forebrain neuronal activity contributes to a host of conditions, including epilepsy, Alzheimer’s disease, and schizophrenia. Here, we present our protocol for differentiating iPSCs into forebrain neural progenitor cells (NPCs) and neurons, whereby neural rosettes are generated from stem cells without dissociation and NPCs purified from rosettes based on their adhesion, resulting in a more rapid generation of pure NPC cultures. Neural progenitor cells can be maintained as long-term cultures, or differentiated into forebrain neurons. This protocol provides a simplified and fast methodology of generating forebrain NPCs and neurons, and enables researchers to generate effective in vitro models to study forebrain disease and neurodevelopment. This protocol can also be easily adapted to generate other neural lineages.
Keywords: iPSCBackground
Induced pluripotent stem cells (iPSCs) are stem cells produced from non-pluripotent source cells and tissues (Shi et al., 2017). Due to their ability to differentiate into a wide range of cell types, they are a promising avenue for improving our understanding of human development and treatment of degenerative diseases (Marchetto et al., 2011). Of particular interest are iPSC-derived models of human forebrain neurons, as these cells are known to mediate higher order brain functions, including consciousness (Baxter and Chiba, 1999), emotion (Morgane et al., 2005), and sleep (Schwartz and Roth, 2008). As a result, deficits in these cells can cause a wide range of neurological disorders, including neurodegenerative diseases like Alzheimer’s (Auld et al., 2002) and Huntington’s (McColgan and Tabrizi, 2018), as well as neurodevelopmental diseases such as autism (Donovan and Basson, 2017) and epilepsy (Heath, 1976). As there are few effective forebrain models for humans, the discovery of iPSCs spurred a rapid push to develop effective protocols to differentiate iPSCs to forebrain neurons (Srikanth and Young-Pearse, 2014). The first protocols that were developed drew upon previous work using embryonic stem cells (ESCs), which relied upon feeder cell cultures. This complicated the procedure and raised concerns about clinical applications. Later protocols were able to generate forebrain neurons without using feeder cells (Bell et al., 2017), with some eliminating all animal generated products entirely (Yuan et al., 2015). It can be difficult to make an all-encompassing statement about the protocols currently used to generate forebrain NPCs, due to the multitude of labs currently generating forebrain neurons and the many variables that can be changed and optimized. However, many of the recently most cited published protocols for the generation of forebrain NPCs and neurons can be divided into two kinds, monolayer and embryoid bodies (EBs) protocols. In an EB based protocol, iPSCs are dissociated and plated in suspension in a neural induction media to allow them to form EBs, which gradually aggregate over 5-7 days (Pasca et al., 2011). These EBs are then transferred to a plate that supports cell attachment, enabling the embryoid bodies to attach to the bottom of the plate and spread out into a neural rosette. From this rosette, neural stem cells (NSCs) arise, which can be passaged to form relatively stable neural progenitors cells (NPCs) (Shi et al., 2012). NPCs can then be plated in a neuronal induction media to give rise to mature neurons (Bell et al., 2017). Monolayer based protocols chiefly differ in that iPSC colonies are maintained as a monolayer during neural induction, and develop directly into rosettes without aggregation (Chandrasekaran et al., 2017). Using either approach, generation of NPCs from iPSCs is typically reported to require 21-30 days, with electrically active neurons requiring an additional 30+ days of differentiation from NPCs, for a total time of 50+ days to generate forebrain neurons from iPSCs (Yuan et al., 2015).
This protocol describes a methodology for generating forebrain neurons from iPSCs, where iPSC colonies are induced to form neural rosettes without mechanical dissociation, and neural progenitor cells are purified from immature clusters of neural cells, known as neural rosettes based on differential adhesion. Neural progenitor cells will not attach to non-adherent plates and aggregate together in a floating mass, while other cells types either adhere or float but do not aggregate with NPCs (Bell et al., 2017). This allows rapid purification of NPCs, has the potential for automation and enables the generation of NPC cultures within 14 days of initiation of differentiation. This modification does not appear to negatively influence the fate of the cells, as we observe uniform staining for key neural progenitor cells markers (Zhang et al., 2010; Venere et al., 2012; Zhang and Jiao, 2015). Indeed, we have found that we are capable of recording electrical activity from neurons consistently in as little as five days of differentiation from NPCs. This protocol can be used to generate forebrain neurons simply and effectively for use in investigating neurodevelopment, the etiology of diseases that affect the forebrain, and drug testing.
Materials and Reagents
- For Cell Culture
- 6-well plate (SARSTEDT, catalog number: 83.3920)
- 35-mm dish (SARSTEDT, catalog number: 83.3900)
- 60-mm dish (SARSTEDT, catalog number: 83.3901.300)
- 100-mm dish (SARSTEDT, catalog number: 83.3902.300)
- Petri dishes (Fisher Scientific, catalog number: FB0875713)
- Coverslips (Fisher, catalog number: 12-545-80)
- Liquid nitrogen (PRAXAIR, catalog number: 7727-37-9)
- iPSCs, either derived from somatic cells or thawed from a frozen aliquot
- TeSRTM-E8TM Media (Stem Cell Technologies, catalog number: 05990)
- BrainPhysTM Neuronal Medium (Stem Cell Technologies, catalog number: 05790)
- Matrigel® (Corning, catalog number: 354277)
- KnockOutTM DMEM/F-12 (Thermo Fisher, catalog number: 12660012)
- DMSO (Sigma-Aldrich, catalog number: C6164)
- StemPro NSC SFM (Thermo Fisher, catalog number: A1050901)
- SM1 Neuronal Supplement (Stem Cell Technologies, catalog number: 05711)
- N2 Supplement-A (Stem Cell Technologies, catalog number: 07152)
- BSA (Gibco, catalog number: 16140071)
- Non-Essential Amino Acid (NEAA) (Gibco, catalog number: 11140050)
- SB431542 (Stem Cell Technologies, catalog number: SB431542)
- Noggin (Gibco, catalog number: PHC1506)
- Laminin (Sigma-Aldrich, catalog number: L2020)
- Gentle Cell Dissociation Reagent (Stem Cell Technologies, catalog number:07174)
- Epidermal growth factor (EGF) (Sigma, catalog number: E9644)
- Fibroblast growth factor (FGF) (Sigma, catalog number: F0291)
- Brain-Derived Neurotrophic Factor (GenScript, catalog number: Z03208-25)
- Glial-Derived Neurotrophic Factor (GenScript, catalog number: Z02927-50)
- Accutase (Sigma-Aldrich, catalog number: SCR005)
- DPBS without CaCl2 and MgCl2 (Sigma-Aldrich, catalog number: D8537)
- Neural Induction Medium 1 (see Recipes)
- Neural Induction Medium 2 (see Recipes)
- Neural Progenitor Media (see Recipes)
- Neuronal Media (see Recipes)
- Culture Dish Coating with Matrigel® (see Recipes)
- For Immunocytochemistry (ICC)
- Glass coverslips (Fisher, catalog number: 12-545-81)
- Microscope slides (Fisher, catalog number: 12-552-3)
- Pipette tips, 1 ml (SARSTEDT,70.1186)
- Pipette tips, 200 µl (SARSTEDT,70.1186)
- Pipette tips, 20 µl (SARSTEDT,70.1186)
- 15-ml conical tube (SARSTEDT, 62.554.002)
- Paraformaldehyde (PFA) (Sigma-Aldrich, catalog number: 252549)
- BSA (Sigma-Aldrich, catalog number: A2058)
- Triton X-100
- DAPI (Thermo Fisher, catalog number: 62248)
- Vectashield® (Vector Labs, catalog number: H-1000)
- Nail Polish (Sally Hansen Insta-Dri Fast-Dry Clear Nail Color)
- Antibodies
Antibodies for iPSCs:- TRA-1-60 (Embryonic Stem Cell Marker Panel, Abcam, catalog number: ab109884)
- SSEA (Embryonic Stem Cell Marker Panel, Abcam, catalog number: ab109884)
- Nanog (Embryonic Stem Cell Marker Panel, Abcam, catalog number: ab109884)
- OCT4 (Stemcell Technologies, catalog number: 60093)
- PAX6 (Stemcell Technologies, catalog number: 60094)
- SOX1 (Stemcell Technologies, catalog number: 60095)
- Nanog (Embryonic Stem Cell Marker Panel, Abcam, catalog number: ab109884)
- Nestin (Stemcell Technologies, catalog number: 60091)
- PAX6 (Stemcell Technologies, catalog number: 60094)
- Tuj1 (Abcam, catalog number: ab14545)
- S100B (Abcam, catalog number: ab52642)
- VGLUT1 (Abcam, catalog number: ab77822)
- GABA (Abcam, catalog number: ab86186)
- GFAP (Abcam, catalog number: ab7260)
- ALEXA 488 anti-mouse (Invitrogen, catalog number: A-11008)
- ALEXA 555 anti-rabbit (Invitrogen, catalog number: A-21422)
- Coating glass coverslips with Poly-ornithine and laminin (see Recipes)
- For Electrophysiology
- Borosilicate pipettes with resistances of 3-6 MΩ (World Precision Instruments, catalog number: 1B150-4)
- Cell strainer (40 µm) (Sigma, catalog number: CLS431750)
- Tetrodotoxin (TTX) (Alomone labs, catalog number: T-550)
- BrainPhysTM Without Phenol Red (Stem Cell Technologies, catalog number: 05791)
- HEPES (Sigma-Aldrich, catalog number: H3375)
- KCl (Sigma-Aldrich, catalog number: 793590)
- Potassium Gluconate (Sigma-Aldrich, catalog number: G4500)
- EGTA (Sigma-Aldrich, catalog number: 324626)
- Mg-ATP (Sigma-Aldrich, catalog number: A9187)
- Creatine phosphate (Sigma-Aldrich, catalog number: CRPHO-RO)
- Guanosine triphosphate (Sigma-Aldrich, catalog number: G8877)
- NMDA (Alomone labs, catalog number: N-170)
- Magnesium Chloride hexahydrate (Sigma, catalog number: M2393)
- Internal pipette solution (see Recipes)
Equipment
- Pipettes (Fisher, catalog number: 4680100)
- Pipette puller (Sutter Instrument, model: P-1000)
- Osmometer (Advanced Instruments, model: 3320)
- Bead bath (Lab Armor, model: M706)
- Fluorescent microscope (Olympus, model: 1X73)
- Recording chamber with six-channel valve controller (Warner Instruments)
- Automatic temperature controller (Warner Instruments, model: TC-324C)
- Micromanipulator (Sutter Instrument, model: MP-225)
- Microelectrode amplifier Multiclamp 770B (Molecular Devices)
- Acquisition system Axon digidata 1550A (Molecular Devices)
- Biological Safety Cabinet Class 2 (Nuaire, Model: NU440400)
- Incubator (Thermo Fisher, catalog number: 51030287)
- Centrifuge (Allegra, model: X-12)
Software
- Clampex 10.5 (Molecular Devices, www.moleculardevices.com)
- GraphPad Prism 7 (GraphPad, www.graphpad.com)
- Excel 2016 (Microsoft, https://products.office.com/en-ca/excel)
Procedure
- Differentiation from iPSCs to Forebrain NSCs (Neural induction)
Note: The following steps are described assuming high-quality iPSCs (see Figure 1) are plated on a 60-mm tissue culture dish that is coated with Matrigel® (for more details, see Recipes and Table 2).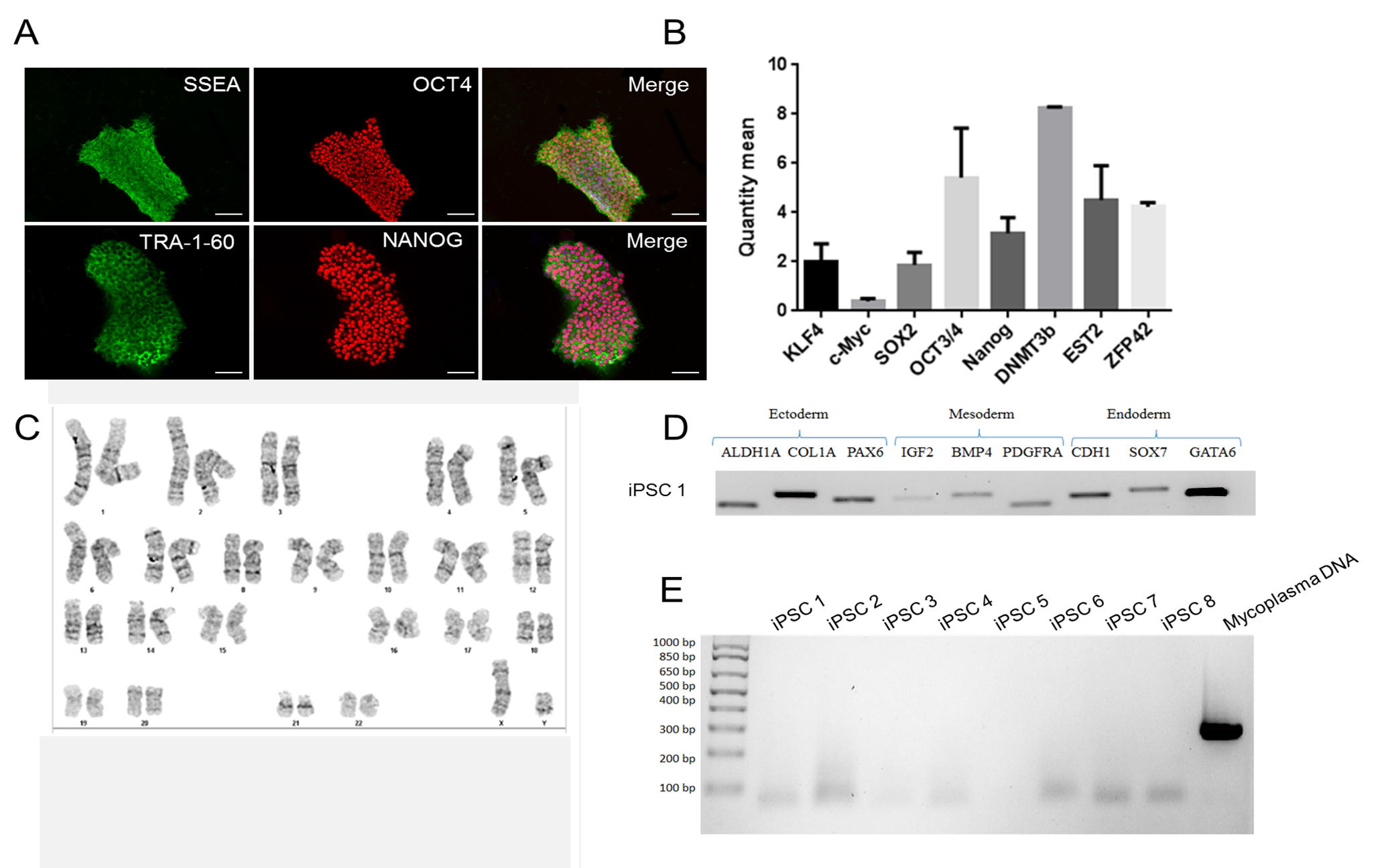
Figure 1. Sample ICC staining for high-quality iPSC cultures. In order for differentiation to proceed effectively, ensure that you begin differentiation with high-quality iPSC cultures. IPSCs should uniformly express pluripotency markers SSEA, OCT4, TRA-1-60, and NANOG (A) when assessed using ICC. Additional pluripotency markers DNMT3b, EST2, and ZFP42 can also be assessed using a qPCR assay (B). IPSCs should be free of karyotypic abnormalities (C), possess the ability to differentiate into all three germ lineages and express characteristic markers of each lineage (D), and test negative for mycoplasma contamination (E). Scale bars represent 100 μm.- Working in a class II biological safety cabinet, use appropriately sized pipettes to plate iPSCs in E8 medium in a 60-mm dish. If starting from a frozen aliquot of iPSC, we recommend plating at least 300,000 cells. This is considered Day 0 of iPSC culture.
- On Day 1 of iPSC culture (15%-25% confluency), aspirate the spent medium to remove non-attached cells, and check the size of colonies. If colonies are approximately 100-200 µm in diameter, they are an appropriate size to begin differentiation. Add 3 ml of complete Neural Induction Medium 1, pre-warmed in a bead bath to each plate using a 5 ml pipette. Return the plates to an incubator maintained at 37 °C, 5% CO2 and atmospheric (~20%) Oxygen. If plates do not contain colonies of sufficient size, add 3 ml of E8 media and check daily until colonies reach the appropriate size.
- On Day 2 (about 48 h after switching to Neural Induction Medium), change the medium by aspirating old medium from each well. Add 3 ml of pre-warmed complete Neural Induction Medium 1 to each plate.
- On Day 4 of neural induction, cells will be reaching confluency. If necessary, mark any colonies with non-neural differentiation. Remove these unwanted colonies with a 200 µl pipette tip. Aspirate the spent medium from each well. Add 3 ml of pre-warmed complete Neural Induction Medium 1 to each plate.
Note: Due to high cell density in the culture from Day 4 onwards, doubling the volume of Neural Induction Medium is very critical for cell nutrition. Also, minimal cell death should be observed from Days 4 to 7 after neural induction. If the color of cells turns yellowish with many floating cells during Days 4 to 7 of neural induction, it indicates that the starting density of iPSCs was too high. In this case, change the Neural Induction Medium every day, remove some colonies and double the volume per well/plate. Ideally, work with these variables to ensure that the media does not continue to turn yellow. - On Day 6 of neural induction, cells should be near maximal confluence. Remove any non-neural differentiated cells that can be observed and add 3 ml of complete Neural Induction Medium into each plate.
- On Day 7 of neural induction, the medium should be switched into Neural Induction Medium 2. Add 3 ml of complete Neural Induction Medium 2 to each plate. The medium should be changed every day for 5 days. For example morphologies, see Figure 2.
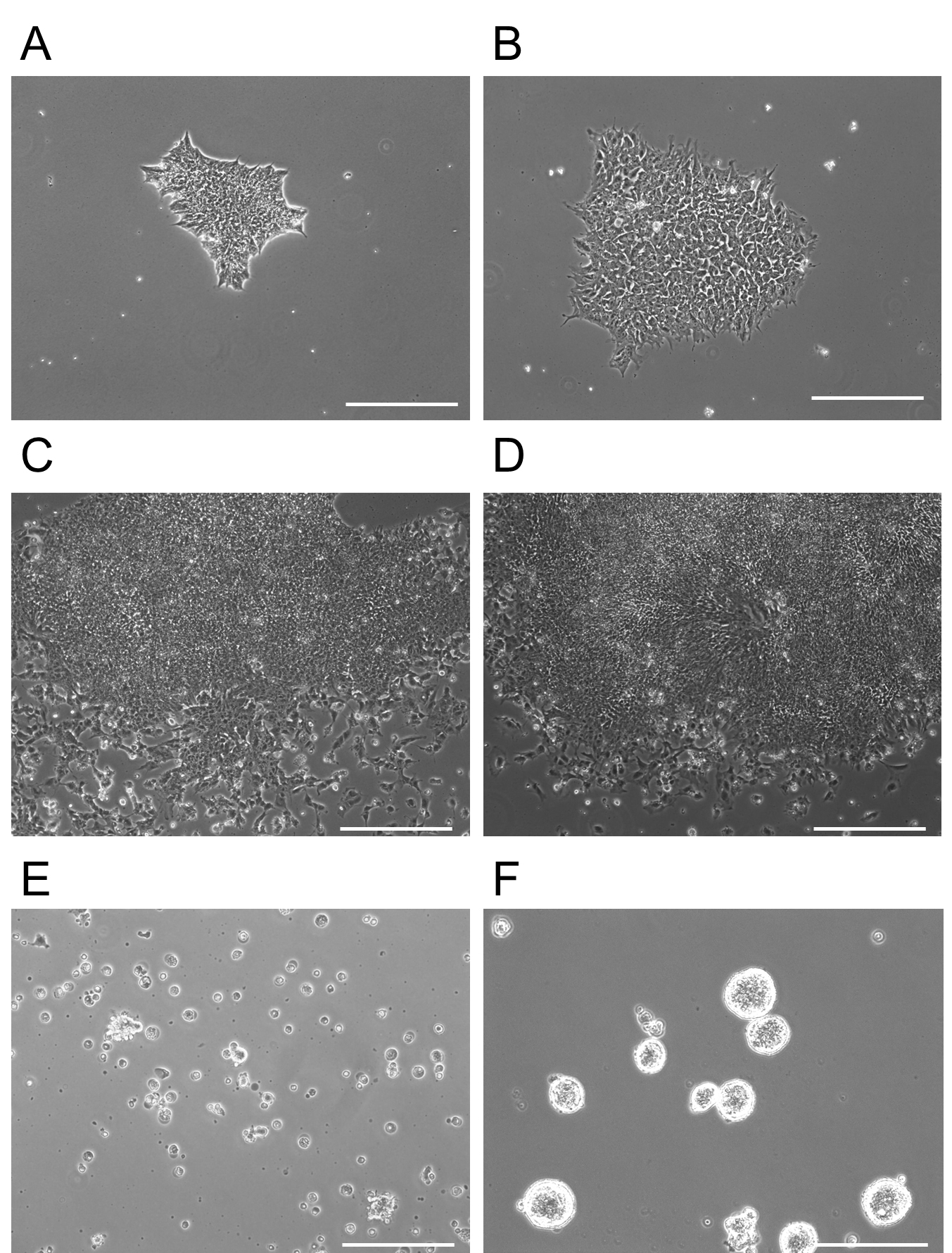
Figure 2. Morphology of iPSCs differentiating into forebrain NPCs. A. Day 0: Showing a single iPSC colony of appropriate size immediately prior to addition of Neural Induction Media 1. B. Day 2: The iPSC colony, which has been treated with Neural Induction Media 1 for 2 days, begins to change cellular morphology and some cells extend processes. C. Day 5: Increased expansion of the colony with some differentiation of outer cells. D. Day 12: Appearance of rosettes in the colony become visible. NSCs are present in high confluence in the middle of these structures. It is at this point that colonies are detached and re-plated on non-adherent plates at D13 for two days. E. D13 immediately after plating on adherent plates. This image shows floating rosette colonies that will continue to proliferate and differentiate in a floating mass. Non-rosette cells either remained on the dish at D12 after chemical release or float as single cells on the non-adherent plates shown in (E). F. At D15, rosette clusters expand in size and are moved to adherent plates. Cell aggregates here are 3-dimensional, but are attached to the plate. Note the purity of the clusters at this point (F). Scale bars represent 130 µm. - Harvest and expansion of NPCs
Note: On Day 12 of neural induction, NPCs are ready to be harvested and expanded.- Aspirate the spent Neural Induction Medium from each plate to be passaged.
- Gently add DPBS without CaCl2 and MgCl2 to each plate twice to rinse the cells.
- Add 1.5 ml of pre-warmed Gentle Cell Dissociation Reagent to each plate and incubate for 5 min at 37 °C until most cells detach from the surface of the culture vessels. Tap plates gently to dislodge cells still attached.
- Use a pipette to gently rinse the surface of the plates with the Gentle Cell Dissociation Reagent already in the plates to detach any remaining cells.
- Using a pipette, transfer the cell suspension to a 15-ml conical tube.
- Add 1 ml of DPBS to each plate to collect residual cells and transfer the cell suspension to the conical tube.
- Gently pipet the cell suspension up and down 3 times with a 5-ml or 10-ml pipette to break up the cell clumps.
- Centrifuge the cells at 300 x g for 5 min.
- Aspirate the supernatant and re-suspend the cells in pre-warmed Neural Progenitor Cell (NPC) Medium (i.e., 10 ml for all cells from each plate).
- Plate the cells suspended in NPC Medium onto a 10 cm Petri dish.
- Culture the cells in a CO2 incubator for 2 days. During this time, NPCs will form aggregations while floating in NPC medium.
- Once aggregates have reached an appropriate size of approximately 70-200 µm, prepare a 10 cm tissue culture dish coated with 5 ml of Matrigel® for at least 1 h. If few aggregates have formed, plate cells in a 60 mm dish instead.
- Using a 5-ml or 10-ml pipette, take up and pass the NPC medium through a cell strainer to collect NPC aggregations.
- Reverse the strainer and pass 10 ml of fresh pre-warmed NPC medium through strainer where the cell aggregates are bound so that they are transferred onto the Matrigel®-coated 10 cm plate.
- Culture the cells in a CO2 incubator to allow for NPC aggregates to attach to the coated dish and migrate and proliferate.
- Change medium every 2-3 days until cells reach confluence and are ready for passaging or cryopreservation. Dissociate using warm accutase at 37 °C for five minutes.
- To assess the purity of NPC culture, fix cells and check for NPC markers using ICC (see Figure 3).
- To cryopreserve NPCs, freeze in an 80/20 mix of FBS/DMSO. Store at -80 °C for use within a few months, or in liquid nitrogen for long-term storage.
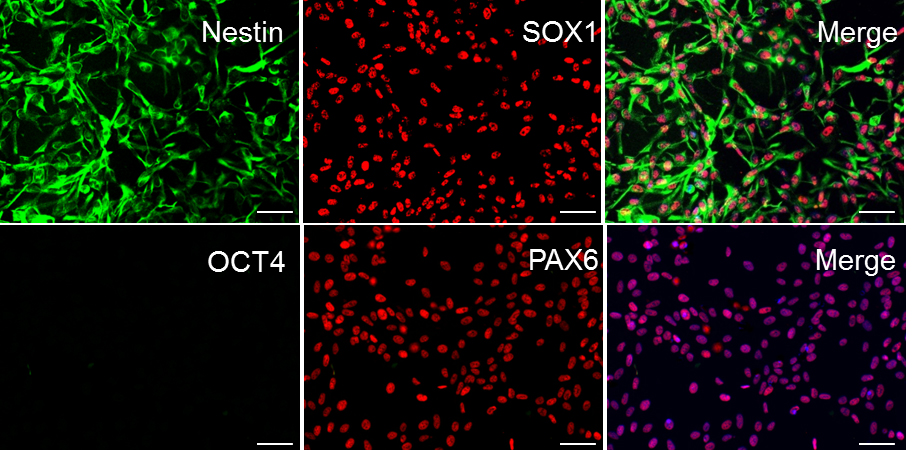
Figure 3. Sample ICC staining for high-quality NPC cultures. In order for differentiation to proceed effectively, ensure that iPSC cultures uniformly express Nestin, SOX1, and PAX6. NPC cultures should have no expression of the pluripotent marker OCT4 (DAPI shown in blue in merge of PAX6 and OCT4; all cells express PAX6, i.e., 100% purity of the culture). Scale bars represent 100 μm.
- Differentiation of forebrain NPCs into neurons
- Plate NPCs on a tissue culture dish that is coated with Matrigel®. Wait until cells have achieved 70%-95% confluency before beginning differentiation.
- Once NPCs have reached desired confluency, aspirate media and replace with an equal volume of Neuronal Media.
- Every 2-3 days, aspirate half of the media in the plate and replace with fresh Neuronal Media.
Note: As some media will be lost to evaporation, you may need to add a little more media than you remove from the plate in order to keep the media volume stable over time. - Continue to change the media until neurons reach the desired stage of development. For example morphologies of developing forebrain neurons, consult Figure 4. For example ICC characterization of forebrain neurons, consult Figure 5.
Notes:- The purity of your line will be very easily detected during this stage of development. Cell lines that contain a high percentage of NPCs will rapidly polarize and form neuronal projections (usually around Days 2-5), whereas lines that contain a high percentage of non NPC cells (Astrocytes, neural crest cells, etc.) will not.
- There is variability in how long neurons in a particular plate will take to reach a certain stage of development depending on line, clone, passage number etc. However, we have found that neurons in good quality cultures consistently achieve the following landmarks by the following number of days into differentiation.
Day 5 = Cells are post-mitotic
Day 15 = Cells have clearly polarized axons and dendrites, and have clearly detectable electrophysiological properties, such as action potentials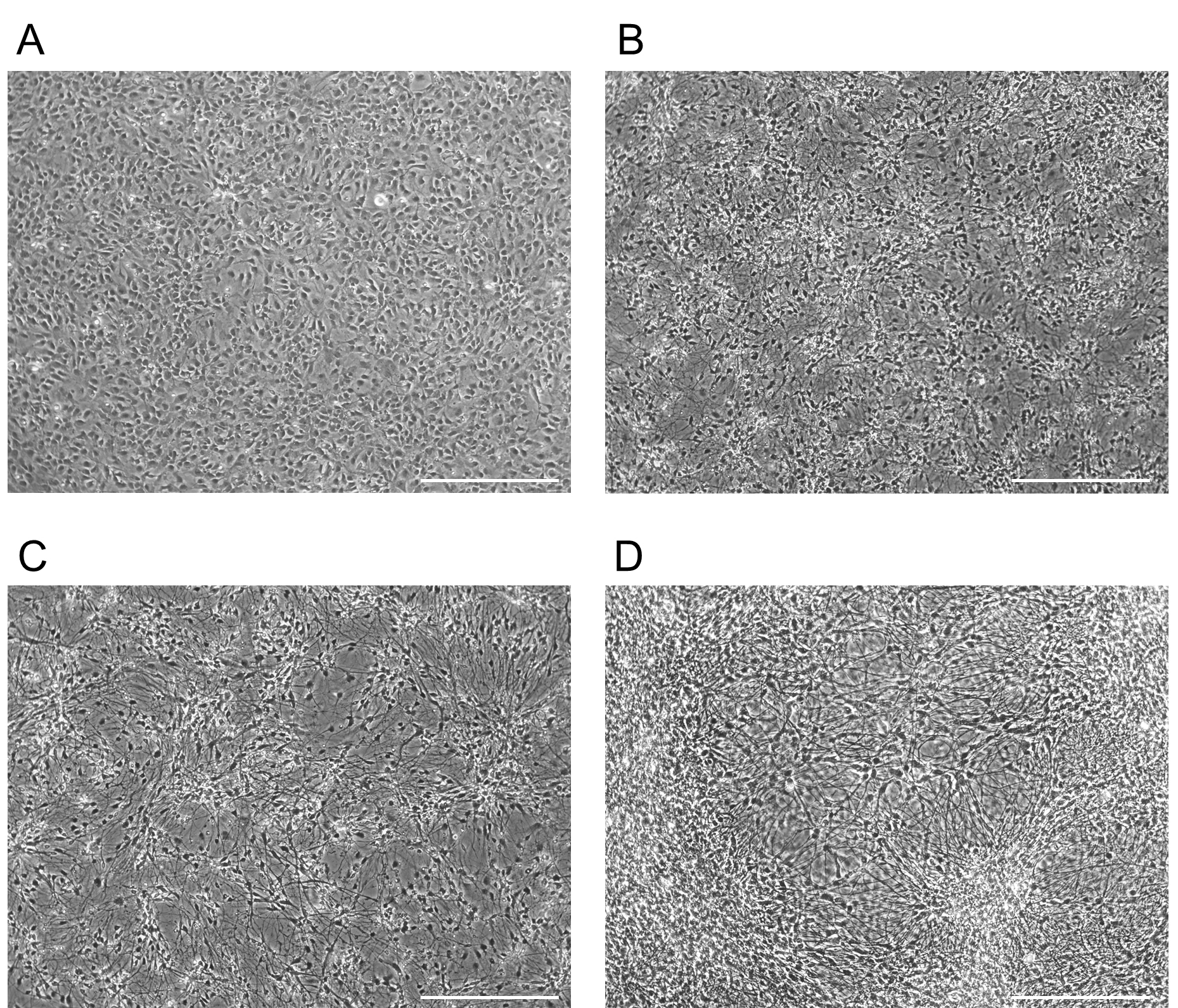
Figure 4. Example morphology for forebrain NPCs differentiating into neurons. Morphology of a forebrain NPC culture differentiating into neurons. Images taken at D0 (A), D5 (B), D15 (C), D30 (D). Scale bars represent 130 µm. - To assess the purity of your neuronal culture, fix cells and check for forebrain markers using ICC. To assess the quality of the neurons produced, ensure that the cells display proper electrophysiological properties (see Data analysis).
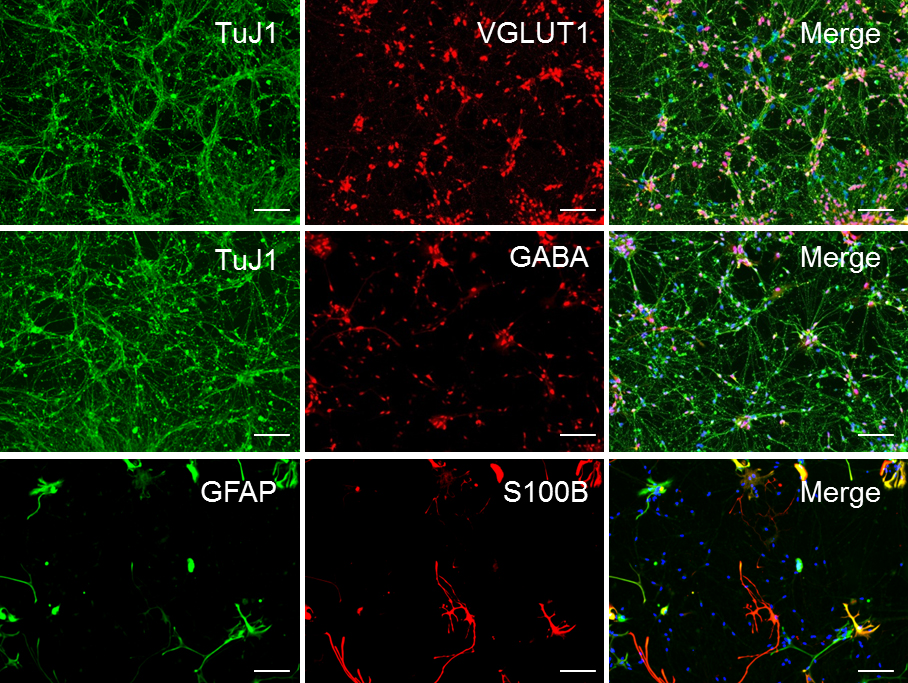
Figure 5. Sample ICC staining for high-quality forebrain neuronal. Representative ICC of forebrain neuronal culture following 30 days of differentiation (D30) from NPCs, demonstrating the relative abundance of glutamatergic, GABAergic, and astrocytic markers in the population. These cultures are approximately 65% glutamatergic, 30% GABAergic, and 5%-10% astroglial. Scale bars represent 50 μm.
- Assessment of culture purity using immunocytochemistry
Note: The following steps are described assuming cells are plated on glass coverslips coated with poly-ornithine and laminin (For more details, see Recipes).- Fix samples in 4% paraformaldehyde (PFA) diluted in PBS. Incubate at room temperature for 15 min.
- Wash samples with PBS (3 x 15 min) at room temperature.
- Permeabilize samples by incubation in PBS + 0.1% Triton X-100 at room temperature for 10 min.
- Aspirate permeabilization buffer and replace with 5% BSA diluted in PBS to block samples. Incubate at room temperature for 60 min.
- Prepare working stocks of primary antibodies by diluting in blocking 5% BSA-PBS. See Table 1 for recommended working dilutions and antibodies for different cells types. Coat coverslips in primary antibody solution and incubate overnight at 2-8 °C.
Table 1. Antibodies used in immunocytochemistry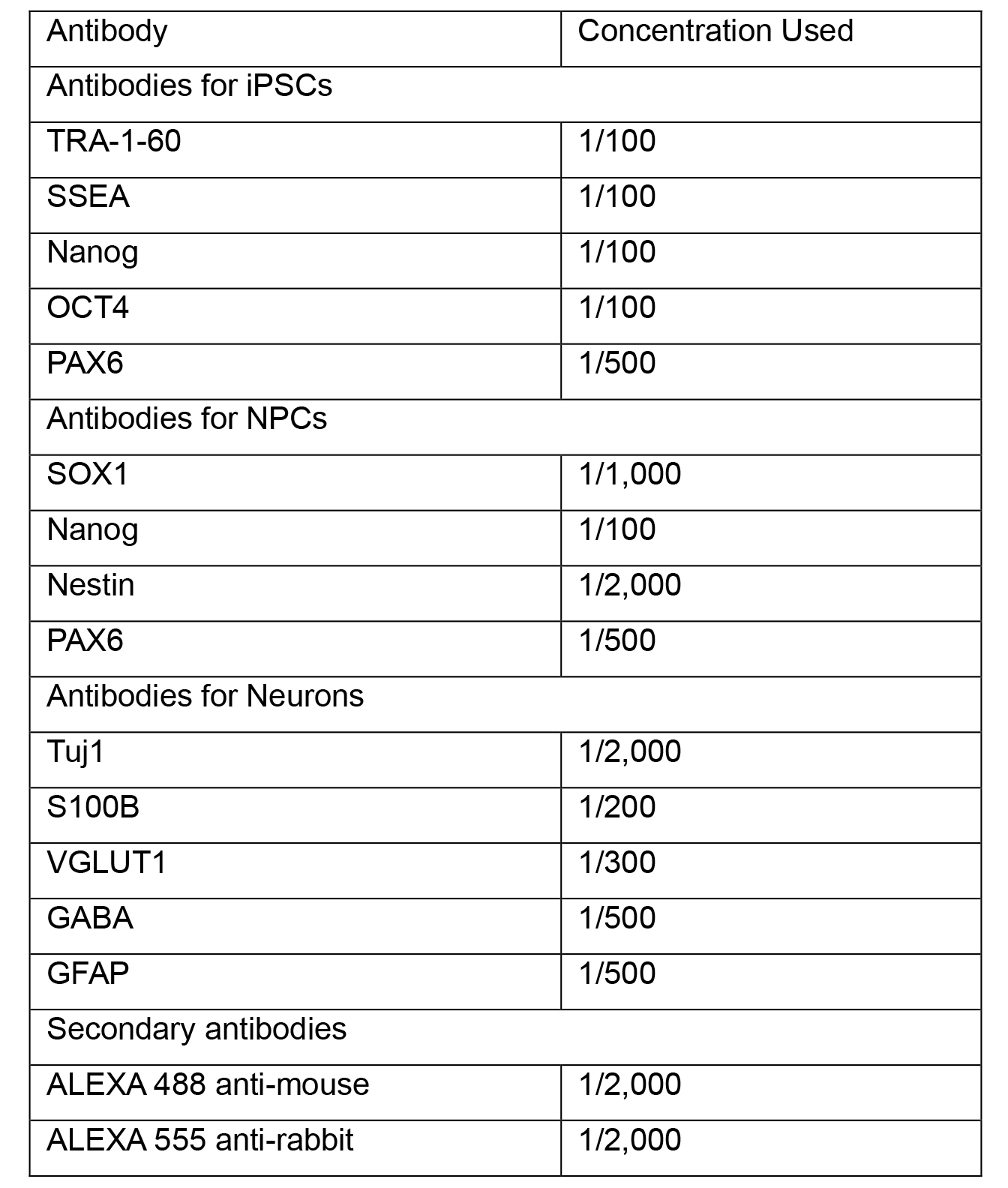
- Wash samples with PBS (3 x 15 min) at room temperature.
- Aspirate PBS and add secondary antibodies diluted in 5% BSA-PBS. See Table 1 for recommended secondary antibodies and dilutions. Incubate coverslips at room temperature, away from light for one hour.
- Wash samples with PBS (3 x 15 min) at room temperature.
- If desired, add DAPI diluted in PBS, incubate for 5 min in room temperature.
- Add a drop of Vectashield® to a glass slide. Carefully use a needle and forceps to transfer the coverslip, cell-side down, to the slide. Seal using nail polish.
- Assessment of neuronal quality using Electrophysiology
- Pull pipettes from glass capillaries. Their resistance should range from 3 to 6 MΩ when filled with the internal pipette solution.
- Transfer individual coverslips containing differentiated human iPSC-derived neurons into a heated recording chamber and continuously perfused (1 ml/min) with BrainPhys Neuronal Medium without phenol bubbled with a mixture of CO2 (5%) and O2 (95%) and maintained at 35 °C using an automatic temperature controller.
- Choose the cells that you will record from.
- Fill the pipette with the internal pipette solution and place it in the electrode holder. Lower the pipette to place it into the external solution. After compensating offsets, approach the pipette to the chosen cell with the help of the remote micromanipulator to form a high resistance cell-attached seal.
- Once the seal is formed and the whole cell configuration is established, compensate series resistance at 80%-90%.
- Wait for 5 to 10 min before starting to record. This allows the cell content to equilibrate with the internal pipette solution.
- For acquisition, set your filter at 2 kHz and your sampling rate at 20 kHz.
- Once whole-cell recording had been established, recordings of fundamental neuronal properties, including rheobase, resting membrane potential, action potential parameters and spontaneous postsynaptic currents can be performed. Add NMDA via pipette. Assessed neurons should be hold neurons in voltage clamp at -70 mV except when examining changes in the resting membrane potential and rheobase, which should be performed in current clamp. Clampex and GraphPad Prism 7 are recommended software to use to display data. For example electrophysiological recordings obtained from cortical neurons, consult Figure 6.
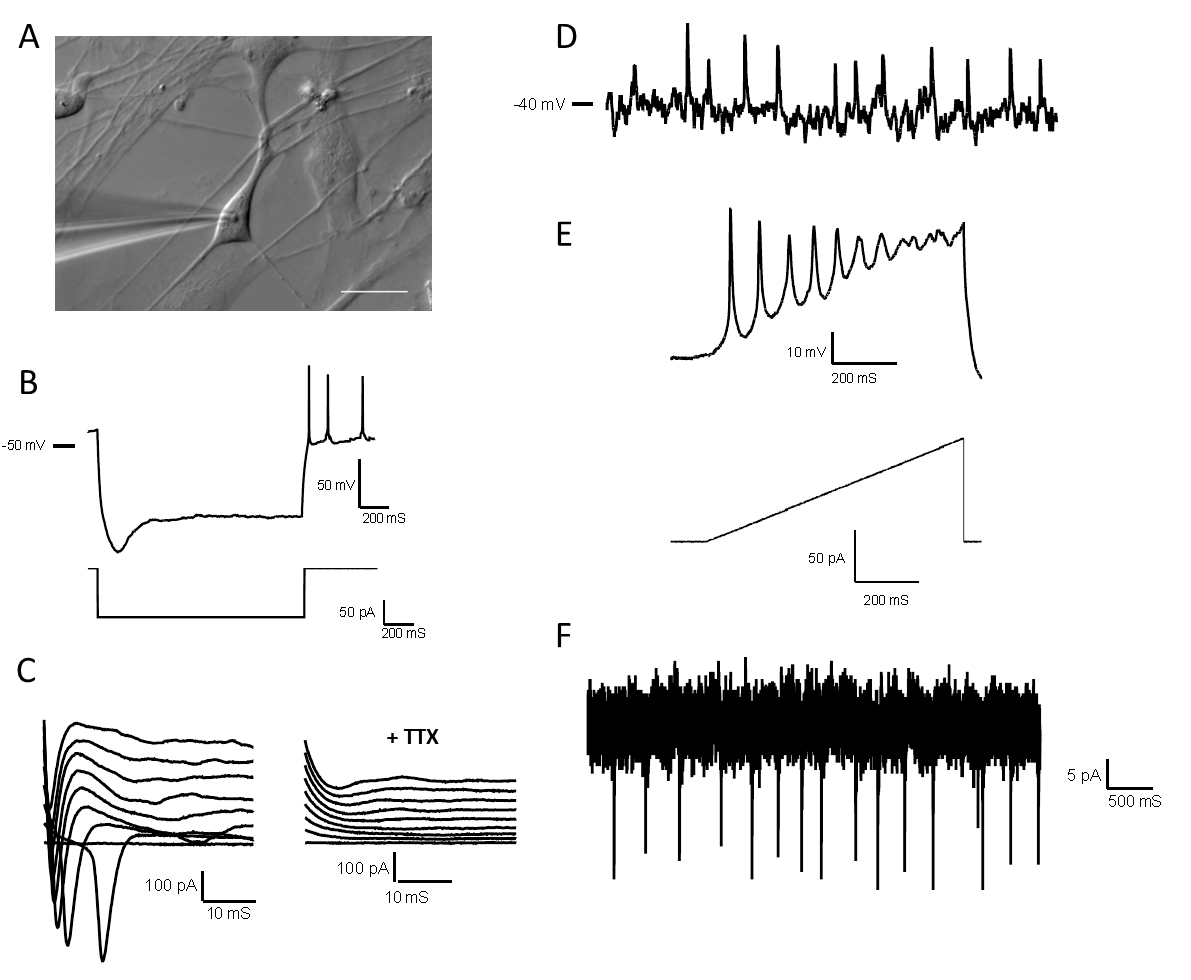
Figure 6. Electrophysiological properties of high-quality forebrain neurons. A. Differential image contrast of a glass microelectrode recording from a single neuron in the whole-cell configuration. Scale bars represent 20 μm. B. A hyperpolarizing pulse showing a depolarizing sag followed by multiple rebound action potentials. C. Left: Representative traces of voltage clamp recordings showing inward Na+ currents; Right: Sodium current traces disappear after tetrodotoxin (TTX) 1 μM application. D. Representative current-clamp recording from a spontaneously active neuron with resting membrane potential -40 mV. E. Representative recording showing action potentials fired by forebrain neurons during a current ramp protocol. F. Representative voltage-clamp recording from a neuron with spontaneous synaptic input. All electrophysiological data was obtained from D14 neurons.
Data analysis
Electrophysiological data should be processed with currents should be filtered at 2 kHz and digitized at 20 kHz. Values should be reported correcting for the nominal membrane potential in voltage- and current-clamp recordings for the calculated 10-mV liquid junction potential.
Recipes
- Media
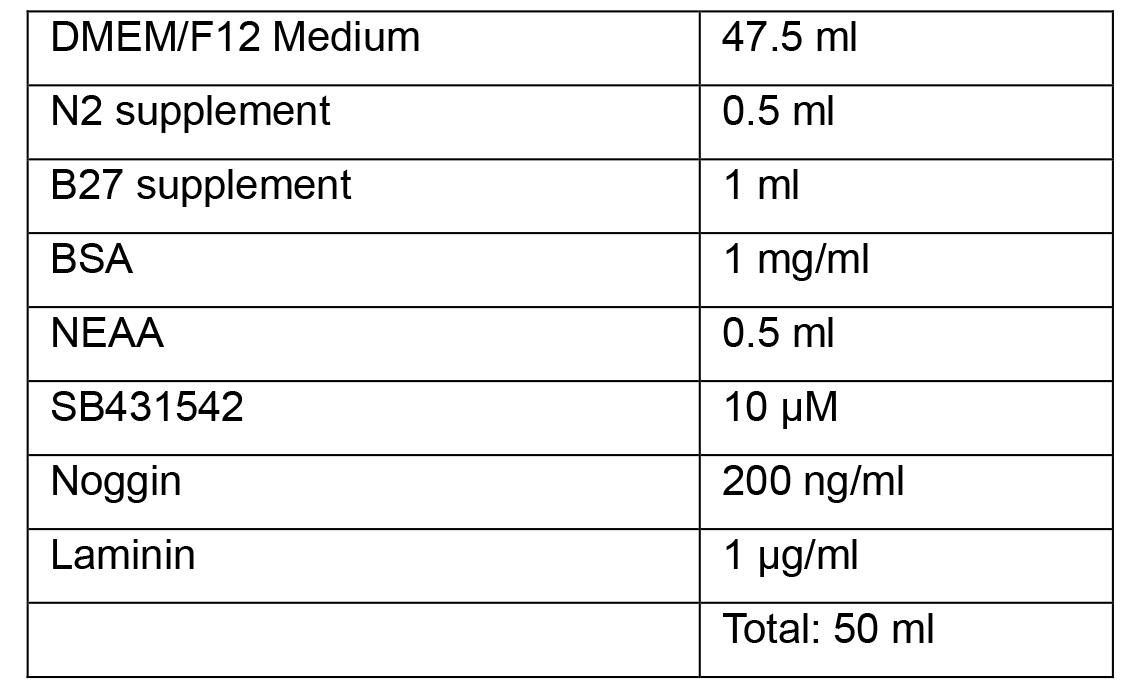
Note: All media as given as recipes for 50 ml as it is possible to prepare this volume in a single 50 ml tube, suitable for warming in a bead or water bath.- Neural Induction Medium 1
- Neural Induction Medium 2

Note: Complete Neural Induction Medium 1 and 2 can be stored at 2-8 °C in the dark for up to 2 weeks. Warm the Neural Induction Medium in a 37 °C water bath for 5-10 min before using. Do not warm the Neural Induction Medium in a 37 °C water bath for longer than 10 min, as this may cause degradation of the medium. - Neural Progenitor Media
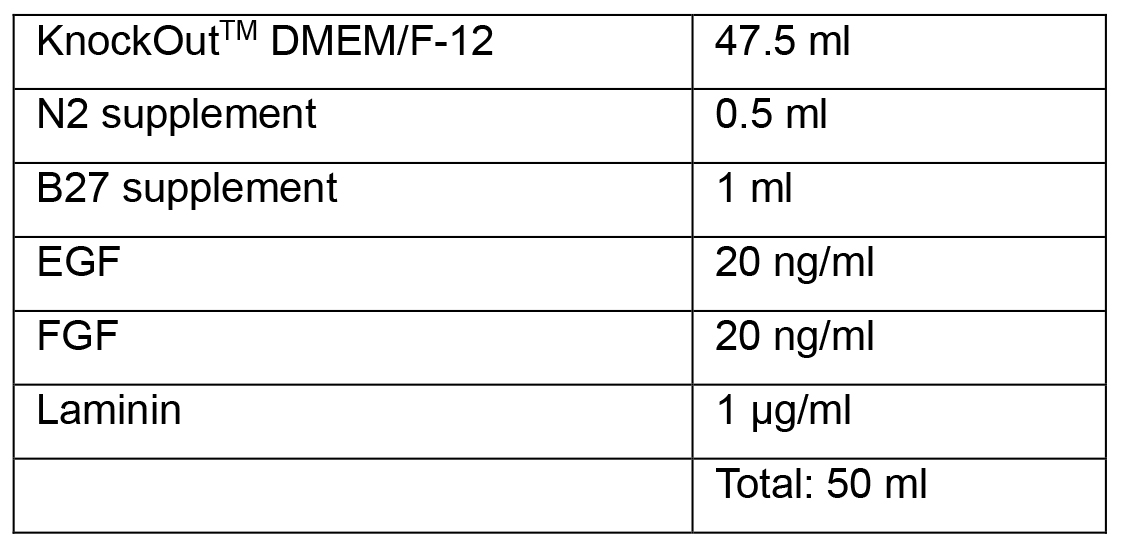
- Neuronal Media
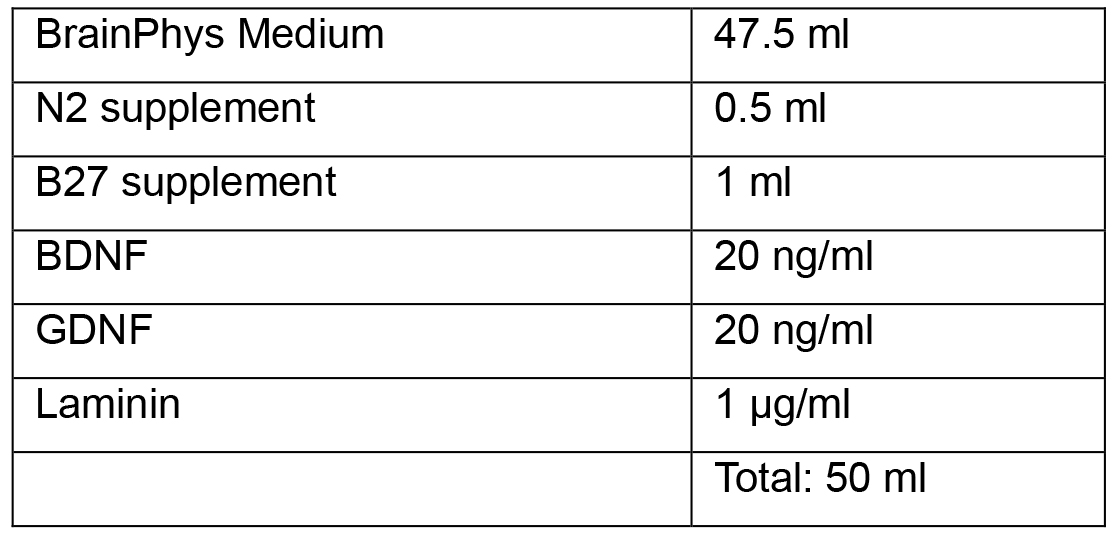
- Neural Induction Medium 1
- Buffers and solutions
- Internal pipette solution
5 mM HEPES
2 mM KCl
136 mM potassium gluconate
5 mM EGTA
5 mM ATP-Mg2+
8 mM creatine phosphate
0.35 mM guanosine triphosphate
The pH is adjusted to 7.2 with KOH and the osmolality is adjusted with distilled water or concentrated potassium gluconate if needed to between 295 and 298 mOsm.
Note: The difference in osmolality between Internal and external solutions should be near 5%. - Culture Dish Coating with Matrigel®
- Thaw a frozen aliquot of Matrigel® (250 μl) from -80 °C by placing it in a 4 °C fridge for 1 h
- To create a working solution, dilute the thawed Matrigel® in 25 ml of cold PBS or media
Note: Diluting Matrigel® in DMEM or other media with a strong coloration will make it easier to determine that the whole dish is evenly covered. - Quickly cover the whole surface of each culture vessel with the appropriate amount matrix solution (Table 2)
- Incubate the culture vessels in a 37 °C, 5% CO2 incubator for at least 1 h
- The culture vessels are now ready for use. Just before use, aspirate the diluted Matrigel® solution from the culture vessels. Cells can be plated directly onto the Matrigel®-coated culture vessels without rinsing
Note: Coated culture vessels can also be stored at 2-8 °C for up to one week. When storing, seal culture vessels with Parafilm® laboratory film to prevent drying. Before using, warm up the coated culture vessels stored at 2-8°C at room temperature for 30 min.
Table 2. Required volume of Matrigel® matrix solution for coating different culture vessels
- Coating glass coverslips with Poly-ornithine and laminin
- Place glass coverslips in a Petri dish or suspension plate
- Sterilize coverslip by exposing plates to UV radiation for 20 min
- Coat coverslips in 100 μl of 50 μg/ml polyorinithine in PBS. Wait two hours
- Aspirate solutions. Wash once using PBS, then coat coverslip in 10 μg/ml laminin diluted in PBS
- Incubate at 37 °C for two hours
- Aspirate solution, coat coverslips in 10% FBS DMEM
- The plates are now ready to use. For best results, use within 24 h of preparation. Before plating cells, wash plates once with PBS, as FBS may influence differentiation
- Internal pipette solution
Acknowledgments
This work was supported by grants from the Canada Research Chairs program and the Canadian Institute of Health Research to CE. Scott Bell is funded by the (Fonds de la recherche en santé du Québec) FRQS, Nuwan Hettige is funded by FRQS, Malvin Jefri is supported by the Government of Malaysia, and Lilit Antonyan is supported by CONACYT (Mexico). This protocol was adapted from previous work (Bell et al., 2017 and 2018).
Competing interests
Carl Ernst is president of ManuStem.com.
Ethics
All work was approved by the Research Ethics Board of the Douglas Hospital.
References
- Auld, D. S., Kornecook, T. J., Bastianetto, S. and Quirion, R. (2002). Alzheimer's disease and the basal forebrain cholinergic system: relations to β-amyloid peptides, cognition, and treatment strategies. Prog Neurobiol 68(3): 209-245.
- Baxter, M. G. and Chiba, A. A. (1999). Cognitive functions of the basal forebrain. Curr Opin Neurobiol 9(2): 178-183.
- Bell, S., Maussion, G., Jefri, M., Peng, H., Theroux, J. F., Silveira, H., Soubannier, V., Wu, H., Hu, P., Galat, E., Torres-Platas, S. G., Boudreau-Pinsonneault, C., O'Leary, L. A., Galat, V., Turecki, G., Durcan, T. M., Fon, E. A., Mechawar, N. and Ernst, C. (2018). Disruption of GRIN2B impairs differentiation in human neurons. Stem Cell Reports 11(1): 183-196.
- Bell, S., Peng, H., Crapper, L., Kolobova, I., Maussion, G., Vasuta, C., Yerko, V., Wong, T. P. and Ernst, C. (2017). A rapid pipeline to model rare neurodevelopmental disorders with simultaneous CRISPR/Cas9 gene editing. Stem Cells Transl Med 6(3): 886-896.
- Chandrasekaran, A., Avci, H. X., Ochalek, A., Rosingh, L. N., Molnar, K., Laszlo, L., Bellak, T., Teglasi, A., Pesti, K., Mike, A., Phanthong, P., Biro, O., Hall, V., Kitiyanant, N., Krause, K. H., Kobolak, J. and Dinnyes, A. (2017). Comparison of 2D and 3D neural induction methods for the generation of neural progenitor cells from human induced pluripotent stem cells. Stem Cell Res 25: 139-151.
- Donovan, A. P. and Basson, M. A. (2017). The neuroanatomy of autism - a developmental perspective. J Anat 230(1): 4-15.
- Heath, R. G. (1976). Brain function in epilepsy: midbrain, medullary, and cerebellar interaction with the rostral forebrain. J Neurol Neurosurg Psychiatry 39(11): 1037-1051.
- Marchetto, M. C., Brennand, K. J., Boyer, L. F. and Gage, F. H. (2011). Induced pluripotent stem cells (iPSCs) and neurological disease modeling: progress and promises. Hum Mol Genet 20(R2): R109-115.
- McColgan, P. and Tabrizi, S. J. (2018). Huntington's disease: a clinical review. Eur J Neurol 25(1): 24-34.
- Morgane, P. J., Galler, J. R. and Mokler, D. J. (2005). A review of systems and networks of the limbic forebrain/limbic midbrain. Prog Neurobiol 75(2): 143-160.
- Pasca, S.P., Portmann, T., Voineagu, I., Yazawa, M., Shcheglovitov, A., Pasca, A.M., Cord, B., Palmer, T.D., Chikahisa, S., Nishino, S., Bernstein, J. A., Hallmayer, J., Geschwind, D. H. and Dolmetsch, R. E. (2011). Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med 17: 1657-1662.
- Schwartz, J. R. and Roth, T. (2008). Neurophysiology of sleep and wakefulness: basic science and clinical implications. Curr Neuropharmacol 6(4): 367-378.
- Shi, Y., Inoue, H., Wu, J. C. and Yamanaka, S. (2017). Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov 16(2): 115-130.
- Shi, Y., Kirwan, P. and Livesey, F. J. (2012). Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat Protoc 7(10): 1836-1846.
- Srikanth, P. and Young-Pearse, T. L. (2014). Stem cells on the brain: modeling neurodevelopmental and neurodegenerative diseases using human induced pluripotent stem cells. J Neurogenet 28(1-2): 5-29.
- Venere, M., Han, Y. G., Bell, R., Song, J. S., Alvarez-Buylla, A. and Blelloch, R. (2012). Sox1 marks an activated neural stem/progenitor cell in the hippocampus. Development 139(21): 3938-3949.
- Yuan, F., Fang, K. H., Cao, S. Y., Qu, Z. Y., Li, Q., Krencik, R., Xu, M., Bhattacharyya, A., Su, Y. W., Zhu, D. Y. and Liu, Y. (2015). Efficient generation of region-specific forebrain neurons from human pluripotent stem cells under highly defined condition. Sci Rep 5: 18550.
- Zhang, J. and Jiao, J. (2015). Molecular biomarkers for embryonic and adult neural stem cell and neurogenesis. Biomed Res Int 2015: 727542.
- Zhang, X., Huang, C.T., Chen, J., Pankratz, M.T., Xi, J., Li, J., Yang, Y., LaVaute, T.M., Li, X.-J., Ayala, M., Bondarenko, G. I., Du, Z. W., Jin, Y., Golos, T. G. and Zhang, S. C. (2010). Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell 7(1): 90-100.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Bell, S., Hettige, N. C., Silveira, H., Peng, H., Wu, H., Jefri, M., Antonyan, L., Zhang, Y., Zhang, X. and Ernst, C. (2019). Differentiation of Human Induced Pluripotent Stem Cells (iPSCs) into an Effective Model of Forebrain Neural Progenitor Cells and Mature Neurons. Bio-protocol 9(5): e3188. DOI: 10.21769/BioProtoc.3188.
Category
Stem Cell > Pluripotent stem cell > Cell differentiation
Neuroscience > Cellular mechanisms > Cell isolation and culture
Cell Biology > Cell isolation and culture > Cell differentiation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.