- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

QsRNA-seq: A protocol for Generating Libraries for High-throughput Sequencing of Small RNAs


Published: Vol 9, Iss 5, Mar 5, 2019 DOI: 10.21769/BioProtoc.3179 Views: 8281
Reviewed by: Gal HaimovichOmar AkilAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






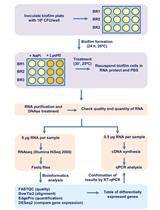

Abstract
Small RNAs (sRNAs) are 20-30 nt long non-coding RNA molecules that regulate essentially all cellular processes. Besides being an intensively studied topic in academic research, sRNAs also hold a promise as clinical biomarkers. While the need for expressional profiling of sRNAs is growing, preparation of sRNA libraries for high-throughput sequencing (HTS) remains technically challenging, due to their small size. The common PAGE-based protocol is time-consuming and inefficient due to material loss, while gel-free protocols generate libraries of insufficient quality. To overcome these shortcomings, we modified the conditions of size-selection by Solid Phase Reversible Immobilization (SPRI) in a way that allows separation of nucleic acids shorter than 100 nt and differing in length by only 20 nt. Implementing the method for preparation of small RNA libraries for HTS resulted in QsRNA-seq, a gel-free, fast and easy-to-perform protocol, amenable to automation, generating very clean libraries that result in high-depth expression data. The protocol also utilizes Unique Molecular Identifiers (UMI) for reduction of library preparation biases and to quantify expression levels. QsRNA-seq provides an excellent solution to the growing needs for small RNA expression profiling for research clinical use.
Keywords: Small RNABackground
Although small RNAs, which are 20-30 nt long regulatory non-coding RNA molecules, constitute a tiny percentage of the total cellular RNA, they have a huge impact on many cellular processes (including, gene regulation, organism development, adaptation to changing environment and so on) (Jost et al., 2011). There are several classes of small RNA (Youngman and Claycomb, 2014) with microRNAs (miRNAs) being the most extensively studied (Bartel, 2009).
Besides being of a great interest for basic research, miRNAs hold promise as diagnostic markers. Due to their exceptional stability, intact miRNAs are present in preserved tumor samples (formalin-fixed paraffin-embedded [FFPE]), which exhibit a near total mRNA degradation (Nagy et al., 2016). Moreover, stable miRNAs circulate in body fluids such as plasma, urine and sputum of healthy and diseased patients (Mitchell et al., 2008). Liquid biopsy of circulating miRNAs can serve as potential markers for a broad range of human diseases especially for diseases for which ordinary biopsies cannot be obtained, such as cardiovascular diseases, and psychiatric diseases (Kim, 2015; Backes et al., 2016).
While assessing the repertoire and abundance of cellular miRNAs became an important aspect in many studies, the tools to do so lag behind. High-throughput sequencing (HTS) is a gold standard method for mRNA profiling, extensively used in both basic research and clinical settings. In striking contrast, HTS profiling of miRNAs is not widespread, as preparation of small RNA libraries remains technically challenging.
To become compatible with Illumina or other HTS platforms, small RNAs are ligated from both sides (5’ and 3’) to oligonucleotide adapters and then reverse transcribed. The resulting small RNA library is then amplified by PCR with primers complementary to the 5’ and 3’ adapters that include sequences recognized by the sequencing machinery (Lu et al., 2007). If small RNAs are not separated from other RNA species, in particular from tRNA, which are highly abundant and close in size (~70 bp), the resulting libraries will be highly contaminated by irrelevant sequences. In addition, 3’ and 5’-adapters ligate to each other, forming adapter-dimers, which contaminate libraries with null sequences. Thus, for generation of a high-quality sRNA library, a preparation protocol must enable separation of small RNA from total RNA and separation of the ligation products from free adapters. Size-selection on Solid Phase Reversible Immobilization (SPRI) paramagnetic beads (Lundin et al., 2010), used in preparation of mRNA and DNA libraries, was not suitable for preparation of small RNA libraries, as the lower limit of size discrimination is 100 nt (Paithankar et al., 1991). The only remaining option, electrophoresis on PAGE gel, is cumbersome, time-consuming, requires technical skills, results in a huge loss of the product and eliminates the possibility of automatization. Preventing adapter-dimer contamination by molecular biology strategies (Kawano et al., 2010; Vigneault et al., 2012) is not a good alternative to size-selection as it does not solve the problem of contamination by irrelevant sequences.
To allow easy size selection of short nucleic acid molecules, we modified the SPRI method by using a combination of two crowding agents, Polyethylene Glycol (PEG) and Isopropanol. This modification enables separation of nucleic acid fragments shorter than 100 nt, differing in length by only 20 nt (Fishman et al., 2018). Exploiting this new separation method, we designed a protocol for preparation of small RNA libraries for high-throughput sequencing based on Lau et al. (2001) and we named the protocol QsRNA-seq. The input material for the protocol is either total RNA or low molecular weight RNA fraction (LMW RNA, < 200 nt). During the protocol three SPRI-based size selection steps are performed, each using different conditions, to allow separation of sRNA from total RNA and of ligation products from free adapters after each ligation (for the protocol outline see the Procedure section below). In addition, the protocol supports the possibility of multiplexing samples by adding a 4-nt barcode to the 5’-adapter. The protocol also supports using unique molecular identifiers (UMI) for reduction of ligation bias (Fuchs et al., 2015) and correction of amplification bias (Kivioja et al., 2011) by adding an 8-nt random sequence to the 5’-adapter as well. To correct the amplification bias, identical small RNA sequences with the same UMI are considered to be amplification products and merged to one sequence (i.e., collapsed) during data processing. By comparing libraries generated from the same RNA using adapters with the same barcode either with UMI or without UMI, we found that adding UMI significantly reduced both biases (Fishman et al., 2018).
QsRNA-seq has several significant advantages over the existing protocols. The entire protocol can be performed in one day and does not require high manual skills. Because it does not require any gel use there is less material lost, which enables small quantities of the input RNA to be analyzed. Moreover, it allows simultaneous processing of many samples as well as automation, turning the preparation of small RNA libraries into a routine procedure. In addition, usage of UMI allows reduction of library preparation biases, resulting in more reliable quantitation. To summarize, QsRNA-seq is an easy-to-use protocol that generates reliable and quantifiable high-throughput small RNA libraries.
Materials and Reagents
- RNase-free plastic pipette tips with filter (10 μl, 20 μl, 200 μl, 1,000 μl)
- 1.7 ml RNase-free microtubes
- 0.2 ml RNase-free PCR tubes
- miRVana miRNA Isolation Kit (Thermo Fisher Scientific, catalog number: AM1560) or TRIzol reagent (Ambion, catalog number: 15596026) with Direct-Zol RNA MiniPrep Plus (Zymo Research, catalog number: R2072)
- RiboLock RNase Inhibitor (Thermo Fisher Scientific, catalog number: EO0381)
- T4 RNA ligase 1 (ssRNA Ligase) (NEB, catalog number: M0204S)
- T4 RNA ligase 2, truncated (NEB, catalog number: M0242S)
- RppH enzyme (NEB, catalog number M0356S)
- QScript Flex cDNA synthesis kit (Quanta, catalog number: 95049-025)
- Phusion High-Fidelity DNA Polymerase (NEB, catalog number: M0530S)
- Agencourt Ampure XP (Beckman Coulter, catalog number: A63881) or SPRI select reagent Kit (Beckman Coulter, catalog number: B23319)
- RNA BR assay kit (Quibit, catalog number: Q10210)
- dsDNA HS assay kit (Quibit, catalog number: Q10210)
- PEG 8000 (NEB, catalog number: B1004)
- Nuclease-free water (Sigma-Aldrich, catalog number: W4502)
- Ethyl Alcohol absolute
- Isopropyl-alcohol (Isopropanol) chemical grade
- Ethidium Bromide (hylabs, catalog number: BP451)
- Gel loading dye purple (6x) (NEB, catalog number: B7024)
- [Optional] Low-range ultra agarose (Bio-Rad, catalog number: 161-3107)
- ATP (NEB, catalog number: P0756S)
- DMSO (NEB, catalog number B0515)
- Tris (Spectrum, catalog number: s1519)
- Boric Acid (Bio-Lab, catalog number: 000201059100)
- EDTA (J.T. Baker, catalog number: 8993-01)
- 50 bp DNA ladder (NEB, catalog number: N3236s)
- Oligonucleotides: all the oligonucleotides were ordered from IDT (www.idtdna.com)
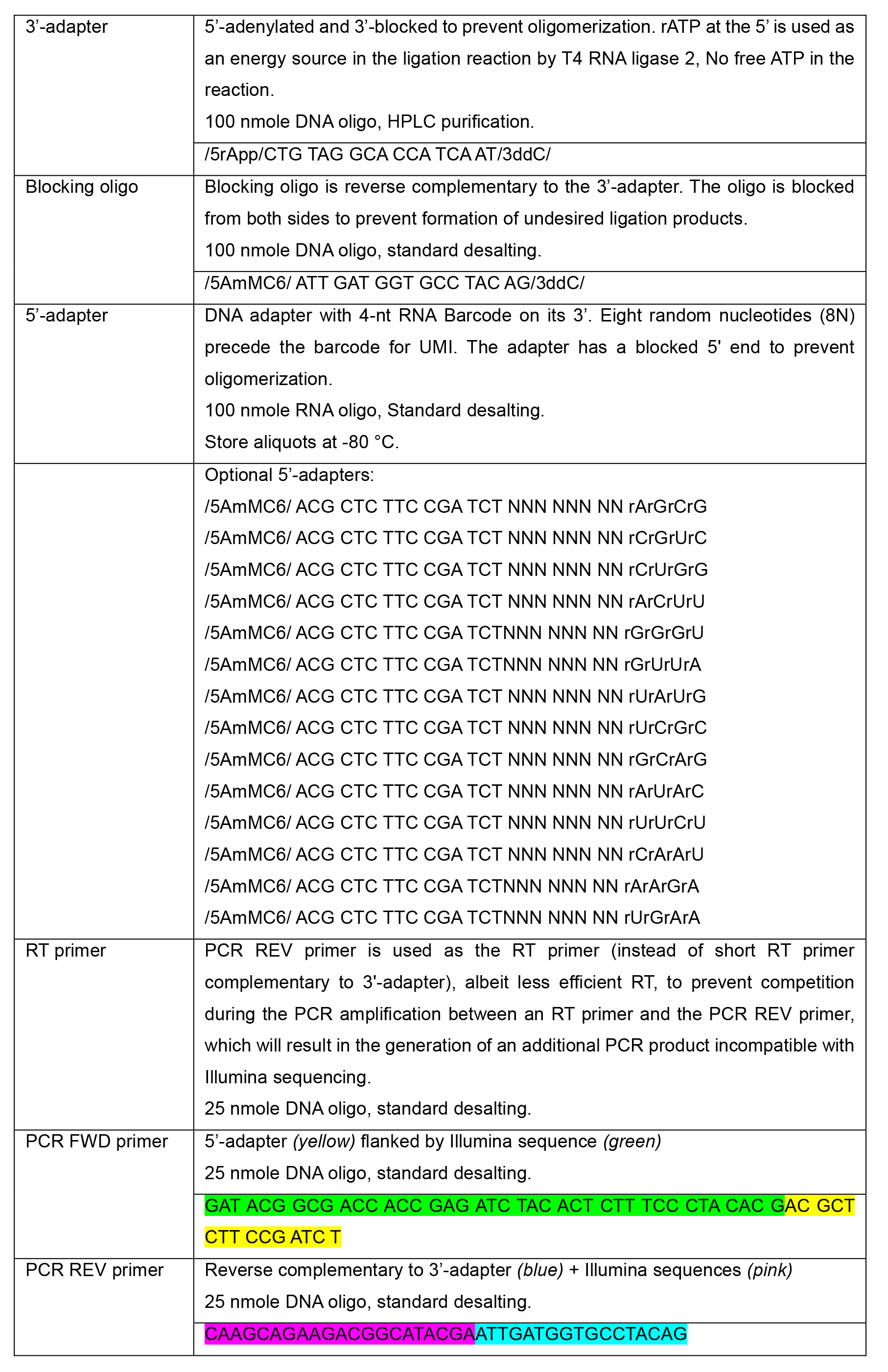
- [Optional] NucleoSpin gel and PCR Clean-up kit (Macherey-Nagel, catalog number: 740609.50) or Gel/PCR DNA fragments Extraction kit (Geneaid, catalog number: DF004)
- 5x TBE (see Recipes)
Equipment
- Micro-pipettors (Gilson, Pipetman)
- DynaMag-2 magnetic stand (Thermo Fisher Scientific, catalog number: 12321D)
- Benchtop microcentrifuge, non-refrigerated (5424 with Rotor FA-45-24-11, Eppendorf, catalog number: 5424000010)
- Vortex mixer, flat head, Vortex-Genie 2 (Scientific Industries, model: SI-0266)
- Cooling block for 1.7 ml tubes (MS, catalog number: MC-0203)
- Agarose gel electrophoresis apparatus
- Electrophoresis power supply
- Gel imager (Bio-Rad, Gel Doc XR+)
- Fluorometer, Quibit 2.0 (Invitrogen, model: Q32866)
- Thermocycler (Biometra, TProfessional)
- Tapestation/Bioanalyzer
Procedure
Protocol outline: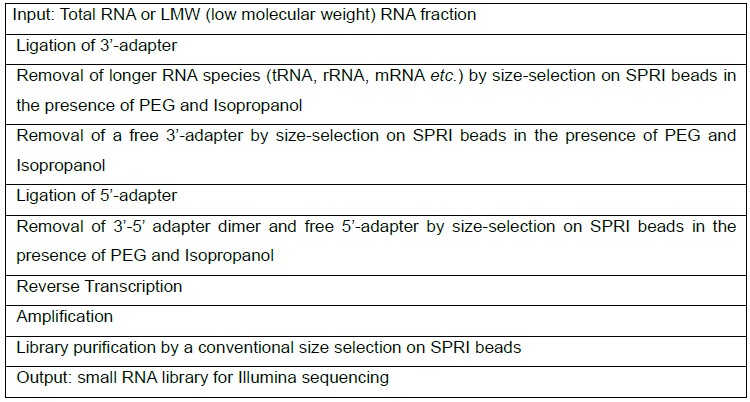
- Preparation of RNA samples
- Prepare your RNA samples using column based RNA extraction kits
Notes:- RNA origin: using this protocol we successfully prepared libraries from C. eleganss, Drosophila, human, Arabidopsis and fungi RNA. Thus we assume it will work for other species as well.
- Column based kits enable non-biased recovery of small RNA. Do not use protocols based on precipitation of RNA by isopropanol, as they result in a poor recovery of small RNA. In our hands, using either MiRVana miRNA isolation kit (Ambion) or Direct-Zol RNA miniprep (Zymo) resulted in excellent libraries.
- Some kits, for example MiRVana kit (Ambion) allow extraction of either total RNA or of separated High molecular weight (HMW) and Low molecular weight (LMW) fractions. Using LMW as starting material results in cleaner libraries.
- Measure sample concentration
Note: We use Qubit Fluorometer, but NanoDrop can be used as well. - Estimate RNA quality by electrophoresis or better by Bioanalyzer/TapeStation
Note: To avoid library contamination by degradation products of longer RNA types, use only high-quality RNA (as indicated by sharp non-degraded rRNA bands if evaluating by gel electrophoresis or RNA integrity number [RIN] > 8.5 if using Bioanalyzer/TapeStation).
- Prepare your RNA samples using column based RNA extraction kits
- Ligation of 3’-adapter
Note: Ligation reaction between sRNA and 5’-Adenylated 3’-adapter is catalyzed by T4 RNA ligase 2 truncated in the absence of free ATP.- Combine in an RNase-free 1.7 ml microtube
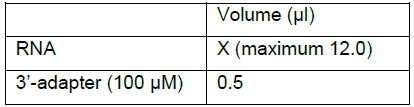
- Mix well by pipetting up and down 6-8 times or by gentle vortex.
- Incubate at 72 °C for 3 min.
- Transfer directly to ice for 2 min.
- Spin down briefly.
Notes:- If you are not limited by RNA quantity, start with 5 μg Total RNA or 0.5 μg of LMW RNA, but it is also possible to start with 1/10 and even 1/100 lesser quantities; we successfully prepared a library from 2 ng of purified pool of synthetic 22 nt oligo. For C. elegans 100 µl worm/egg pellet generates ~200 µg total RNA.
- Excess of adapter promotes generation of adapter dimers. 0.5 μl of 100 μM 3’-adapter can be used for 5 μg Total RNA or 0.5 μg LMW RNA. For lower starting quantities dilute both the 3’-adapter and the 5’-adapter accordingly.
- It is important to transfer the samples into ice immediately after the denaturation steps to prevent reformation of secondary structures.
- Add to the sample
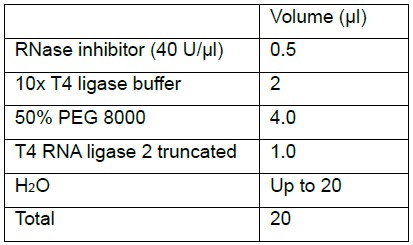
- Mix well by pipetting up and down 6-8 times or by gentle vortex.
- Incubate at 25 °C for 1 h.
Notes:- T4 Ligase 2 truncated arrives with 50% PEG in a separate tube.
- Add RNase inhibitor first to prevent RNA degradation.
- Combine in an RNase-free 1.7 ml microtube
- Dephosphorylation (optional)
This step is required only if small RNAs with triphosphate structures at the 5’-end are needed (instead of monophosphate), such as secondary siRNA in C. elegans and retroviral infection-derived miRNA in mammalians. RNA ligases are not able to ligate triphosphorylated 5’ ends. RppH enzyme removes two terminal phosphates, generating monophosphate 5’-termini, which can then be ligated to the 5’-adapter.- To the ligation reaction add 1 μl of RppH enzyme (NEB, 5 U/μl).
- Incubate for 30 min at 37 °C.
- 1st SPRI-based size selection
Note: During this step molecules > 60 nt, comprising longer RNA species, including tRNA, whether adapter-ligated or not, are bound to beads, while adapter-ligated sRNA (37-45 bp) and free 3’-adapter (18 bp) stay in the supernatant. Following magnetic separation, the supernatant is transferred into a new tube while the beads are discarded.- Add 40 μl of RNase-free H2O to the 20 μl of ligation reaction for a final volume of 60 μl.
- Add 67 μl of Agencourt Ampure XP beads and 78 μl of 100% isopropanol, to obtain a final concentrations of 7.5% PEG and 38% isopropanol (for explanations regarding the concentrations of the crowding agents PEG and Isopropanol see Table 2 below).
- Mix well by pipetting up and down 6-8 times or by gentle vortex.
- Incubate at room temperature for 5 min. Place the reaction tube on a magnet stand and allow the beads to settle for 7 min.
- Carefully transfer the supernatant to a new 1.7 ml tube (discard the beads).
Notes:- Agencourt Ampure XP are paramagnetic SPRI beads in a solution containing 20% PEG.
- Optimal conditions for size-selection depend on the length of the fragments being separated. The conditions above are optimized for separation of adapter-ligated sRNA (37-45 bp) from fragments > 60 bp. If preparing a library from longer RNA or/and using custom adapters with a different length choose the optimum conditions for the 1st size-selection step using Table 2 from the appendix protocol below.
- Beckman currently manufactures two types of SPRI beads: Ampure XP and SPRI-select. Both beads are in 20% PEG. SPRI-select is RNase free certified and pass more stringent Quality Assurance (QA), to ensure reproducibility, however, they are much more expensive. In our hands, both bead types produced identical results.
- Since precision is important, avoid carryover.
- Aliquot SPRI beads into 1.7 ml tubes to prevent contamination (1 ml beads suffices for 3 libraries).
- 2nd SPRI-based size selection
During this step adapter-ligated-sRNA (37-45 nt) are bound to beads, while free sRNA (19-27 nt) and 3’-adapter (18 nt) stay in the supernatant. Following magnetic separation, the supernatant is removed and discarded while adapter-ligated sRNAs are eluted from beads.- To the supernatant from the previous step add 86 µl of Agencourt Ampure XP beads and 144 µl 100% isopropanol, to obtain the final concentrations of 7.5% PEG and 51% isopropanol.
- Mix well by pipetting up and down 6-8 times or by gentle vortex.
- Incubate at room temperature for 5 min.
- Place the tube on a magnet stand and allow the beads to settle for 7 min.
- Carefully remove and discard the clear supernatant.
- Proceed according to wash and elution protocol below.
- Elute in 19 μl RNase-free H2O.
- Wash and elution protocol
- With the tube still on the magnet stand, add 500 μl of 85% non-denatured ethanol (freshly made from 100% ethanol), without disturbing the beads.
- Incubate at room temperature for 30 s.
- Carefully remove the ethanol without disturbing the beads and discard it.
- Repeat the ethanol wash.
- Let the beads air dry for 5 min. Aspirate residual ethanol and let it dry for additional 2 min. Make sure no residual ethanol is present before proceeding to elution.
- Remove the tube from the magnet stand and add the desired volume of molecular biology grade RNase-free water directly on the beads.
- Gently mix by pipetting the entire volume 10 times.
- Incubate at room temperature for 2 min.
- Spin down briefly.
- Place the microtube on the magnetic stand and allow the beads to settle for 2 min or until the supernatant turns completely clear.
- Transfer the desired volume of the clear supernatant containing small RNA into a new 1.7 ml microtube.
- Ligation of the 5’-adapter
Note: Ligation reaction between 3’-ligated-sRNA and the 5’-adapter is catalyzed by T4 RNA ligase 1 in the presence of ATP.- Combine
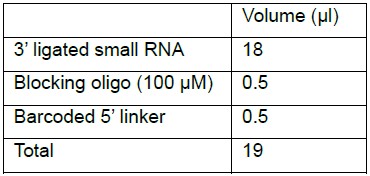
- Mix well by pipetting up and down 6-8 times or by gentle vortex.
- Incubate at 72 °C for 3 min.
- Transfer directly to ice for 2 min.
- Spin down briefly.
- Prepare the ligation mix:
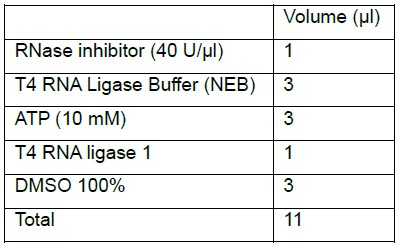
- Vortex and spin down the ligation mix.
- Add 11 μl of ligation mix to the tube with the small RNA sample.
- Incubate at 37 °C for 1 h.
- Incubate at 95 °C for 30 s.
- Transfer directly to ice for 2 min.
- Spin down briefly.
Notes: - Combine
- Blocking oligo hybridizes with the free 3’-adapter, turning it into dsDNA, which is not a substrate for RNA ligase, thus preventing generation of adapter-dimer.
- Samples are heated to 95 °C to release the blocking oligo that might have annealed to the 3’-adapter bound to the small RNA. Free oligos will be removed by SPRI separation. If not released and removed, the oligo may compete with the reverse primer during the RT step and PCR reaction (the oligo is blocked, so no product will be generated, but the yield might be compromised).
- Aliquot ATP to prevent its degradation. Thaw the ATP solution at the bench and not in your palm.
- 3rd SPRI-based size-selection
Note: During this step 3’-5’ ligated small RNA (64-72 nt) are bound to the beads, while adapter-dimer (45 nt), free 5’-adapter (27 nt) and blocking oligo (18 nt) stay in the supernatant. Following magnetic separation, the supernatant is removed and discarded while the 3’-5’ ligated sRNAs are eluted from the beads.- Add 20 μl of RNase-free H2O to 30 μl of the ligation reaction for a final volume of 50 μl.
- Add 68 μl of Agencourt Ampure XP beads and 64 μl of 100% isopropanol, to obtain final concentrations of 7.5% PEG and 35 % isopropanol.
- Mix well by gently pipetting up and down 10 times.
- Incubate at room temperature for 5 min.
- Place the tube on the magnet stand and allow the beads to settle for 10 min.
- Carefully remove and discard the clear supernatant.
- Proceed according to the above wash and elution protocol (Procedure F).
- Elute in 15 μl H2O.
- Transfer 12.5 μl of 3’-5’ linked-small RNA to a PCR tube.
- Reverse transcription
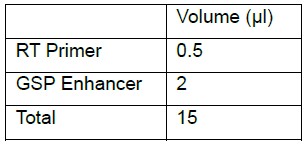
Note: The RT protocol below is compatible with qScript Flex cDNA synthesis kit (Quanta). If using another kit for RT follow the kit’s instructions. In our hands, Invitrogen Superscript III and Superscript II resulted in an inferior yield.- Add to a tube with 12.5 μl of 3’-5’ linked-small RNA:
- Gently pipette the entire volume up and down 6-8 times.
- Spin down briefly.
- Transfer to a PCR machine.
- Incubate at 65 °C for 5 min.
- Cool to 42 °C.
- Add to the sample at 42 °C:

- Gently pipette the entire volume up and down 6-8 times.
- Incubate at 42 °C for 60 min.
- Incubate at 85 °C for 5 min.
- Cool to 10 °C.
- Proceed to PCR amplification.
- GSP Enhancer is a component of qScript Flex cDNA synthesis kit used for RT performed with specific primers.
- Samples can be kept at -20 °C for up to a week.
- Add to a tube with 12.5 μl of 3’-5’ linked-small RNA:
- Pilot cDNA amplification
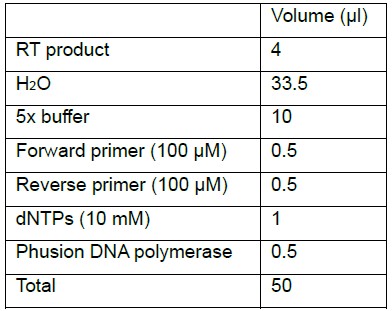
Note: The aim of this step is to determine the optimal number of amplification cycles. After the system has been calibrated, pilot PCR step can be omitted. In this case, perform the main PCR directly with the known optimal cycle number.- Combine in a PCR tube:
- Perform PCR for 23 cycles.
- Pause the thermocycler after cycles: 11, 14, 17, 20, and 23; open the tubes without removing them from the cycler and withdraw 7 μl for analysis.
- PCR conditions:
Notes:98 °C 30 s Cycles 1-4 98 °C 10 s 50 °C 30 s 72 °C 15 s Cycles 5-20 98 °C 10 s 64 °C 30 s 72 °C 15 s 72 °C 2 min 10 °C hold - The number of cycles can be raised if using small starting RNA quantities.
- Aliquot dNTPs to prevent freeze-thaw cycle degradation.
- RT product can constitute no more than 1/10 volume of a PCR reaction.
- Combine in a PCR tube:
- Pilot gel electrophoresis
Note: Samples can be checked by TapeStation/Bioanalyzer instead.- Cast 3.0% agarose/TBE gel.
- Load 5 μl of each one of the withdrawn samples.
- Run samples at 80 V for 1.5 h.
- Take a picture.
- Use Purple or Orange loading dyes (NEB) as they produce no UV visible bands masking the library.
- Use TBE buffer instead of TAE, as it allows a better resolution of small fragments.
- Evaluation of the PCR product
Note: Evaluate the PCR products to determine the optimal PCR cycle number for the main PCR. The optimal number of cycles results in a clearly visible but not too thick/smeary product band and the best library/artifact bands ratio (for typical PCR results see Figure 1).
Possible PCR products: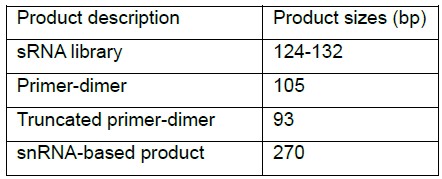
Note: Truncated primer-dimer originates from annealing of a reverse primer to one of random 8N sequences during RT or PCR. It lacks barcode and a part of the 8N sequence.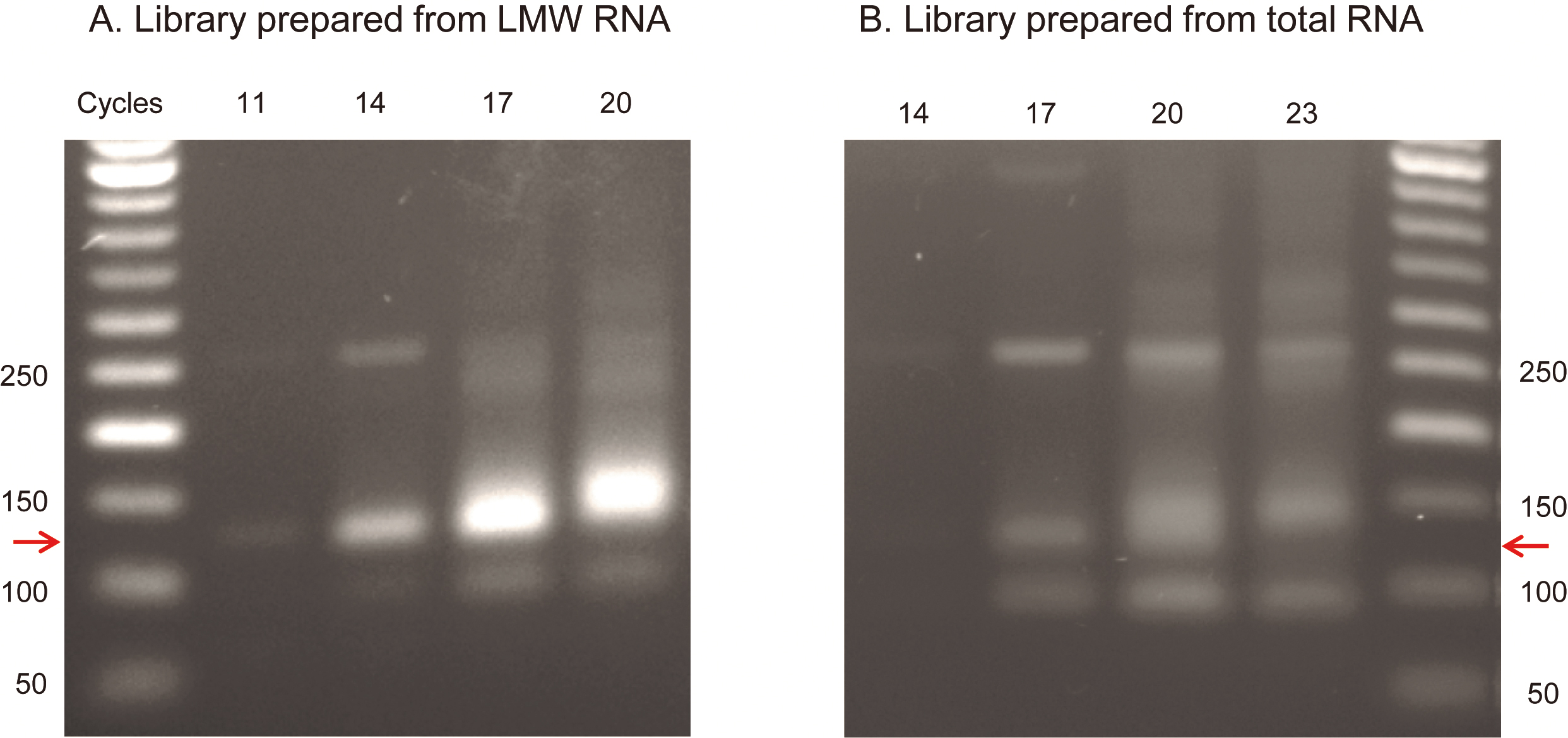
Figure 1. Example of a Pilot PCR electrophoresis. A. Library prepared from 1 µg of LMW RNA fraction extracted from C. elegans worms synchronized to L4 developmental stage was amplified for 11, 14, 17 and 20 cycles. B. Library prepared from 1 µg of total RNA extracted from C. elegans worms synchronized to L4 developmental stage was amplified for 14, 17, 20 and 23 cycles. Five µl of each amplification product was electrophoresed on 3% agarose-TBE gel. The sRNA Library product is indicated by a red arrow. The ~130 bp library product is visible starting at cycle 11 in (A) and cycle 14 in (B). The upper ~270 bp band originates from snRNA and the lower 93 bp band originates from truncated adapter-dimer. Based on these results the chosen number of cycles for the main PCR was 14 cycles for library (A) and 17 cycles for library (B). - Main cDNA amplification
Prepare PCR reaction as above and amplify with the predetermined optimal number of cycles.
Note: If skipping the pilot PCR, take an aliquot after the amplification for quality check before proceeding to library purification. - Cleaning PCR product
Notes:- Most contaminating bands, except for a primer-dimer, can be efficiently removed by a conventional double-side size-selection using SPRI beads in 20% PEG (for typical cleaning output see Figure 2).
- A 105 bp long primer-dimer is inefficiently removed by SPRI beads. Low level of contamination by primer-dimer is acceptable, but in a case of a prominent primer-dimer band, we suggest cleaning the PCR product by gel-extraction.
- Removal of bands > 200 bp
- Transfer 50 μl of the PCR product to a 1.7 ml tube.
- Add beads as follows:
- For libraries predominantly contaminated with higher bands (x1.4V, x2.0V), add 70 μl.
- For libraries predominantly contaminated with lower bands (x1.2V, x1.8V), add 60 μl.
- Mix the total reaction volume by pipetting 10 times.
- Incubate at room temperature for 5 min.
- Place the tube on the magnetic stand and allow the beads to settle for 7 min.
- Transfer the entire clear supernatant to a new tube (discard the beads).
- Removal of bands < 100 bp
- Add 30 μl beads to the supernatant from the previous step.
- Mix the total reaction volume by pipetting 10 times.
- Incubate at room temperature for 5 min.
- Place the reaction vessels on the magnet stand and allow the beads to settle for 7 min.
- Carefully remove and discard the clear supernatant.
- Proceed according to wash and elution protocol above (Procedure F).
- Elute with 20 μl H2O.
- Examples of sRNA libraries before and after cleaning are presented in Figure 2.
- Cast 3.5% agarose or 3.0% low-range ultra agarose (Bio-Rad) gel in TBE.
- Electrophorese sample at 80 V for 2.5 h.
- Excise the library band and weight the slice.
- Chop the slice finely (use separate razor for each library).
- Extract using gel extraction kit.
Note: In our hands, NucleoSpin gel and PCR Clean-up kit (Macherey-Nagel) and Gel/PCR DNA fragments Extraction kit (Geneaid) resulted in a satisfactory yield. - Elute with 15 μl H2O/elution buffer.
- Final product validation
- Measure product concentration.
- Validate the library purity by TapeStation/Bioanalyzer.
- Library storage
Once the library is ready for high-throughput sequencing (HTS), store library at -20 °C until use.
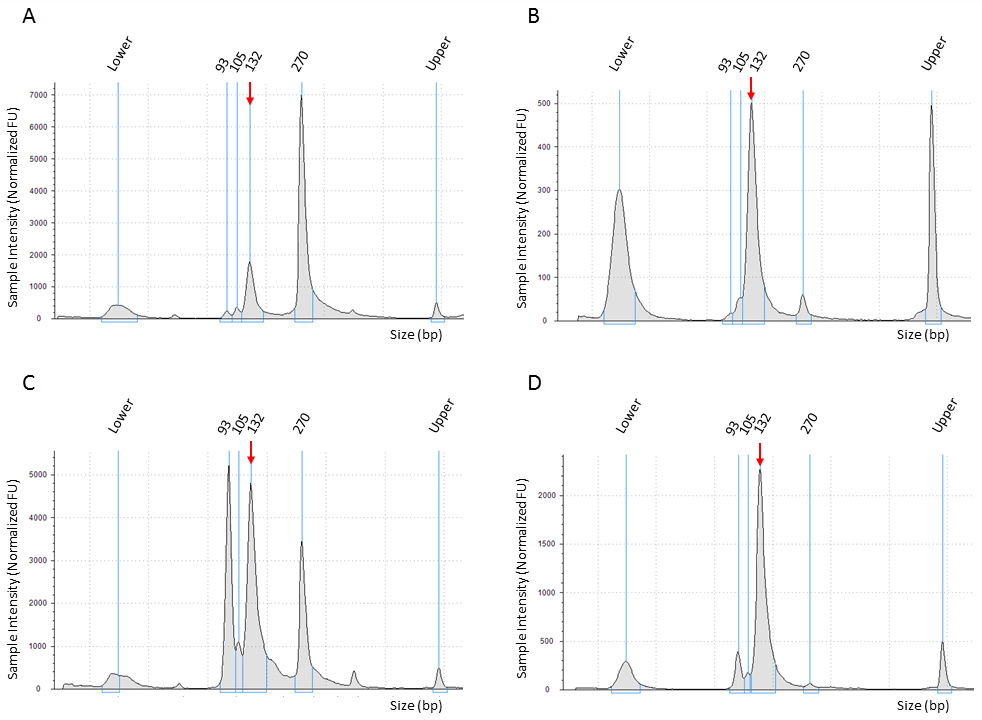
Figure 2. Removal of library contaminants by size-selection on SPRI beads. TapeStation traces of PCR products before (left panels: A, C) and after (right panels: B, D) cleaning using SPRI beads. Libraries were prepared from 5 µg total RNA extracted from C. elegans synchronized to L4 developmental stage. Upper row (A-B): The major contaminant is 270 bp long constituting ~65% of the total PCR product. Cleaning by double side size selection using 1.4x V beads followed by 2.0x V beads reduced the contaminant to below 10%. Lower row (C-D): The major contaminant is 93 bp long constituting ~30% of the total PCR product. Cleaning by double side size selection using 1.2x V beads followed by 1.8x V beads reduced the contaminant to below 10%. sRNA library corresponding peaks are marked by red arrows. Peak sizes are indicated. The outer peaks titled “Lower” and “Upper” are 25 nt and 1,500 nt molecular size markers, respectively.
Cleaning by gel-extraction (optional)
Note: Clean by gel-extraction instead of SPRI beads in a case of a heavy contamination with primer-dimer.
Appendix protocol: Fine-tuning of binding conditions for different sizes of ligated molecules
Size selection conditions of the main protocol above are suited for ligation of 19-27 nt long small RNA to 18 nt long 3’-adapter and 27 nt long 5’-adapter. As binding efficiency of a molecule is dependent on its size, if preparing a library from longer RNA molecules or if using custom adapters of a different length, adjustment of the conditions may be required for one or all size-selection steps.
- Calculate the sizes of the fragments that are needed to be separated (see Table 1 for example)
Table 1. Sizes of ingredients, intermediates and products of small RNA library preparation process described in the main protocol above. Numbers in brackets correspond to predominantly miRNA libraries, such as in human.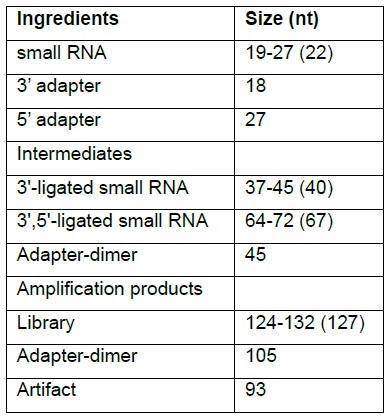
- Determine the optimal separation conditions for the calculated sizes using Table 2.
Table 2. Conditions for SPRI-based separation of short RNA molecules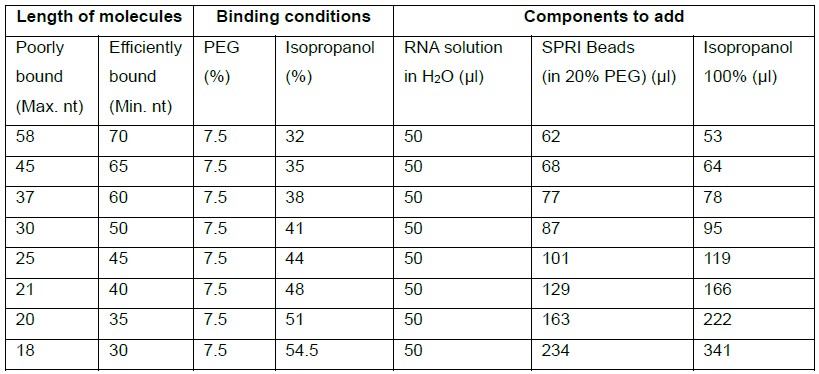
The table presents approximate sizes of fragments that a given combination of PEG and isopropanol allows to separate, i.e., a minimal length of a longer fragment (that is bound to the beads) and the maximal length of a shorter fragment (that stays in the supernatant). Poor binding is defined as < 5%, efficient binding is defined as > 50%. Note that the PEG concentration is always 7.5% and that the volume of RNA in H2O is 50 μl.
Examples of Table 2 usage
Example 1: At the 3rd SPRI selection step of the protocol above we need to separate the 3’-5’-adapter-ligated sRNAs (64-72 bp) from adapter dimers (45 bp). According to Table 2 the suitable conditions, i.e., conditions that allow efficient binding of 64 nt long fragments and poor binding of 45 nt long fragments, are 7.5% PEG and 35% isopropanol. To obtain these conditions we need to bring the sample to a volume of 50 μl by adding H2O and then to add 68 μl 20% PEG (beads solution) and 64 μl isopropanol.
If the RNA solution already contains PEG, the quantity of PEG needed to be added would need to be recalculated.
Example 2: At the 1st SPRI selection of the protocol above, according to Table 2 we need to add 77 μl of 20% PEG to 50 μl RNA in water to obtain a final concentration of 7.5% PEG. However, the ligation reaction already contains 4 μl of 50% PEG, which equals to 10 μl of 20% PEG. Therefore, the amount of added PEG has to be reduced by 10 μl (67 μl instead of 77 μl), in parallel the volume of H2O was increased by 10 μl (40 μl instead of 30 μl).
Similar adjustments have to be done for both PEG and Isopropanol when cleaning the fragment of interest first from higher and then from lower bands (double-side selection).
Example 3: At the 2nd SPRI selection, for separation of the ligated sRNA from free adapter, 163 μl of 20% PEG and 222 μl isopropanol are needed to obtain a final concentration of 7.5% PEG and 51% isopropanol. However, the sample already contains 77 μl of 20% PEG and 78 μl isopropanol added at the 1st size-selection, thus only 86 μl of beads solution and 144 μl 100% isopropanol are needed to be added.
For each combination of PEG and isopropanol, binding efficiency of a fragment longer than the minimal length listed in the table will be higher, while that of a fragment shorter than the maximal length will be lower than the value appearing in the table. Better separation between the fragments can be achieved on the expense of recovery. Pure longer fragment can be obtained if using slightly lower isopropanol concentration, while pure shorter fragment can be obtained by double-side size-selection if removing longer fragment at higher isopropanol concentration.
Example 4: When separating 37 nt and 58 nt long fragments, to obtain pure 58 nt fragment perform binding with 38% isopropanol. At this concentration, some of the 58 nt fragments will remain unbound, but not the 37 nt fragments. In order to obtain pure 37 nt, first perform binding using 44% isopropanol. All 58 nt fragments and some of the 37 nt fragments will be removed from the solution. The remaining pure 37 nt long fragment is next recovered from the solution using 51% isopropanol. If wishing to obtain both fragments, perform the first binding using 41% isopropanol and the second binding using 51%. Upon these conditions, each of the fragments will be slightly contaminated by the other. For additional information see Fishman et al. (2018).
Testing binding conditions other than the ones listed in Table 2- Calculate quantities of Isopropanol and beads (in 20% PEG) to be added.
According to the equation:
where,
V is total volume
X is volume of nucleic acid solution
P is desired concentration of PEG (in %)
Q is desired concentration of (%) isopropanol
A total volume of binding solution is equal to: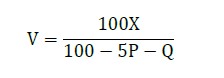
- Substitute P and Q for the desired concentrations of PEG and Isopropanol, respectively and X for the volume of nucleic acid solution (we recommend 50 μl) and calculate a total volume of binding solution (V).
- Next, calculate the volumes of beads and Isopropanol that are equal to 5PV/100 and QV/100, respectively.
- Measure binding efficiency
- Prepare solution of 100 ng of synthetic single-stranded DNA oligonucleotide in 50 μl H2O in a 1.7 ml microtube.
- Add beads and Isopropanol at volumes determined using the calculation method above.
- Mix thoroughly.
- Incubate at room temperature for 5 min.
- Separate on a magnet stand for 7 min.
- Carefully remove the supernatant and wash the beads twice with 85% ethanol according to wash and elution protocol. Elute in 20 μl H2O.
- Measure oligonucleotide concentrations of the input oligonucleotide solution and the eluted sample by Qubit® Fluorometer using Qubit® ssDNA Assay Kit (Molecular probes). Calculate the binding efficiency as the percentage of the output oligonucleotide from the input quantity.
- Calculate quantities of Isopropanol and beads (in 20% PEG) to be added.
Recipes
- 5x TBE
Dissolve 54 g Tris base (FW = 121.14) and 27.5 g boric acid (FW = 61.83) in 900 ml deionized water
Add 20 ml of 0.5 M EDTA (pH 8.0)
Adjust the solution to a final volume of 1 L with deionized water
Notes:- The pH must be ~8.3 (do not adjust pH!)
- Discard if precipitate was formed.
- Prepare and run agarose gel in 1x TBE.
Acknowledgments
We thank Prof. emeritus Vladimir Lin from the Technion Faculty of Mathematics for his help in development of the equation for calculating volumes of PEG and Isopropanol.
This work was funded by The Israeli Centers of Research Excellence (I-CORE) program, (Center No. 1796/12 to ATL), The Israel Science Foundation (grant Nos. 644/13 and 927/18 to ATL), and Israel Cancer Research Fund (ICRF). This protocol was adapted from Fishman et al. (2018).
Competing interests
PCT Patent Application No. PCT/IL2017/050761, entitled “Methods for isolation and quantification of short nucleic acid molecules”, authored by A. F. and A. T. L. was filed by Technion Research and Developmental Foundation on 6 July 2017. The application claims priority from US Provisional Application No. 62/359,245.
References
- Backes, C., Meese, E. and Keller, A. (2016). Specific miRNA disease biomarkers in blood, serum and plasma: challenges and prospects. Mol Diagn Ther 20(6): 509-518.
- Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136(2): 215-233.
- Fishman, A., Light, D. and Lamm, A. T. (2018). QsRNA-seq: a method for high-throughput profiling and quantifying small RNAs. Genome Biol 19(1): 113.
- Fuchs, R. T., Sun, Z., Zhuang, F. and Robb, G. B. (2015). Bias in ligation-based small RNA sequencing library construction is determined by adaptor and RNA structure. PLoS One 10(5): e0126049.
- Jost, D., Nowojewski, A. and Levine, E. (2011). Small RNA biology is systems biology. BMB Rep 44(1): 11-21.
- Kawano, M., Kawazu, C., Lizio, M., Kawaji, H., Carninci, P., Suzuki, H. and Hayashizaki, Y. (2010). Reduction of non-insert sequence reads by dimer eliminator LNA oligonucleotide for small RNA deep sequencing. Biotechniques 49(4): 751-755.
- Kim, Y. K. (2015). Extracellular microRNAs as biomarkers in human disease. Chonnam Med J 51(2): 51-57.
- Kivioja, T., Vaharautio, A., Karlsson, K., Bonke, M., Enge, M., Linnarsson, S. and Taipale, J. (2011). Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 9(1): 72-74.
- Lau, N. C., Lim, L. P., Weinstein, E. G. and Bartel, D. P. (2001). An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294(5543): 858-862.
- Lu, C., Meyers, B. C. and Green, P. J. (2007). Construction of small RNA cDNA libraries for deep sequencing. Methods 43(2): 110-117.
- Lundin, S., Stranneheim, H., Pettersson, E., Klevebring, D. and Lundeberg, J. (2010). Increased throughput by parallelization of library preparation for massive sequencing. PLoS One 5(4): e10029.
- Mitchell, P. S., Parkin, R. K., Kroh, E. M., Fritz, B. R., Wyman, S. K., Pogosova-Agadjanyan, E. L., Peterson, A., Noteboom, J., O'Briant, K. C., Allen, A., Lin, D. W., Urban, N., Drescher, C. W., Knudsen, B. S., Stirewalt, D. L., Gentleman, R., Vessella, R. L., Nelson, P. S., Martin, D. B. and Tewari, M. (2008). Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A 105(30): 10513-10518.
- Nagy, Z. B., Wichmann, B., Kalmar, A., Bartak, B. K., Tulassay, Z. and Molnar, B. (2016). miRNA Isolation from FFPET Specimen: A technical comparison of miRNA and total RNA isolation methods. Pathol Oncol Res 22(3): 505-513.
- Paithankar, K. R. and Prasad, K. S. (1991). Precipitation of DNA by polyethylene glycol and ethanol. Nucleic Acids Res 19(6): 1346.
- Vigneault, F., Ter-Ovanesyan, D., Alon, S., Eminaga, S., D, C. C., Seidman, J. G., Eisenberg, E. and G, M. C. (2012). High-throughput multiplex sequencing of miRNA. Curr Protoc Hum Genet Chapter 11: Unit 11.12. 1-10.
- Youngman, E. M. and Claycomb, J. M. (2014). From early lessons to new frontiers: the worm as a treasure trove of small RNA biology. Front Genet 5: 416.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Fishman, A. and Lamm, A. T. (2019). QsRNA-seq: A protocol for Generating Libraries for High-throughput Sequencing of Small RNAs. Bio-protocol 9(5): e3179. DOI: 10.21769/BioProtoc.3179.
Category
Molecular Biology > RNA > RNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.