- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

On-demand Labeling of SNAP-tagged Viral Protein for Pulse-Chase Imaging, Quench-Pulse-Chase Imaging, and Nanoscopy-based Inspection of Cell Lysates


Published: Vol 9, Iss 4, Feb 20, 2019 DOI: 10.21769/BioProtoc.3177 Views: 6977
Reviewed by: Vamseedhar RayaproluJolene RamseyKristin L. Shingler
Original research article
The authors used this protocol in:

Jun 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Advanced labeling technologies allow researchers to study protein turnover inside intact cells and to track the labeled protein in downstream applications. In the context of a viral infection, the combination of imaging and fluorescent labeling of viral proteins sheds light on their biological activity and interaction with the host cell. Initial approaches have fused fluorescent proteins such as green fluorescent protein (GFP) to the viral protein-of-interest. In contrast, self-labeling enzyme tags such as the commercial SNAP-tag, a modified version of human O6-alkylguanine-DNA-alkyltransferase, covalently link synthetic ligands, which users can add on demand. The first two protocols presented here build on previously published protocols for fluorescent labeling in pulse-chase and quench-pulse-chase experiments; the combination of fluorescent labeling with advanced light microscopy visualizes the dynamic turnover of the SNAP-tagged viral protein in intact mammalian cells. A third protocol also outlines how to inspect cellular lysates microscopically for detergent-resistant assemblies of the labeled viral protein. These protocols showcase the flexibility of the SNAP-based labeling system for tracking a viral protein-of-interest in live cells, intact fixed cells, and cell lysates. Moreover, the protocols employ recently developed commercial microscopes (e.g., Airyscan microscopy) that balance resolution, speed, phototoxicity, photobleaching, and ease-of-use.
Keywords: Fusion proteinsBackground
To better understand the role of a viral protein during the infectious life cycle, we have adapted existing strategies that facilitate the determination of intracellular protein location, the assessment of protein dynamics in intact cells, and the continued tracking of the viral protein after lysis of the host cell. The first two protocols build on previously published protocols for on-demand SNAP-labeling and the analysis of protein turnover (Bodor et al., 2012). In previous work, our laboratory has fused a viral protein-of-interest, nonstructural protein 3 (nsP3) of Chikungunya virus (CHIKV), to the SNAP-tag, a modified form of a 20-kDa monomeric DNA repair enzyme (Remenyi et al., 2017 and 2018). During SNAP-labeling, the addition of a synthetic O6-benzylguanine (BG) derivative results in a covalent bond between a reactive cysteine residue in the SNAP-tag and the BG-probe (Keppler et al., 2004a and 2004b).
We have also combined SNAP-labeling with a protocol for inspecting cell lysates via light microscopy, which enables visualization of detergent-resistant protein assemblies. Similar approaches have allowed researchers to detect stable granular assemblies of GFP-tagged stress-granule proteins in cell lysates (Jain et al., 2016; Wheeler et al., 2017). Stress granules are assemblies of RNA and protein (RNPs), which form under conditions of cellular stress (Kedersha et al., 2005). It is now possible to isolate the more stable stress granule core from both yeast and mammalian cells (Jain et al., 2016). SNAP-labeling offers an alternative way of tracking tagged viral proteins that may be present in similar subcellular assemblies. Hence, Protocol 3 may not only be useful to study the biochemical nature of viral proteins but also to track any cellular protein that resides in non-membranous organelles such as RNPs and stress granules. For example, integration of the SNAP-tag into the development of cell lines that produce fluorescently tagged stress granules (Kedersha et al., 2008) could increase experimental flexibility during dynamic and quantitative imaging of these cellular sub-compartments.
Our three protocols also take advantage of recently developed commercial imaging systems for multi-color fluorescence microscopy. We analyze labeled samples in Protocol 1 with live-cell imaging (Figure 1) and thus recommend following general procedures for controlling temperature, reducing phototoxicity, limiting photobleaching, and maintaining cell viability (Frigault et al., 2009). The chosen 2018 Nikon Ti-E2 system allowed us to image a large field-of-view, lessen focus drift with a proprietary Perfect Focus System (PFS), and record multiple positions with the motorized stage. Moreover, illumination with an LED and light exposures not exceeding 1 s allowed for gentler imaging compared to the typical imaging setup of a laser scanning confocal microscope.
For the analysis of labeled protein in Protocols 2 and 3, we chose a confocal imaging setup with proprietary ZEISS Airyscan technology (Figure 1), which is a commercial version of the ‘image scanning microscopy’ approach (Muller and Enderlein, 2010; Sheppard et al., 2013). Airyscan microscopy represents one of the recent innovations in fluorescence super-resolution microscopy, also referred to as nanoscopy (Li et al., 2018). The Airyscan technology improves system resolution with an improved detector design, which features a 32-channel detector array (Huff, 2015). With a new 2D super-resolution mode for Airyscan, this detection approach can now enhance resolution 2-fold while lowering the required fluorescence intensity to obtain high-quality images (Huff et al., 2017). The Airyscan system allowed us to stain our samples with fluorescent dyes from the SNAP product range (e.g., SNAP-Cell® 647-SiR and SNAP-Cell® TMR-Star) and detect them even at low levels during the recovery phase of the quench-pulse-chase protocol. Increased sensitivity also helped in the nanoscopy-based inspection of cellular lysate in Protocol 3 and allowed the visualization of granular structures made up of the SNAP-tagged viral protein. The combination of SNAP-based labeling and innovative detection via Airyscan has the potential to further bioimaging with higher resolution, sensitivity, and user-friendliness.
Figure 1. Overview of experimental protocols. This flowchart outlines the essential steps in Protocols 1, 2, and 3. For the definition of ‘stable CHIKV cells’, see Protocol 1, Procedure section.
Protocol 1: Pulse-chase experiments for long-term imaging with Nikon Ti2-E
Materials and Reagents
- 10-cm Petri dish (Corning, catalog number: 430167)
- Micropipette tips, serological pipettes, pipette aids, and microtubes for liquid handling
- TipOne® 1,000 µl XL (catalog number: S1122-1830)
- TipOne® 200 µl (catalog number: S1120-8810)
- TipOne® 20 µl filter tips (catalog number: S1120-1810) or SARSTEDT pipette tip 10 µl (catalog number: 70.1130.600)
- FisherbrandTM 5 ml serological pipets (catalog number: 13-676-10H)
- FisherbrandTM 10 ml serological pipets (catalog number: 13-676-10J)
- FisherbrandTM 25 ml serological pipets (catalog number: 13-676-10K)
- Drummond Pipet-Aid XL (catalog number: 4-000-205)
- Microtube 1.5 ml (SARSTEDT, catalog number: 72.690.001)
- Nunc 35-mm glass bottom (#1.5 or 0.16-0.19 mm thickness) dishes with 27 mm viewing area (Thermo Fisher Scientific, catalog number: 150682)
Notes:- Only open packages inside a biosafety cabinet and reseal any remaining dishes to avoid contamination.
- Use any dish or slide format that is compatible with an inverted microscope. Match the thickness of the glass bottom with the suggested thickness found on the microscope’s objective (typically #1.5 or 0.16-0.19 mm thickness). We prefer the dishes listed above because of their large viewing area. The cell lines used in this protocol can grow on glass substrates, but surface treatment (e.g., Poly-Lysine coating) may be necessary for other cell lines.
- HuH-7 cell line (Japanese Collection of Research Bioresources [JCBR], catalog number: JCBR 0403)
Note: HuH-7 is a well-differentiated hepatocyte-derived cellular carcinoma cell line. It was originally taken from a liver tumor in a Japanese male in 1982 (Nakabayashi et al., 1982). The cells used in the creation of this protocol were obtained from John McLauchlan (Centre for Virus Research, Glasgow). - A plasmid that encodes SNAPf®, a SNAP-tag protein (NEB, catalog number: N9183S; or as part of the SNAP-Cell® Starter Kit, NEB, catalog number: E9100S)
- For initial validation: plasmid that encodes pSNAPf-Cox8A control plasmid (also part of the SNAP-Cell® Starter Kit, NEB, catalog number: E9100S)
- Trypsin EDTA solution (Sigma, catalog number: T3924-500ML)
- Specific reagents for our cell line (for basic tissue culture), which we derived from the hepatoma cell line HuH-7
- Dulbecco's modified Eagle's medium (Sigma, catalog number: D6429-500ML)
- 100% Fetal Calf Serum (Gibco, catalog number: 10500-064)
- Gibco MEM Nonessential amino acids solution (100x), store at 4 °C up to 24 months from the date of manufacture (Thermo Fisher Scientific, catalog number: 11140050)
- Gibco HEPES (N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid) buffer (100x), store at 4 °C up to 24 months from the date of manufacture (Thermo Fisher Scientific, catalog number: 15630130)
- FluoroBrite DMEM, store at 4 °C up to 24 months from the date of manufacture (Thermo Fisher Scientific, catalog number: A1896701)
- Cell-permeable SNAP-Cell® 647-SiR (New England Biolabs, catalog number: S9102S)
Notes:- Package contains 30 nmol of the substrate. Resuspend with 50 µl of sterile dimethyl sulfoxide (DMSO, Fisher BioreagentsTM, catalog number: BP231-100) to make up a stock solution, which can be stored at -20 °C. We have used stock solutions that have been stored for up to 3 years. However, the manufacturer’s recommended shelf-life is three months dissolved in DMSO and two years dry.
- NEB also offers cell-impermeable SNAP probes (‘Cell Surface’ Probes); use these probes for studies of viral proteins that accumulate at the surface of host cells.
- Optional: Red fluorescent or orange fluorescent cellular dye (e.g., MitoTrackerTM Orange CMTMRos, Thermo Fisher Scientific, catalog number: M7510)
Note: To prepare a stock solution, dissolve lyophilized MitoTrackerTM probe in DMSO to a final concentration of 1 mM and store frozen and protected from light. - Molecular Probes Invitrogen ProLong Live Antifade Reagent, for live cell imaging (Thermo Fisher Scientific, catalog number: P36975)
Notes:- Store at 2-8 °C for short-term storage and ≤ -20 °C for long-term storage.
- According to the manufacturer’s instructions, use the product within 30 days when stored at 2-8 °C. When stored at ≤ -20 °C, the product is stable for at least six months with up to four freeze-thaw cycles.
- Glutamax-I (Thermo Fisher Scientific, catalog number: 35050061)
- Live cell imaging solution (see Recipes)
- Tissue culture medium containing serum (see Recipes)
Equipment
- Air-displacement micropipettes
- Starlab ErgoOne® 100-1,000 µl Single-Channel Pipette (catalog number: S7110-1000) or Gilson F123602 PIPETMAN Classic Pipet P1000 (Fisher Scientific, catalog number: 10387322)
- Starlab ErgoOne® 20-200 µl Single-Channel Pipette (catalog number: S7100-2200) or Gilson F123615 PIPETMAN Classic Pipet P100 (Fisher Scientific, catalog number: 10442412)
- Starlab ErgoOne® 2-20 µl Single-Channel Pipette (catalog number: S7100-0220) or Starlab ErgoOne® 0.1-2.5 µl Single-Channel Pipette (catalog number: S7100-0125) or Gilson F144801 PIPETMAN Classic Pipet P2 (Fisher Scientific, catalog number: 10635313) or Gilson F123600 PIPETMAN Classic Pipet P20 (Fisher Scientific, catalog number: 10082012)
- Equipment for basic cell culture techniques and aseptic procedures, i.e.,
- Biosafety cabinet (e.g., Thermo Scientific Holten Safe 2010 Model 1.2, catalog number: 8207071100)
- Humidified incubator set to 37 °C (Panasonic Incusafe, catalog number: MCO-20AIC)
- -20 °C freezer (Labcold, catalog number: RLCF1520)
- Water bath (e.g., Clifton, catalog number: NE2-8D; however, any water bath set to 37 °C may be used)
- Table-top microcentrifuge, 4 °C to room temperature (RT), max speed ≥ 12,000 x g (Eppendorf, catalog number: 5424R)
Note: For spinning down precipitate that may form during extended storage of SNAP probes. - Live-cell imaging microscope for long-term imaging. For example, a Nikon Ti2-E inverted widefield microscope (for a similar setup, contact your local Nikon representative to create a customized order for the microscope system)
- Nikon Ti2-E inverted microscope stand
- Motorized stage (standard Ti2 encoded motorized XY stage)
- Lumencor Spectra X LED light source
- CFI Plan Apo Lambda 60x oil/1.4 NA objective
- Photometric Prime 95B sCMOS monochrome camera
- Semrock 32 mm filter (Green) for Nikon Ti2-E: GFP-4050B Filter cube with Ex 466/40 single-band bandpass filter (product code: FF01-466/40-25), DM495 dichroic beamsplitter (product code: FF495-Di03-25x36), BA525/50 single-band bandpass filter (product code: FF03-525/50-25)
- Semrock 32 mm filter (Red) for Nikon Ti2-E: Cy3-4040C Filter cube with Ex 531/40 single-band bandpass filter (product code: FF01-531/40-25), Dichroic Mirror DM 562 (product code: FF562-Di03-25x36, Barrier Filter: BA 593/40 (product code: FF01-593/40-25)
- Semrock 32 mm filter (Far-Red) for Nikon Ti2-E: Cy5-4040C Filter cube with Ex 628/40 single-band bandpass filter (product ID: FF02-628/40-25), Single Band Emitter (product ID: FF01-692/40-25), Single Band Dichroic (product ID: FF660-Di02-25x36)
- Okolab stage top incubator, catalog number: H301-NIKON-NZ100/200/500-N. Set to 37 °C with 5% CO2 (any manufacturer and model that can be mounted on a Nikon Ti2 motorized stage will do)
- Perfect Focus System (PFS)
- The above list is not exhaustive but rather lists the essential components that have been useful in the creation of Protocol 1. Consult with your local Nikon representative for the remaining components of a customized microscope system.
- We prefer to image cells in open tissue-culture dishes with a coverslip-like glass bottom, which necessitates an inverted microscope stand.
- As Protocol 1 is a powerful approach for long-term imaging of the labeled protein, it is important to keep cells in focus and correct for focus drift caused by thermal and mechanical conditions. This live-cell imaging system included Nikon’s fourth-generation Perfect Focus System (PFS), which monitors the partial reflection of a low-power infrared laser beam on the interface between the dish’s glass bottom and liquid media above. This feature provides continuous and real-time focus drift correction.
- A large (25 mm x 25 mm) field of view (FOV) enables increased data throughput and capture of additional cells that would be outside the normal FOV.
- We preferred an LED light source over laser-based illumination. LED illumination limited photobleaching and phototoxicity; ‘gentle’ imaging was essential during acquisition of five-dimensional datasets (5-D: multi-color 3-D Z-stacks over time).
- A motorized microscope stage and the ability to record multiple positions during one experiment further increased the throughput of the imaging system.
Software
- Nikon NIS Elements AR imaging software
Nikon NIS Elements AR imaging software controls all components of the microscope. Use NIS Elements to set-up parameters for acquisition of 5-D datasets. Users can also create rendered videos within NIS Elements and save individual frames of 5-D data. For our widefield images, we also used a Richardson-Lucy algorithm (set to 10 iterations) to deconvolve datasets within the Elements software.
Procedure
A published labeling procedure forms the basis of the protocol described here (Bodor et al., 2012). We build on this protocol by also describing a live-cell imaging setup that is suitable for long-term examination of protein turnover in five dimensions (i.e., 3-D multi-color fluorescence microscopy over time).
Note: Carry out all liquid handling steps that involve live cells inside the biosafety cabinet. Only use sterile pipettors, serological pipets, micropipettors, microtubes, and tips.
- Production of stable CHIKV cells
We have previously used the HuH-7 hepatoma cell line to derive cells that stably harbor a modified CHIKV replicon; we designed this replicon to encode a fusion protein of CHIKV nsP3 and the SNAP-tag (Remenyi et al., 2018). We will refer to the cells from this secondary cell line as ‘stable CHIKV cells’ in the rest of the protocol. These cells also constitutively express the green fluorescent protein ZsGreen.- Use standard molecular cloning methods to generate N-, C-terminal, or internal SNAP-tag fusions. In our experimental system, we inserted the SNAP-tag within the C-terminal region of CHIKV nsP3 and added a flexible linker of Glycine amino acids at the N-terminal and C-terminal junctions (Remenyi et al., 2017). A plasmid that encodes SNAPf®, a SNAP-tag protein, is available from NEB (N9183S; or as part of the SNAP-Cell® Starter Kit, E9100S).
- After the SNAP-sequence has been inserted into a viral genome, evaluate whether modified viruses or replicons remain viable and whether the tagged protein can carry out the same biological function as the untagged protein. The methods for verification will vary depending on the virus and the protein-of-interest.
- Maintain cells in preferred tissue-culture format: we routinely passage our stable CHIKV cells in 10-cm Petri dishes.
- We have developed this protocol with the stable CHIKV cell line, which we derived from HuH-7 cells. Other cell lines that support prolonged replication of non-cytotoxic CHIKV replicons include the C2C12 (mouse myoblast) cell line (Remenyi et al., 2018) and BHK-21 (baby hamster kidney) cell line (Utt et al., 2015). However, we have not yet validated Protocols 1-3 in these cell lines.
- Detach stable CHIKV cells from the growth surface by adding enough trypsin to cover the cells that are attached to the surface of the respective culturing vessel (e.g., 1-2 ml of trypsin for 10-cm dishes) and seed into a 35-mm dish with coverslip bottom. Prepare a second and third dish containing positive and negative control.
- As a negative control, we seed naïve cells (known not to express SNAP-tag protein) in a second dish.
- NEB’s SNAP-Cell Starter Kit also contains a positive control plasmid (pSNAPf-Cox8A Control Plasmid), which can be transfected into cells to produce SNAP-tagged cytochrome c oxidase with a well-characterized mitochondrial localization. Thus, the third dish may contain cells transfected with pSNAPf-Cox8A control plasmid or any plasmid encoding a SNAP-tagged protein with well-characterized subcellular localization.
- For beginners, we recommend handling only three dishes at a time (one dish for the sample, one dish for the negative control, and one dish for the positive control) during the labeling phase of this protocol. Advanced users requiring higher throughput may consider 35-mm imaging dishes with four compartments (e.g., ibidi µ-Dish 35 mm Quad, catalog number 80416) instead of using individual dishes. Carry out simultaneous experiments (e.g., two sample conditions, one negative control and one positive control) in the subdivisions of the dish.
- The exact amount of trypsin needed to dissociate adherent cells is dependent on the cell type and age of cells.
- We adjust the cell seeding density depending on the length of our desired chase period and the duplication time of the cell line we use for the experiment.
- We typically aim for 60% to 80% confluency. For example, assuming a doubling time of 24 h and a desired chase period of 24 h, we would seed our SNAP-tagged cell line to 20% confluency, stain with SNAP-reagents the following day (= pulse, at 40% confluency) and image during a 24-h chase (allowing cells to reach 80% confluency during live-cell imaging).
- Incubate cells under standard growth conditions (i.e., 37 °C at 5% CO2) overnight.
- On the next day, take the frozen stock solution of SNAP-Cell® 647 SiR from the -20 °C freezer and thaw at room temperature.
- In a 1.5-ml microtube, dilute thawed SNAP-Cell® 647 SiR; perform a 1:1,000 (final concentration of 0.6 µM) dilution in complete cell culture media. Final volume should be at least 0.6 ml to cover the dish area above the coverslip. Users can increase the volume to 1 ml to reduce the risk of drying out the cells. Vortex briefly (for ≥ 5 s) or pipet the diluted labeling solution up and down (ten times).
- SNAP-Cell® probes are cell-permeable. Our protocols label a fusion protein of CHIKV nsP3 and the SNAP-tag. This fusion protein localizes to intracellular compartments, and hence we only stain with cell-permeable probes.
- Other probes from NEB include SNAP-Cell® 505 (green fluorescent), SNAP-Cell® Oregon Green (green fluorescent), and SNAP-Cell® TMR-Star (red fluorescent). Optimize final concentrations for different SNAP-tagged proteins and cell lines; factors like protein abundance and non-specific binding may vary depending on the tagged protein or cell line.
- According to the manufacturer, optimal substrate concentrations range from 1 to 20 µM, with best results usually obtained at concentrations between 1 and 5 µM. We have found that even 0.6 µM of SNAP-Cell® SiR provided sufficient staining. We do not prepare more media than we expect to consume within one hour. Always include a negative control (naïve cells known not to express SNAP-tag protein) when optimizing labeling conditions.
Note: Recipe for three dishes (using 0.6 ml in Step 7, scale accordingly for more dishes): 1998 µl Complete Cell Culture Media; 2 µl SNAP-Cell® 647 SiR.
- Spin the diluted labeling solution for 5 min at maximum speed (≥ 12,000 x g) to remove possible insoluble fluorescent debris. Take care not to disturb the pellet when removing supernatant (which may be invisible).
- Replace the medium on stable CHIKV cells with 0.6-1 ml of SNAP-tag labeling medium (pre-heated to 37 °C in a water bath). Incubate for 15 min at 37 °C, 5% CO2.
Note: At this step, the benzyl group on the SNAP-Cell® 647 SiR substrate will covalently link to the SNAP-tag and release guanine. We found that a 15-min incubation gave us optimal labeling. According to the manufacturer, optimal reaction times range from 5 to 30 min, respectively, depending on experimental conditions and expression levels of the SNAP-tagged protein. - During this 15-min incubation period, prepare a cold water bath containing ice and water. Remove a frozen aliquot of ProLong Live reagent from the -20 °C freezer and thaw the aliquot in the cold water bath. Do not exceed 37 °C while thawing or using the reagent. We keep the reagent in the ice bath until Step 10.
- Wash the cells three times each with 2 ml of tissue culture medium containing serum (pre-heated to 37 °C).
- Replace the regular cell-culture media with 2 ml of fresh live cell imaging solution, consisting of FluoroBrite DMEM, supplemented with fetal bovine serum (at a final concentration of 10%), HEPES, Glutamax-I, Nonessential Amino Acids, and ProLong Live Antifade Reagent (see Recipes).
- Place cells back into the incubator after the final wash. Incubate for another 30 min, 37 °C at 5% CO2.
Notes:- The primary purpose of this step is to reduce the background staining of the SNAP reagent. The background staining of some SNAP probes, such as SNAP Cell® TMR-Star can be problematic in some cell lines whereas less background staining is observed in others (Cole, 2014). If the background staining is an issue, we recommend reducing the labeling time, concentration of probe, or increasing the number of washes. Step 10 also starts the incubation with ProLong Live reagent.
- The manufacturer’s instructions of the ProLong Live reagent suggest incubating cells in the dark for 15 min to 2 h.
- In our experience, by the time the sample dish reaches the microscope, final imaging settings have been set up, and the actual image acquisition starts, the total incubation period of cells with media containing ProLong Live will be at least 1.5 h (~40 min for processing three dishes in Steps 10-12, ~20 min for transferring dishes from tissue culture facility to microscope, and ~30 min for setting up imaging conditions).
- Wash cells as described in Step 9 with regular cell-culture media. Replace media with 2 ml of live cell imaging media (made up in Step 10).
Note: We supplement the live cell imaging media with ProLong Live solution at this step if live-cell imaging does not exceed 24 h. The manufacturer does not recommend leaving ProLong Live solution on live cells for more than 24 h. We also calculate the chase period from the completion of this step, since it marks the last time point at which the fluorescent substrate can label SNAP-tagged proteins. - Optional: Stain cells with red fluorescent or orange fluorescent cellular dye (e.g., MitoTrackerTM Orange CMTMRos).
- Dilute 1 mM MitoTrackerTM stock solution to the final working concentration (25-500 nM) in ‘live cell imaging buffer’.
- Remove media from dishes and add pre-warmed (37 °C) staining solution containing MitoTrackerTM probe.
- Return dishes to the humidified incubator and incubate for 15-45 min, at 37 °C with 5% CO2.
- After incubation period is complete, replace staining solution with fresh pre-warmed ‘live cell imaging buffer’.
- Transfer the three dishes (sample, positive control, and negative control) to microscope area for live-cell imaging with Nikon Ti2-E system. The live-cell imaging setup for SNAP-tagged cells is similar to standard configurations for live-cell fluorescence imaging.
- We recommend Nikon’s resource on ‘live cell imaging’ for an introduction on the appropriate microscope setup for timelapse imaging.
- For additional resources, contact your local Nikon representative for NIS Elements Training handouts on ‘Advanced Acquisition’ modes (i.e., Multi-channel, Multi-point, Timelapse, and Z-stack)
- For alternative live-cell imaging setups, refer to Bodor et al. (2012).
- We used a widefield imaging setup for extended imaging of the same field-of-view. We obtained high-quality results with a Nikon Ti2-E system.
- Several factors determined our preference for this system, namely (i) the Ti2-E is equipped with a unique perfect focus system (PFS) that automatically corrects focus drift in real time during a prolonged period of imaging (ii) imaging with an LED light source allows for gentler imaging compared to laser-based confocal systems (iii) multipoint Z-stacks can be acquired quickly as a result of faster device movement and image acquisition (iv) quick acquisition reduces overall light exposure and subsequent phototoxicity (v) the Ti2-E provides a large field of view (FOV), which captures a large amount of cells within one FOV, and (vi) multi-point acquisitions further increase the throughput of the system.
- Image cells with the preferred imaging system
- SNAP Cell® 647-SiR should have an excitation maximum at 645 nm and an emission maximum at 661 nm.
- With the Nikon Ti2-E inverted microscope, we use standard filter settings for the Cy5 dye. Stable CHIKV cells also endogenously express the green fluorescent ZsGreen reporter protein, which has an excitation maximum of 493 nm and an emission peak at 505 nm (image with a standard GFP filter set).
- The advantage of using the far-red SNAP Cell® 647-SiR is that additional labeling with a red fluorescent cellular dye (e.g., MitoTrackerTM Orange) and imaging with filter settings for Cy3 dye is possible. Figure 2 shows representative images from a timelapse series, in which we set the microscope to take Z-stacks every 15 min for a total of 24 h.
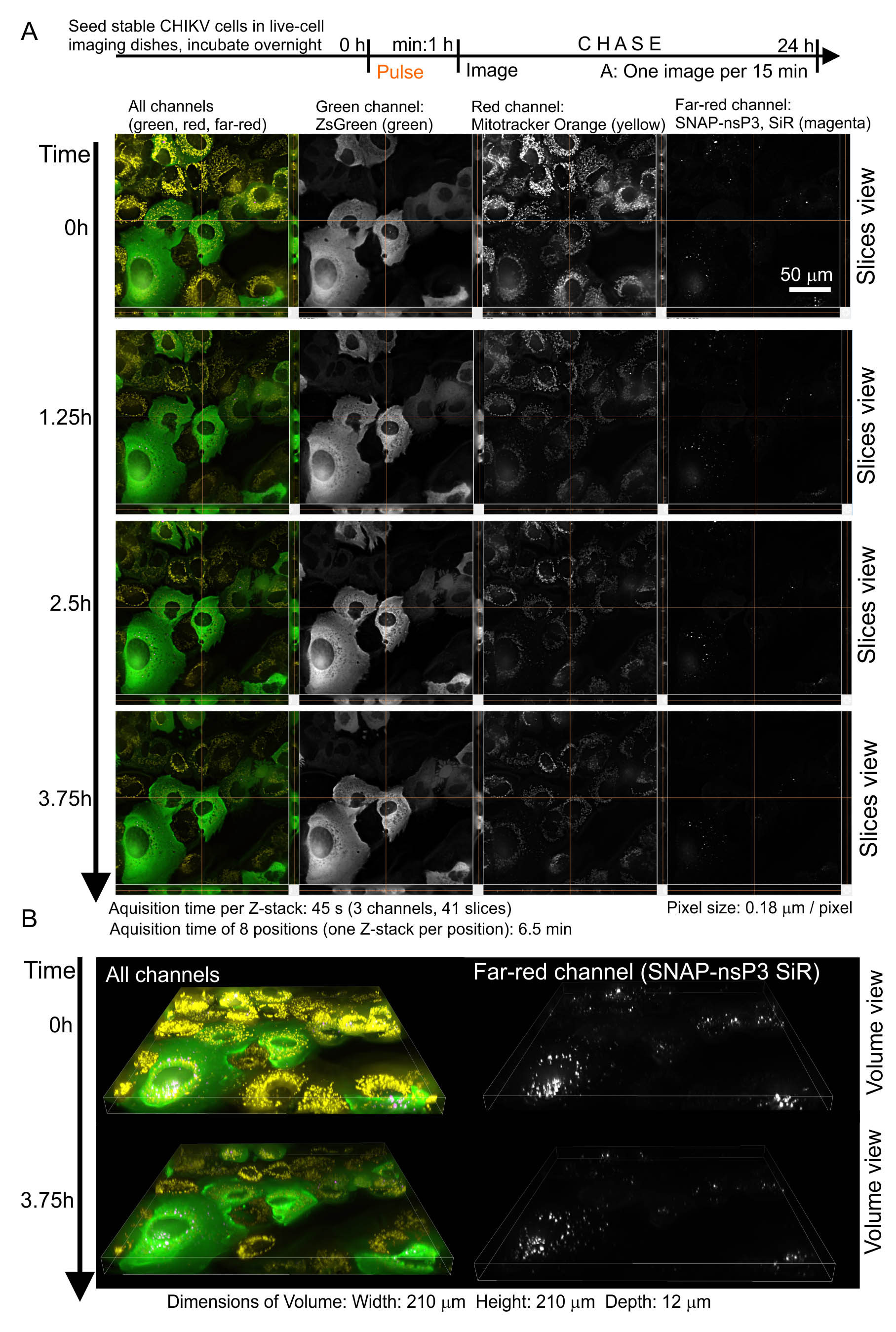
Figure 2. Combination of 5-D imaging and pulse-chase experiments. A. We only show selected frames from a multi-position timelapse series, in which the microscope acquired Z-stacks every 15 min at eight positions. In this setup, each Z-stack (composed of 41 slices) was completed within 45 s, whereas it took 6.5 min to obtain eight positions. The images on the left display all channels with pseudo-colors (green: ZsGreen, yellow: MitoTrackerTM Orange, magenta: SNAP-nsP3). Note that granular structures labeled at 0 h were still present at 3.75 h. This continued presence indicated that these structures remained stable for hours. B. Selected frames from the same timelapse series, this time presented in volume view. The images on the left display all channels with pseudo-colors (green: ZsGreen, yellow: MitoTrackerTM Orange, magenta: SNAP-nsP3).
Recipes
- Live cell imaging solution (enough to add 2 ml of solution to three 35-mm dishes in Steps 10, 12, and optional Step 13)
2.7 ml Fetal Calf Serum
270 µl HEPES
270 µl Glutamax-I
270 µl MEM Nonessential Amino Acids
270-540 µl ProLong Live Antifade Reagent
22.96-23.22 ml FluoroBrite DMEM
The total volume of live cell imaging solution: 27 ml - Tissue culture medium containing serum
500 ml Dulbecco's modified Eagle's medium
56.5 ml Fetal Calf Serum
5.6 ml MEM Nonessential amino acids solution
5.6 ml HEPES
Protocol 2: Quench-pulse-chase experiments paired with ZEISS LSM 880 Airyscan Microscopy
Materials and Reagents
- Micropipette tips, serological pipettes, pipette aids, and microtubes for liquid handling
- TipOne® 1,000 µl XL filter tips (catalog number: S1122-1830)
- TipOne® 200 µl filter tips (catalog number: S1120-8810)
- TipOne® 20 µl filter tips (catalog number: S1120-1810) or SARSTEDT pipette tip 10 µl (catalog number: 70.1130.600)
- FisherbrandTM 5 ml serological pipets (catalog number: 13-676-10H)
- FisherbrandTM 10 ml serological pipets (catalog number: 13-676-10J)
- FisherbrandTM 25 ml serological pipets (catalog number: 13-676-10K)
- Drummond Pipet-Aid XL (catalog number: 4-000-205)
- Microtube 1.5 ml (SARSTEDT, catalog number: 72.690.001)
- 15 ml Centrifuge Tubes, Conical, Sterile (Starlab, catalog number: E1415-0200)
- Glass slide (Academy, catalog number: N/A142)
- Filter paper (Whatman®, catalog number: 1001025)
- 24-well plates (CytoOne®, catalog number: CC7682-7524)
- Clean, sterile 13 mm coverslips (# 1.5) (Academy, catalog number: NPS16/1818)
- Parafilm® M (Bemis, catalog number: PM-996)
- Aluminum foil (Caterwrap, catalog number: AKL-300-030M)
- 10-cm dishes
- Stable CHIKV cells (see Protocol 1)
- Specific reagents for our cell line (for basic tissue culture), which we derived from the hepatoma cell line HuH-7:
- Dulbecco's modified Eagle's medium (Sigma, catalog number: D6429-500ML)
- 100% Fetal Calf Serum (Gibco, catalog number: 10500-064)
- Gibco MEM Non-essential amino acids solution (100x), store at 4 °C up to 24 months from the date of manufacture (Thermo Fisher Scientific, catalog number: 11140050)
- Gibco HEPES (N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid) buffer (100x), store at 4 °C up to 24 months from the date of manufacture (Thermo Fisher Scientific, catalog number: 15630130)
- Trypsin EDTA (Sigma, catalog number: T3924-500ML)
- PBS, Phosphate Buffered Saline (VWR Lifescience, catalog number: E404-100TABS)
- Antibody for immunolabeling of the SNAP-tagged protein
Note: We use a rabbit antibody produced in-house that detects CHIKV nsP3 (Remenyi et al., 2017 and 2018). - Goat anti-rabbit IgG (H+L) Cross-Absorbed Secondary Antibody, DyLight® 405 (Thermo Fisher Scientific, Invitrogen, catalog number: 35551)
Note: The combination of Dylight® 405, ZsGreen, and TMR-Star provides effective color separation and sensitivity for three-color imaging. - SNAP-Cell® TMR-Star (New England Biolabs, catalog number: S9105S; or as part of the SNAP-Cell® Starter Kit, catalog number: E9100S)
Note: Prepare stock solution as described for SNAP-Cell® 647-SiR. - SNAP-Cell® Block (bromothenylpteridine, BTP, New England Biolabs, catalog number: S9106S; or as part of the SNAP-Cell® Starter Kit, catalog number: E9100S). Storage: -20 °C for at least three years dry or three months as a stock solution dissolved in DMSO
- Fixative, 4% Formaldehyde in phosphate buffered saline (PBS), pH 6.9
Note: For the detailed protocol how to prepare the fixative, see: https://www.rndsystems.com/resources/protocols/protocol-making-4-formaldehyde-solution-pbs. Briefly, prepare the fixative from Paraformaldehyde powder (Fisher Chemical, catalog number: T353-500). Dissolve paraformaldehyde powder in 1x sterile PBS (made from tablets) (VWR Lifescience, catalog number: E404-100TABS). Adjust the pH to 6.9 with diluted Hydrochloric Acid (HCl) (Fisher Chemical, catalog number: H/1100/PB17). For long-term storage, keep 15-ml aliquots at -20 °C. When needed, thaw aliquots (vortex thoroughly to remove any precipitation) and use for each experiment. Store any remaining solution for up to 1 month at 4 °C. - Mounting Medium (ProLong Diamond Antifade Mountant, catalog number: P36965)
Note: We use ProLong Diamond because it protects both fluorescent dyes (in our setup: SiR + DyLight® 405) and fluorescent proteins (in our setup: ZsGreen) from fading. - Complete Cell Culture Media (see Recipes)
Equipment
- Micropipettes
- Starlab ErgoOne® 100-1,000 µl Single-Channel Pipette (catalog number: S7110-1000) or Gilson F123602 PIPETMAN Classic Pipet P1000 (Fisher Scientific, catalog number: 10387322)
- Starlab ErgoOne® 20-200 µl Single-Channel Pipette (catalog number: S7100-2200) or Gilson F123615 PIPETMAN Classic Pipet P100 (Fisher Scientific, catalog number: 10442412)
- Starlab ErgoOne® 2-20 µl Single-Channel Pipette (catalog number: S7100-0220) or Starlab ErgoOne® 0.1-2.5 µl Single-Channel Pipette (catalog number: S7100-0125) or Gilson F144801 PIPETMAN Classic Pipet P2 (Fisher Scientific, catalog number: 10635313) or Gilson F123600 PIPETMAN Classic Pipet P20 (Fisher Scientific, catalog number: 10082012)
- Metal tweezers with fine tips for lifting and handling glass coverslips (EMS, catalog number: 78316-1)
- Equipment for basic cell culture techniques and aseptic procedures, i.e.,
- Biosafety cabinet (e.g., Thermo Scientific Holten Safe 2010 Model 1.2, catalog number: 8207071100)
- Humidified incubator set to 37 °C (Panasonic Incusafe, catalog number: MCO-20AIC)
- -20 °C freezer (Labcold, catalog number: RLCF1520)
- Water bath (e.g., Clifton, catalog number: NE2-8D; however, any water bath set to 37 °C may be used)
- Table-top microcentrifuge, 4 °C to room temperature (RT), max speed ≥ 12,000 x g (Eppendorf, model: 5424R)
Note: For spinning down precipitate that may form during extended storage of SNAP probes. - Safety glasses (any manufacturer or model that protects users from formaldehyde splashes will do)
- Confocal laser scanning microscope. For example, a customized ZEISS LSM 880 system that includes the following essential components (for a similar setup, contact your local ZEISS representative for exact ordering information as product codes may differ from customer to customer and country to country):
- Axio Imager Z2 stand, motorized (upright system), product ID: 430000-9902-000
- Motorized Stage, Scanning Stage 130x85 STEP, product ID: 432033-9902-00
- Mounting Frame 160x116 f/ Slides 76x26, product ID: 432315-0000-000
- Scan module LSM 880, product ID: 000000-1994-956
- Support f/scan module LSM (Imager Tube), product ID: 000000-1265-660
- Stepper motor control f, 2 Axes SMC2009, product ID: 432929-9011-000
- Real-time controller standard, product ID: 000000-2031-918
- Objective C PApo 63x/1.4 Oil DIC UV-IR, product ID: 421782-9900-799
- 488 Vis Laser, Laser Argon Multiline 25 mW, product ID: 000000-2086-081
- 561 Vis Laser, Laser 561 nm for LSM 710, product ID: 00000-1410-117
- 633 Vis Laser, Laser Rack LSM 880 incl. 633 Laser, product ID: 000000-2085-478
- Airyscan SR module GaAsP for LSM, product ID: 000000-2058-580
- Emission filter BP495-550 + LP570 for Airyscan, product ID: 000000-2070-488
- Emission filter BP570-620 + LP645 for Airyscan, product ID: 000000-2070-489
- Double BP 420-480 + BP495-620 for Airyscan, product ID: 000000-2095-049
- Double BP 465-505 + LP525 for Airyscan, product ID: 000000-2095-051
- Double BP 420-480 + LP605 for Airyscan, product ID: 000000-2095-052
- User PC advanced Z55A highend, product ID: 000000-2142-968
- ‘Airyscan Fast’ illumination module upgrade for 1x LSM 880 system
- The above list is not exhaustive but only lists essential components that we have found useful for our protocols. Consult with ZEISS about additional components to complete the microscope system (e.g., joystick for stage, hardware license keys, beam splitter, switching mirror, and nosepiece).
- Follow the manufacturer’s recommendations for applying all necessary Airyscan settings within the ZEN microscope software. Our LSM 880 Airyscan microscope could provide a maximum lateral resolution of 140 nm and axial resolution of 400 nm for a fluorophore emitting at 480 nm (images processed with ZEN Black software). The resolution can increase even further to 120 nm XY and 350 nm Z resolution with the use of ZEN Blue software.
- Our original system (LSM 880 with Airyscan) was retroactively upgraded to an ‘Airyscan Fast’ system, whereas today’s systems can be customized with an ‘Airyscan Fast’ module from the start. The ‘Airyscan Fast’ illumination system allows simultaneous illumination of 4 pixels simultaneously to allow very fast and gentle imaging of samples, including the option to scan at super-resolution with 1.5x resolution improvement in XY & Z, imaging at up to 19 images/second with 512 x 512 pixels, 27 images per second at 480 x 480, and 6 images per second at 1024 x 1024.
Software
- ZEISS ZEN software (ZEN 2.3 system HWL for FAST Airyscan)
Note: ZEISS ZEN software drives all components of the LSM 880 system, including setup of Airyscan imaging. ZEN software can also acquire and process raw super-resolution image datasets; save processed image files as ‘.czi’ files, which can be exported to bioimaging analysis software (e.g., Icy software). Saving in this file format ensures the preservation of all the metadata associated with each imaging experiment. - Icy bioimaging software
Note: Bioimaging analysis software. We recommend the free software Icy to visualize, annotate and quantify bioimaging datasets, which can be imported from ZEISS ZEN software packages (de Chaumont et al., 2012). We found that Icy has an intuitive user interface.
Procedure
We use a published labeling protocol (Bodor et al., 2012) to analyze ‘new’ (i.e., freshly translated) pools of the SNAP-tagged viral protein (SNAP-nsP3). By combining the labeling approach with a sensitive detection method, ZEISS LSM 880 Airyscan microscopy, we can visualize the re-emergence of ‘new’ protein and the intracellular sites where ‘new’ proteins accumulate. In the first step (called ‘quench’), a nonfluorescent SNAP-substrate covalently binds to the pool of SNAP-nsP3 present at the onset of an experiment (Figure 3, diagram). After a given amount of time (chase), a second, fluorescent substrate (pulse) labels the cells as described in Protocol 1 (Figure 3, diagram). The pulse only stains the protein pool synthesized during the chase period. Thus, ZEISS LSM 880 Airyscan microscopy will only make this ‘new’ (i.e., freshly translated) pool visible. The total pool of SNAP-nsP3 (pulsed pool + quench pool) can be stained with a standard immunofluorescence assay approach, which reveals the quenched pool that would otherwise remain undetected (Figure 3).
Note: Carry out all liquid handling steps that involve live cells or formaldehyde-containing wells inside the biosafety cabinet. Only use sterile pipettors, pipets, micropipettors, microtubes, and tips when working with live cells. Wear safety glasses when handling formaldehyde solutions.
- Place sterile coverslips in separate wells of a 24-well plate.
Notes:- Adjust the total number of wells according to the number of ‘post-chase’ timepoints. Also include a positive control (e.g., a well without any SNAP Cell® Block added) and negative control (e.g., a well without any chase period).
- In this protocol: two post-chase timepoints (3 h, 6 h) and two controls (four wells in total).
- Detach stable CHIKV cells from growth surface by adding enough trypsin solution (pre-heated to 37 °C in a water bath) to cover the cells that are attached to the surface of the respective culturing vessel (e.g., 1-2 ml of trypsin solution for 10-cm dishes) and seed stable CHIKV cells in wells from Step 1.
Seeding density will again depend on the total duration of the experiment and the doubling time of cells. We aim for about ~80% confluency at the time of fixation of the 0-h, 3-h, and 6-h samples. Thus, we also seed fewer cells in the 3-h and 6-h wells to account for the longer incubation times compared to the 0-h well (10-15% less in 3-h well and 20-30% less in 6-h well). - Incubate at 37 °C, 5% CO2 overnight.
- On the next day, take the frozen stock solution of SNAP Cell® Block from the -20 °C freezer and thaw at room temperature. In a 1.5-ml microtube, dilute SNAP Cell® Block to a final concentration of 2 µM in complete media. Vortex briefly (for ≥ 5 s). Prepare > 200 µl per coverslip. Use diluted reagent within the hour.
- Replace media on cells with 200 µl of the SNAP Cell® Block diluted solution (pre-heated to 37 °C in a water bath) per well (three wells in total). Do not change media on the fourth well (this will serve as the ‘positive control’). Incubate for 30 min.
- Wash cells three times with 1 ml of complete media (pre-heated to 37 °C in a water bath; this washes away any free substrate, which would interfere with downstream applications). Remove media after the last wash step.
- Add 1 ml of complete media (pre-heated to 37 °C in a water bath) to each well and place cells into a tissue-culture incubator for 30 min.
- Wash cells as in Step 6. To prevent the cells from drying out, do not remove the wash media after the last wash step.
- Prepare a new 24-well plate and add 1 ml of 4% formaldehyde fixative to two of the wells.
- Use forceps to transfer one of the ‘quenched’ coverslips and the ‘unblocked’ coverslip to the new plate and submerge in formaldehyde (make sure the side with the layer of cells remains up). The fixed cells on the ‘quenched’ coverslip will serve as the ‘no chase’ control. The cells on the ‘unblocked’ coverslip will serve as a positive control for SNAP-reagent staining.
- Place the original plate, which contains the remaining coverslips, back into the tissue-culture incubator. Incubate cells for the desired chase period of 3 h.
- Place the new plate containing fixed coverslips at 4 °C for storage until all coverslips for all chase periods have been collected. Seal edges of the plate during storage to avoid excessive evaporation (strips of Parafilm M work well).
Note: If the quench was indeed complete, no labeling should occur during the ‘pulse’ period. If Airyscan microscopy can still detect unquenched SNAP-tagged protein, this indicates that the available SNAP Cell® Block reagent did not fully quench the pre-existing pool. Repeat experiments with an increased concentration of SNAP Cell® Block or with a prolonged incubation time until experimental conditions lead to complete quenching of SNAP-tagged protein.
- After three hours, transfer one coverslip to formaldehyde-containing well as described in Step 10 (remove the plate from 4 °C storage and add 1 ml of 4% formaldehyde to a third well). The fixed cells on this coverslip will serve as the ‘3-h chase’ sample.
- Place the original plate, which contains the remaining coverslip, back into the tissue culture incubator. Incubate cells for another three hours. Place plate that holds fixed ‘no chase’ and ‘3-h chase’ coverslips back to 4 °C for storage.
- Repeat Steps 13 and 14 (transfer coverslips into a fourth, formaldehyde-containing well). The fixed cells on this last coverslip will serve as the ‘6-h chase’ sample. Incubate for 30 min at room temperature or for 1-2 h at 4 °C to complete the fixation process of ‘6-h chase’ sample.
- In the biosafety cabinet, wash ‘No chase’, ‘No quench’, ‘3-h chase’, and ‘6-h chase’ controls three times with PBS to remove the formaldehyde fixative.
Note: At this point, users also have the option to handle 24-well plates outside a biosafety cabinet as the cells have already undergone chemical fixation. However, we still prefer to carry out Steps 17, 19, and 20 inside a biosafety cabinet to avoid the contamination of stock solutions and cell culture media. - Take the frozen stock solution of SNAP Cell® TMR-Star from -20 °C freezer and thaw at room temperature. In a 1.5-ml microtube, dilute SNAP-Cell® TMR-Star; perform a 1:600 (final concentration of 1 µM) dilution in complete cell culture media. Prepare at least 200 µl per well to cover the entire coverslip. Vortex briefly (for ≥ 5 s) or pipet the diluted labeling solution up and down (ten times).
Notes:- This protocol should also be compatible with SNAP-Cell® 647 SiR. Use the same labeling conditions as described in Protocol 1 (i.e., 1:1,000 dilution, 15 min incubation).
- Recipe for four coverslips in 24-well plate, scale according to the number of wells:
998.4 µl Complete Cell Culture Media
1.6 µl SNAP-Cell® TMR-Star
- Spin diluted labeling solution for 5 min at maximum speed (≥ 12,000 x g) to remove possible insoluble fluorescent debris. Take care not to disturb the pellet when removing supernatant (which may be invisible).
- Replace the medium on stable CHIKV cells with 200 µl of SNAP-tag labeling medium. Incubate for 15 min at 37 °C, 5% CO2. Protect samples from excessive light exposure during Steps 20-23 (e.g., wrap 24-well plate in aluminum foil).
Note: We found that a 15-min incubation gave us optimal labeling. According to the manufacturer, optimal reaction times range from 5 to 30 min, respectively, depending on experimental conditions and expression levels of the SNAP-tagged protein. - Wash the cells three times each with 1 ml of tissue culture medium containing serum (pre-heated to 37 °C).
- Use a preferred immunofluorescence assay protocol to stain the pool of total tagged protein. At this point, users can handle 24-well plates outside the biosafety cabinet–critical reagents in our case: primary antibodies from rabbit antiserum against nsP3 and dye-conjugated secondary antibodies (anti-rabbit DyLight® 405).
- Mount the immunolabeled coverslips in preferred mounting medium (e.g., ProLong Diamond Antifade Mountant):
- Place a droplet of mountant on a glass slide, remove immunostained coverslips from 24-well plate using forceps, and slowly lower coverslip (cell side down) onto the droplet.
- Use tissue or filter paper to wipe off any excess mountant. We usually fit two to three coverslips on a standard glass slide.
- Allow mountant to cure at room temperature and in the dark (overnight or longer). We store cured slides at 4 °C.
- Image with ZEISS LSM 880 Airyscan microscopy
- SNAP Cell® TMR-Star should have an excitation maximum at 554 nm and an emission maximum at 580 nm. Set up three-color imaging with a blue, green, and red channel. Refer to ZEN software manuals for set-up of ZEISS LSM 880 Airyscan microscopy.
- Upon completion of image acquisition and processing, we use Icy bioimaging software to open saved and processed Airyscan data, which we saved in the .czi file format. Figure 3 shows representative images from a three-color imaging experiment, including screenshots of the Icy histogram viewer for each channel.
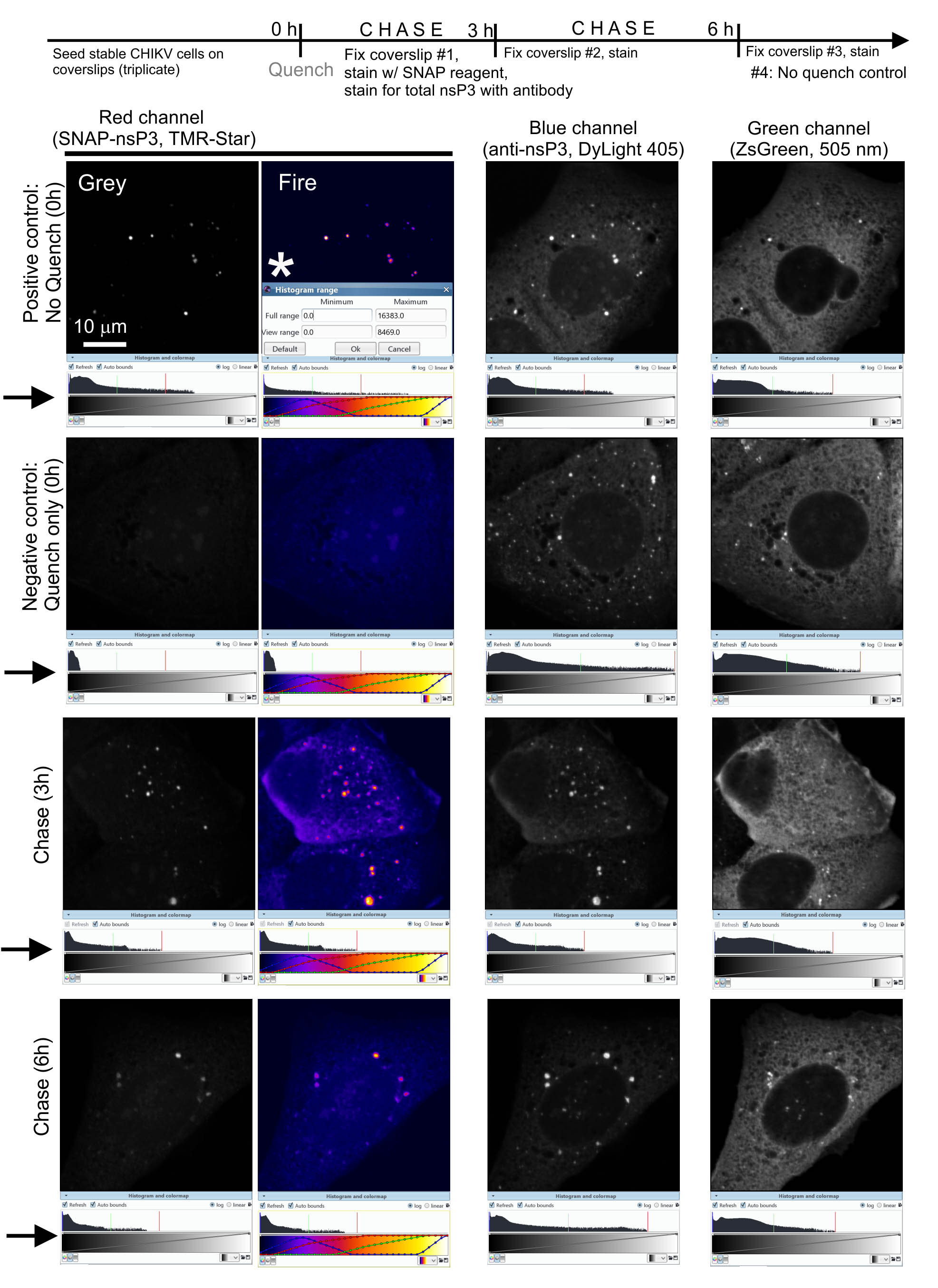
Figure 3. Airyscan microscopy after chase period reveals pools of unblocked viral protein. Representative images from a quench-pulse-chase experiment. Note that the co-distribution of the total nsP3 pool (stained with anti-nsP3 antibody) and the new pool of nsP3 (stained with TMR-Star) suggested a lack of spatial separation of old and new pools of SNAP-nsP3. We set up the microscope for three-color imaging in the blue, green, and red channels. We adjusted image contrast within the Icy platform by dragging the adjustable bounds of the histogram viewer (marked by arrows), which enhanced the contrast in the selected channel without altering the data. We chose a viewing range that provided the best contrast for SNAP-nsP3 channel at the 3-h and 6-h timepoints. We also used the same viewing range to display the data from the positive and negative controls (‘Histogram range’ window, marked by asterisk *, view range minimum of 0 and maximum of 8469.0 pixel intensity values). Note that by applying the colormap ‘Fire’ (within Icy software) we could better display low-intensity granular structures.
Recipes
- Complete Cell Culture Media
500 ml Dulbecco's modified Eagle's medium
56.5 ml Fetal Calf Serum
5.6 ml MEM Nonessential amino acids solution
5.6 ml HEPES
Protocol 3: Tracking SNAP-tagged viral protein assemblies in cell lysates with ZEISS LSM 880 Airyscan microscopy
Materials and Reagents
- Micropipette tips, serological pipettes, pipette aids, and microtubes for liquid handling:
- TipOne® 1,000 µl XL filter tips (catalog number: S1122-1830)
- TipOne® 200 µl filter tips (catalog number: S1120-8810)
- TipOne® 20 µl filter tips (catalog number: S1120-1810) or SARSTEDT pipette tip 10 µl (catalog number: 70.1130.600)
- FisherbrandTM 5 ml serological pipets (catalog number: 13-676-10H)
- FisherbrandTM 10 ml serological pipets (catalog number: 13-676-10J)
- FisherbrandTM 25 ml serological pipets (catalog number: 13-676-10K)
- Drummond Pipet-Aid XL (catalog number: 4-000-205)
- 1.5-ml Microtube (SARSTEDT, catalog number: 72.690.001)
- 15 ml Centrifuge Tubes, Conical, Sterile (Starlab, catalog number: E1415-0200)
- Parafilm® M (Bemis, catalog number: PM996)
- Aluminum foil (Caterwrap, catalog number: AKL-300-030M)
- Moistened paper (e.g., cut sheets of blot absorbent filter paper, Biorad, catalog number: 1703965)
- 10-cm Petri dish (Corning, catalog number: 430167)
- Ibidi 2-well µ-slide, an all-in-one chamber slide with polymer coverslip and ibiTreat surface for optimal cell adhesion (Ibidi, catalog number: 80286)
Note: The IbiTreat surface modification makes the polymer coverslip surface hydrophilic. Alternative approaches to increase the adhesiveness of coverslip surfaces (i.e., Poly-Lysine treatment) and coated glass coverslips may be used (Wheeler et al., 2017). - Greiner CELLSTAR multiwall culture plates, 6-well (Merck/Sigma Aldrich, catalog number: Greiner 657160)
- Stable CHIKV cells (see Protocol 1)
- Trypsin EDTA (Sigma, catalog number: T3924-500ML)
- Materials and reagents for basic tissue culture (see Protocol 1 and 2)
- SNAP-Cell® TMR-Star (New England Biolabs, catalog number: S9105S; or as part of the SNAP-Cell® Starter Kit, catalog number: E9100S), Prepare stock solution as described for SNAP-Cell® 647-SiR
- PBS, Phosphate Buffered Saline (VWR Lifescience, catalog number: E404-100TABS)
- 4% Formaldehyde (see Protocol 2 Step 15)
- Triton X-100 (Sigma, catalog number: T924-500ml)
- KCl (Fisher Chemical, catalog number: P/4240/53)
- NaCl (Fisher Chemical, catalog number: S/3160/60)
- Magnesium chloride (MgCl2) hexahydrate 99.0-101.0%, VWR Chemicals, catalog number: 25108.260)
- Glycerol (Fisher Chemical, catalog number: G/0650/17)
- Piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) (Sigma, catalog number: P1851-100G)
- Leupeptin, hemisulfate salt ≥ 85% by HPLC (Sigma, catalog number: L8511-5MG)
- Pepstatin A (Sigma, catalog number: P5318-5MG)
- Aprotinin, from bovine lung (Sigma, catalog number: A6279-5ML)
- AEBSF (Pefabloc, Sigma, catalog number: 76307-100MG)
- NaOH (Fisher Chemical, catalog number: S/4920/60)
- Home-made Glasgow Lysis Buffer (GLB) containing protease inhibitors (see Recipes)
- 1x GLB, combine the stock solutions, Glycerol, Triton-X, and protease inhibitors (see Recipes)
- This protocol uses our laboratory’s preferred lysis buffer. We have not tried other lysis buffers that use NP-40 or SDS as detergents. However, note that 0.5% NP-40 is a component of the stress granule lysis buffer used to prepare cell lysates for stress granule core isolation (Wheeler et al., 2017).
- Stock solutions for GLB (see Recipes)
- Stock solutions of protease inhibitors (see Recipes)
- 1x GLB (see Recipes)
- Tissue culture medium containing serum (see Recipes)
Note: For #14-24, alternatives of equal purity are also suitable.
Equipment
- Micropipettes
- Starlab ErgoOne® 100-1,000 µl Single-Channel Pipette (catalog number: S7110-1000) or Gilson F123602 PIPETMAN Classic Pipet P1000 (Fisher Scientific, catalog number: 10387322)
- Starlab ErgoOne® 20-200 µl Single-Channel Pipette (catalog number: S7100-2200) or Gilson F123615 PIPETMAN Classic Pipet P100 (Fisher Scientific, catalog number: 10442412)
- Starlab ErgoOne® 2-20 µl Single-Channel Pipette (catalog number: S7100-0220) or Starlab ErgoOne® 0.1-2.5 µl Single-Channel Pipette (catalog number: S7100-0125) or Gilson F144801 PIPETMAN Classic Pipet P2 (Fisher Scientific, catalog number: 10635313) or Gilson F123600 PIPETMAN Classic Pipet P20 (Fisher Scientific, catalog number: 10082012)
- Cell scrapers (Fisher Scientific, catalog number: 08-100-241)
- Equipment for basic cell culture techniques and aseptic procedures, i.e.,
- Biosafety cabinet (e.g., Thermo Scientific Holten Safe 2010 Model 1.2, catalog number: 8207071100)
- Humidified incubator set to 37 °C (Panasonic Incusafe, catalog number: MCO-20AIC)
- -20 °C freezer (Labcold, catalog number: RLCF1520)
- Water bath (e.g., Clifton, catalog number: NE2-8D; however, any type of water bath set to 37 °C is appropriate)
- Table-top microcentrifuge, 4 °C to room temperature (RT), max speed ≥ 12,000 x g (Eppendorf, 5424R)
Note: For spinning down precipitate that may form during extended storage of SNAP probes. - Fume hood (any manufacturer or model that will protect users from formaldehyde fumes will do)
- Confocal laser scanning microscope. For example, a customized ZEISS LSM 880 system that includes the following essential components (for a similar setup, contact your local ZEISS representative for exact ordering information as product codes may differ from customer to customer and country to country):
- Axio Imager Z2 stand, motorized (upright system), product ID: 430000-9902-000
- Motorized Stage, Scanning Stage 130x85 STEP, product ID: 432033-9902-00
- Mounting Frame 160x116 f/ Slides 76x26, product ID: 432315-0000-000
- Scan module LSM 880, product ID: 000000-1994-956
- Support f/scan module LSM (Imager Tube), product ID: 000000-1265-660
- Stepper motor control f, 2 Axes SMC2009, product ID: 432929-9011-000
- Real-time controller standard, product ID: 000000-2031-918
- Objective C PApo 63x/1.4 Oil DIC UV-IR, product ID: 421782-9900-799
- 488 Vis Laser, Laser Argon Multiline 25 mW, product ID: 000000-2086-081
- 561 Vis Laser, Laser 561nm for LSM 710, product ID: 00000-1410-117
- 633 Vis Laser, Laser Rack LSM 880 incl. 633 Laser, product ID: 000000-2085-478
- Airyscan SR module GaAsP for LSM, product ID: 000000-2058-580
- Emission filter BP495-550 + LP570 for Airyscan, product ID: 000000-2070-488
- Emission filter BP570-620 + LP645 for Airyscan, product ID: 000000-2070-489
- Double BP 420-480 + BP495-620 for Airyscan, product ID: 000000-2095-049
- Double BP 465-505 + LP525 for Airyscan, product ID: 000000-2095-051
- Double BP 420-480 + LP605 for Airyscan, product ID: 000000-2095-052
- User PC advanced Z55A highend, product ID: 000000-2142-968
- Transmitted light detector T-PMT, product ID: 000000-2014-999
- ‘Airyscan Fast’ illumination module upgrade for 1x LSM 880 system
- The above list is not exhaustive but only lists essential components that we have found useful for our protocols. Consult with ZEISS about additional components to complete the microscope system (e.g., joystick for stage, hardware license keys, beam splitter, switching mirror and nosepiece).
- Follow the manufacturer’s recommendations for applying all necessary Airyscan settings within the ZEN microscope software. Our Airyscan microscope could provide a maximum lateral resolution of 140 nm and axial resolution of 400 nm for a fluorophore emitting at 480 nm (images processed with ZEN Black software). Airyscan resolution can increase even further to 120 nm XY and 350 nm Z resolution with the use of ZEN Blue software.
- Our original system (LSM 880 with Airyscan) was retroactively upgraded to an ‘Airyscan Fast’ system, whereas today’s systems can be customized with an ‘Airyscan Fast’ module from the start. The ‘Airyscan Fast’ illumination system allows simultaneous illumination of 4 pixels simultaneously to allow very fast and gentle imaging of samples, including the option to scan at super resolution with 1.5x resolution improvement in XY & Z, imaging at up to 19 images/second with 512 x 512 pixels, 27 images per second at 480 x 480, and 6 images per second at 1024 x 1024.
Software
- ZEISS ZEN software (ZEN 2.3 system HWL for FAST Airyscan)
Note: ZEISS ZEN software drives all components of the LSM 880 system, including setup of Airyscan imaging. ZEN software can also acquire and process raw super-resolution image datasets; save processed image files as ‘.czi’ files, which can be exported to bioimaging analysis software (e.g., Icy software). Saving in this file format ensures the preservation of all the metadata associated with each imaging experiment. - Icy bioimaging software
Note: Bioimaging analysis software. We recommend the free software Icy to visualize, annotate and quantify bioimaging datasets, which can be imported from ZEISS ZEN software packages (de Chaumont et al., 2012). We found that Icy has an intuitive user interface.
Procedure
This protocol uses fluorescence light microscopy to reveal stable assemblies of SNAP-nsP3 protein that persist in cell lysates. It is partly based on an isolation protocol (Wheeler et al., 2017) that has been used to determine the proteome and substructure of stress granules (Jain et al., 2016). Our protocol adds additional flexibility in labeling (by using the SNAP labeling system) and detection (by using the sensitive ZEISS LSM 880 Airyscan confocal imaging system).
Note: Carry out all liquid handling steps that involve live cells inside the biosafety cabinet. Only use sterile pipettors, pipets, micropipettors, microtubes, and tips. After cell lysis, users can handle samples outside the biosafety cabinet. Wear safety glasses when handling formaldehyde solutions.
- Detach stable CHIKV cells with trypsin and seed in at least one well of a 6-well microtiter plate. Also, detach and seed naïve cells, which do not express SNAP-tagged proteins, in at least one well; these cells will serve as a negative control.
- Incubate cells under standard growth conditions (i.e., 37 °C at 5% CO2) overnight.
- On the next day, take frozen SNAP Cell® TMR-Star from -20 °C freezer and thaw at room temperature. In a 1.5-ml microtube, dilute SNAP-Cell® TMR-Star 1:600 (final concentration of 1 µM) dilution in complete cell culture media. Prepare at least 1 ml per well to cover the entire area of the well. Vortex briefly (for ≥ 5 s) or pipet the diluted labeling solution up and down (ten times). We have only validated this protocol with the SNAP-Cell® TMR-Star reagent. However, the protocol should also be compatible with SNAP-Cell® 647 SiR. Use the same labeling conditions as described in Protocol 1 (i.e., 1:1,000 dilution, 15 min incubation).
Note: Recipe for two dishes, scale accordingly for additional dishes:
1996.8 µl Complete Cell Culture Media
3.2 µl SNAP-Cell® TMR-Star - Spin diluted labeling solution for 5 min at maximum speed (≥ 12,000 x g) to remove possible insoluble fluorescent debris. Take care not to disturb the pellet when removing supernatant (which may be invisible).
- Replace the medium on stable CHIKV cells with 1 ml of SNAP-tag labeling medium (pre-heated to 37 °C in a water bath). Also, replace medium in the ‘negative control’ well. Incubate for 15 min at 37 °C, 5% CO2.
Note: We found that a 15-min incubation gave us optimal labeling. According to the manufacturer, optimal reaction times range from 5 to 30 min, respectively, depending on experimental conditions and expression levels of the SNAP-tagged protein. - Wash the cells three times each with 2 ml of tissue culture medium containing serum (pre-heated to 37 °C in a water bath). Do not remove media after the final wash.
- Place cells back into the humidified incubator for another 45-60 min.
We use an extended wash-out period (compared to Protocol 1) to reduce the non-specific background-binding further. Moreover, we do not add ProLong Gold Live in this protocol, as live-cell imaging is only limited to an optional quality-control-step in Step 8. - Optional: Confirm fluorescent staining of SNAP-nsP3 with microscopy
- A basic widefield fluorescent microscope equipped with a 10x or 20x objective is sufficient to evaluate the quality of SNAP-labeling through the microscope eyepiece. If documentation of staining quality is needed, take pictures with a connected camera. Also, confirm the absence of staining in the well that contains negative control.
- We recommend this step when testing labeling conditions for the first time. Perform imaging quickly and proceed to Step 9 as soon as possible to limit the deterioration of cell health. If this is not logistically possible, we recommend proceeding to Step 9 directly.
- Replace complete cell media with 1 ml PBS and use separate cell scrapers to detach cells from growth area in each well. Transfer the resulting cell suspension to a 1.5 ml microtube. Pellet cells at 1,500 x g, 3 min at room temperature. Remove supernatant.
Optional pause point: We freeze pellets at ≤ -20 °C if we want to carry out lysis at a later time. - Lyse pellets by adding 300 µl of ice-cold Glasgow Lysis buffer containing protease inhibitors (see Recipes). Ensure complete re-suspension of pellet through repeated pipetting, flicking the tube, or vortexing. If pellet came from the freezer, thaw on ice for 5 min before adding lysis buffer.
- Vortex Lysates for 30 s. Place on ice for 30 s. We return samples to ice in between vortexing cycles to prevent an extended incubation period at room temperature. Alternatively, lysates can be vortexed in a cold room.
- Repeat Step 11 three times.
- Spin at 850 x g for 5 min at 4 °C to remove remaining cellular debris.
Note: We do not further purify assemblies of SNAP-nsP3 after this step but instead use the crude supernatant in the microscopic analysis. It would be interesting to test whether subsequent centrifugation at 18,000 x g can pellet assemblies of SNAP-nsP3; this step is essential in the isolation of a pure population of stress granule cores (Wheeler et al., 2017). After this step, users can handle cell lysates outside a biosafety cabinet. - Transfer the entire volume of supernatant to a two-well Ibidi chambered plastic slide. Discard pelleted cellular debris.
Note: We have noticed that the supernatant may appear turbid at this step. Proteins that are components of RNPs are known to undergo liquid-liquid phase transitions, and turbidity is due to the formation of small protein-rich droplets (Molliex et al., 2015). However, we have not yet tested whether purified SNAP-nsP3 can form similar droplets in solution. - Incubate overnight at 4 °C in a humidified environment in the dark.
- To obtain humidified conditions, we cover the Ibidi slide with the supplied plastic cover and place the slide in a 10-cm Petri dish that also contains moistened paper (e.g., cut sheets of Western blotting filter).
- We further seal dishes with Parafilm M and wrap them in aluminum foil to prevent light exposure. We incubate overnight to give assemblies of SNAP-nsP3 enough time to settle to the bottom of the chamber.
- Future experiments may determine the minimum duration for deposition of SNAP-nsP3 to the bottom of the chambered slide; this information would be useful for reducing the overall time required to complete Protocol 3.
- In a fume hood, add 1 ml of 4% formaldehyde to wells containing cell lysates. Incubate for one hour at room temperature.
Note: Slowly dispense formaldehyde with a micropipette. We add this step to fix assemblies of SNAP-nsP3; fixation reduces the likelihood that the assemblies detach from the well surface during subsequent washing steps. - Wash wells three times with PBS. Ensure that wells do not dry out in between washes.
- Transfer slide to microscopy system of choice:
As described in Protocol 2, we use the ZEISS LSM 880 imaging system operated in Airyscan mode. Use the appropriate filter settings for TMR-Star, which should have an excitation maximum at 554 nm and an emission maximum at 580 nm. Also, acquire an image in brightfield mode. This image can provide an additional record of the examined biostructure. - We recommend taking at least three images for each experiment:
We acquire images with the 63x objective. Export processed Airyscan files to Icy bioimaging software. Adjust the image contrast within the Icy software by dragging the boundaries of the viewing range in the software’s histogram viewer (see Figure 4, arrows). We prefer viewing the SNAP-nsP3 channel with the ‘Fire’ colormap. Use Icy’s controls to zoom into regions-of-interest. See Figure 4 for representative images.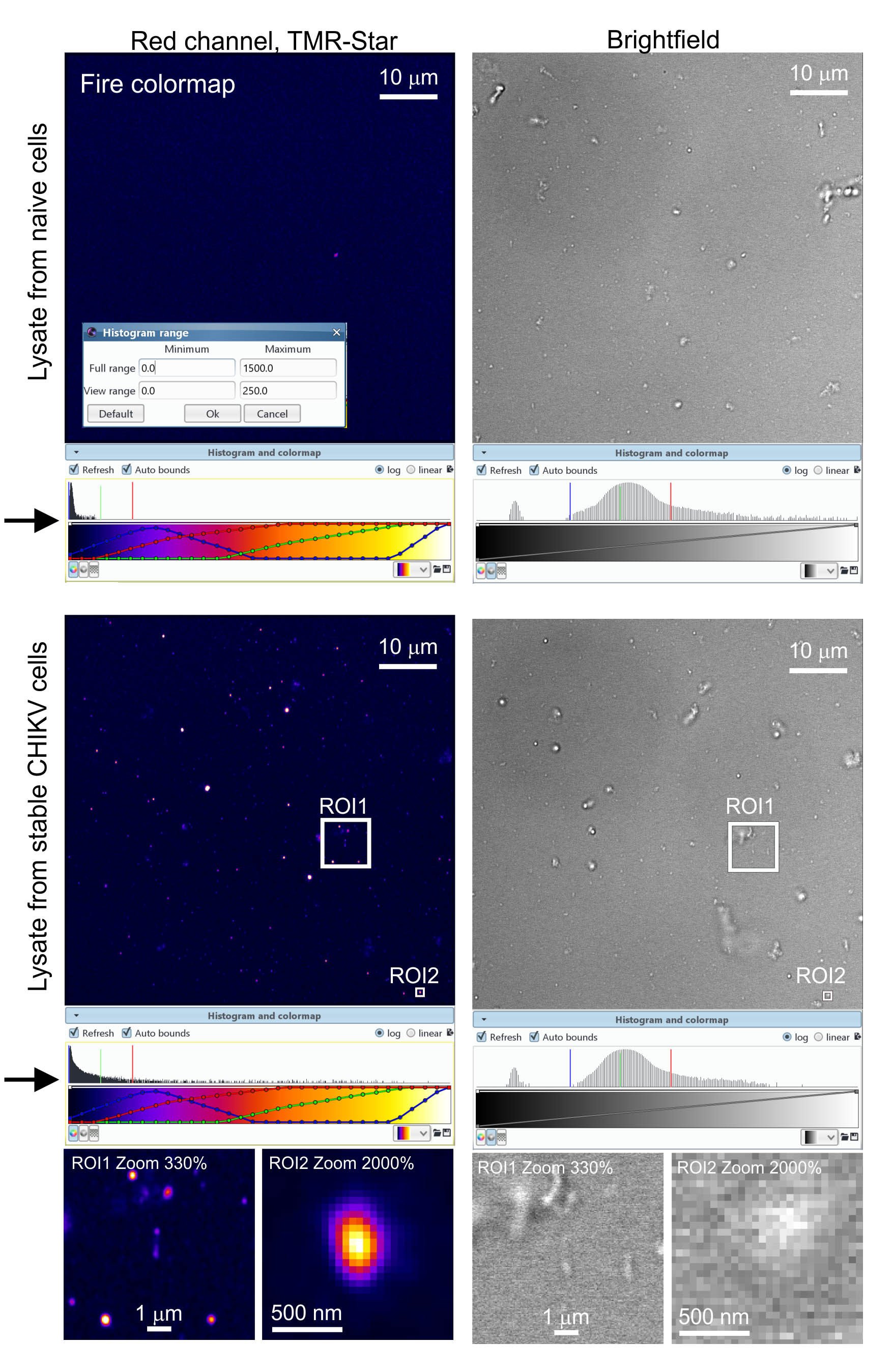
Figure 4. Analysis of cell lysates with Airyscan microscopy. We processed samples according to Protocol 3 and detected granular structures containing TMR-Star-labelled SNAP-nsP3. We chose the ‘Fire’ colormap in Icy to display signals from the TMR-Star staining. We also adjusted image contrast by dragging the adjustable bounds of the histogram viewer (marked by arrows). We acquired images at a zoom factor of 1 with a 63x objective. During image processing, we also magnified regions-of-interest (ROI) by increasing the digital zoom within the Icy software (i.e., by a factor of 3.3 for ROI1 and 20 for ROI2). Lastly, we also acquired a brightfield channel to provide a reference and reveal all contrast-producing structures present in the lysate.
Recipes
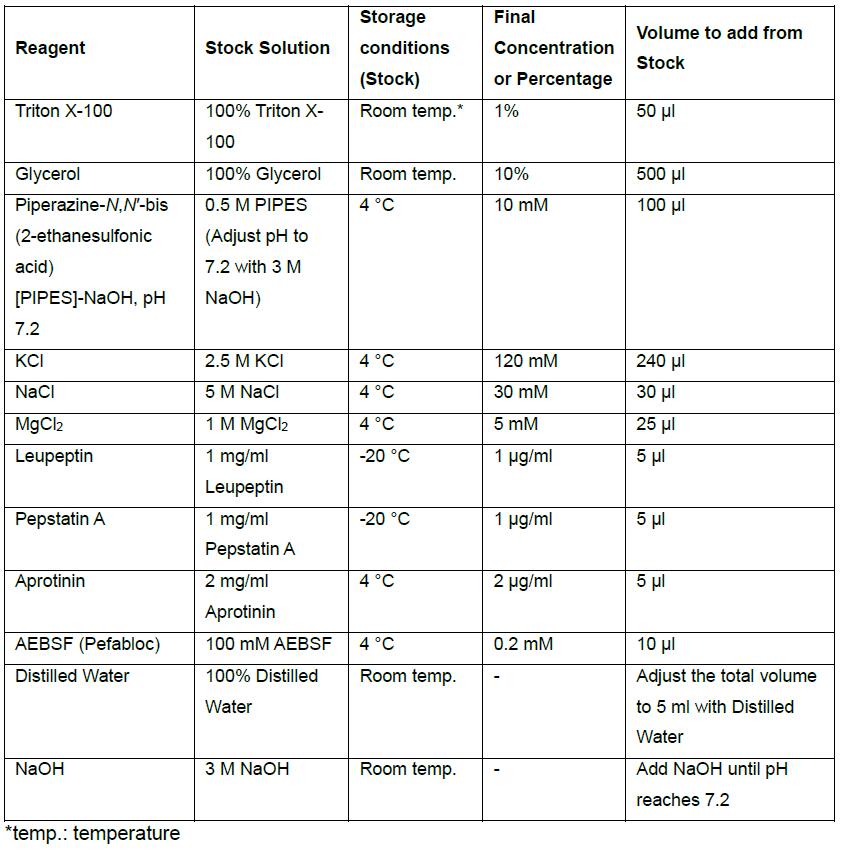
- Home-made Glasgow Lysis Buffer (GLB) with protease inhibitors, 5 ml volume
- Stock solutions for GLB
Add the appropriate amount of solids to distilled H2O to make stock solutions of PIPES (adjust pH to 7.2 with 3 M NaOH), KCl, NaCl, MgCl2 at concentration listed in the table. Store these stock solutions at 4 °C. - Stock solutions of protease inhibitors, store in the following manner:
Leupeptin 1 mg/ml at -20 °C
Pepstatin A 1 mg/ml at -20 °C
Aprotinin 2 mg/ml at 4 °C
AEBSF (Pefabloc) 100 mM at 4 °C - 1x GLB
Combine the stock solutions, Glycerol, Triton-X, and protease inhibitors according to the table. Adjust volume to 5 ml with distilled H2O and pH to 7.2 with 3 M NaOH
Note: Use fresh 1x GLB for each experiment. - Tissue culture medium containing serum
500 ml Dulbecco's modified Eagle's medium
56.5 ml Fetal Calf Serum
5.6 ml MEM Nonessential amino acids solution
5.6 ml HEPES
Data analysis
We consider the unprocessed image files that microscope users save in either the Nikon NIS Elements AR software (.nd2 file format) or ZEISS ZEN software (.czi file format) to be ‘raw’ data. Thus, data analysis in these protocols was for qualitative purposes and made up exclusively of image visualization and digital processing of acquired images (e.g., adjusting brightness, contrast, and pseudo-colors) in either Nikon NIS Elements AR software or Icy bioimaging software. Although it is also possible to extract quantitative data from these images, our initial method development, which focused on the application of new labeling and imaging methods, did not include these types of bioimage-informatics approaches. Future studies may benefit greatly from incorporating rigorous bioimage-informatics techniques to the protocol described here.
Acknowledgments
A Wellcome Trust Investigator Award funded this work (WT 096670, awarded to Mark Harris). Purchase of shared equipment was made possible by a Wellcome Trust Multi-user equipment award (Zeiss LSM 880 instrument, WT104918MA, ‘Multifunctional imaging of living cells for biomedical sciences’). The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication. We thank Dr. Sally Boxall and the Bio-imaging Facility within the Faculty of Biological Sciences of the University of Leeds for access and help with Airyscan microscopes. The authors acknowledge Gina Gamble and Dr. Kate Lewis for their help using the Nikon Ti2-E Inverted microscope. We recognize the previous study by Bodor et al. (2012) which described the pulse-chase and quench-pulse-chase approaches. We are grateful to Mark B. Carascal and Dr. Samuel Ko for comments during the revision of the manuscript.
Competing interests
The authors declare that no conflicts of interest or competing interests exist.
References
- Bodor, D. L., Rodriguez, M. G., Moreno, N. and Jansen, L. E. (2012). Analysis of protein turnover by quantitative SNAP-based pulse-chase imaging. Curr Protoc Cell Biol Chapter 8: Unit8.8.
- Cole, N. B. (2014). Site-specific protein labeling with SNAP-tags. Curr Protoc Protein Sci 73: Unit 30.1.
- de Chaumont, F., Dallongeville, S., Chenouard, N., Herve, N., Pop, S., Provoost, T., Meas-Yedid, V., Pankajakshan, P., Lecomte, T., Le Montagner, Y., Lagache, T., Dufour, A. and Olivo-Marin, J. C. (2012). Icy: an open bioimage informatics platform for extended reproducible research. Nat Methods 9(7): 690-696.
- Frigault, M. M., Lacoste, J., Swift, J. L. and Brown, C. M. (2009). Live-cell microscopy–tips and tools. J Cell Sci 122(Pt 6): 753-767.
- Huff, J., Bergter, A., Birkenbeil, J., Kleppe, I., Engelmann, R. and Krzic, U. (2017). The new 2D Superresolution mode for ZEISS Airyscan. Nat Methods 14: 1223.
- Huff, J. (2015). The Airyscan detector from ZEISS: confocal imaging with improved signal-to-noise ratio and super-resolution. Nat Methods 12: 1205.
- Jain, S., Wheeler, J. R., Walters, R. W., Agrawal, A., Barsic, A. and Parker, R. (2016). ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 164(3): 487-498.
- Kedersha, N., Stoecklin, G., Ayodele, M., Yacono, P., Lykke-Andersen, J., Fritzler, M. J., Scheuner, D., Kaufman, R. J., Golan, D. E. and Anderson, P. (2005). Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 169(6): 871-884.
- Kedersha, N., Tisdale, S., Hickman, T. and Anderson, P. (2008). Real-time and quantitative imaging of mammalian stress granules and processing bodies. Methods Enzymol 448: 521-552.
- Keppler, A., Kindermann, M., Gendreizig, S., Pick, H., Vogel, H. and Johnsson, K. (2004a). Labeling of fusion proteins of O6-alkylguanine-DNA alkyltransferase with small molecules in vivo and in vitro. Methods 32(4): 437-444.
- Keppler, A., Pick, H., Arrivoli, C., Vogel, H. and Johnsson, K. (2004b). Labeling of fusion proteins with synthetic fluorophores in live cells. Proc Natl Acad Sci U S A 101(27): 9955-9959.
- Li, C., Kuang, C. and Liu, X. (2018). Prospects for fluorescence nanoscopy. ACS Nano 12(5): 4081-4085.
- Molliex, A., Temirov, J., Lee, J., Coughlin, M., Kanagaraj, A. P., Kim, H. J., Mittag, T. and Taylor, J. P. (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163(1): 123-133.
- Muller, C. B. and Enderlein, J. (2010). Image scanning microscopy. Phys Rev Lett 104(19): 198101.
- Nakabayashi, H., Taketa, K., Miyano, K., Yamane, T., Sato, J. (1982). Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 42(9): 3858-63.
- Remenyi, R., Gao, Y., Hughes, R. E., Curd, A., Zothner, C., Peckham, M., Merits, A. and Harris, M. (2018). Persistent replication of a chikungunya virus replicon in human cells is associated with presence of stable cytoplasmic granules containing nonstructural protein 3. J Virol 92(16).
- Remenyi, R., Roberts, G. C., Zothner, C., Merits, A. and Harris, M. (2017). SNAP-tagged chikungunya virus replicons improve visualisation of non-structural protein 3 by fluorescence microscopy. Sci Rep 7(1): 5682.
- Sheppard, C. J., Mehta, S. B. and Heintzmann, R. (2013). Superresolution by image scanning microscopy using pixel reassignment. Opt Lett 38(15): 2889-2892.
- Utt, A., Das, P. K., Varjak, M., Lulla, V., Lulla, A., Merits, A. (2015). Mutations conferring a noncytotoxic phenotype on chikungunya virus replicons compromise enzymatic properties of nonstructural protein 2. J Virol 89(6): 3145-3162.
- Wheeler, J. R., Jain, S., Khong, A. and Parker, R. (2017). Isolation of yeast and mammalian stress granule cores. Methods 126: 12-17.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Remenyi, R., Li, R. and Harris, M. (2019). On-demand Labeling of SNAP-tagged Viral Protein for Pulse-Chase Imaging, Quench-Pulse-Chase Imaging, and Nanoscopy-based Inspection of Cell Lysates. Bio-protocol 9(4): e3177. DOI: 10.21769/BioProtoc.3177.
Category
Cell Biology > Cell imaging > Fluorescence
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.