- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation of Neural Stem Cells from the Embryonic Mouse Hippocampus for in vitro Growth or Engraftment into a Host Tissue



Published: Vol 9, Iss 4, Feb 20, 2019 DOI: 10.21769/BioProtoc.3165 Views: 10102
Reviewed by: Xi FengNolwenn PasquetKhyati Hitesh Shah
Original research article
The authors used this protocol in:

Feb 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
For both stem cell research and treatment of the central nervous system disorders, neural stem/progenitor cells (NSPCs) represent an important breakthrough tool. In the expanded stem cell-based therapy use, NSPCs not only provide a powerful cell source for neural cell replacement but a useful model for developmental biology research. Despite numerous approaches were described for isolation of NSPCs from either fetal or adult brain, the main issue remains in extending cell survival following isolation. Here we provide a simple and affordable protocol for making viable NSPCs from the fetal mouse hippocampi, which are capable of maintaining the high viability in a 2D monolayer cell culture or generating 3D neuro-spheroids of cell aggregates. Further, we describe the detailed method for engraftment of embryonic NSPCs onto a host hippocampal tissue for promoting multilinear cell differentiation and maturation within endogenous environment. Our experimental data demonstrate that embryonic NSPCs isolated using this approach show the high viability (above 88%). Within a host tissue, these cells were capable of differentiating to the main neural subpopulations (principal neurons, oligodendrocytes, astroglia). Finally, NSPC-derived neurons demonstrated matured functional properties (electrophysiological activity), becoming functionally integrated into the host hippocampal circuits within a couple of weeks after engraftment.
Keywords: Embryonic neural stem/progenitor cells (NSPCs)Background
Multipotent stem cells of neural origin, neural stem/progenitor cells (NSPCs), represent a versatile tool for enabling nerve cell replacement in a number of neurological disorders characterized by the loss of specialized neural subpopulations. The unlimited lifespan and directed NSPC differentiation into the key neural subtypes (neurons, astrocytes, and oligodendrocytes) make these cells the most attractive candidate for emerging stem cell-based therapies for presently incurable brain disorders (Kopach and Pivneva, 2018). Stem cells exist in the developing and mature nervous system and can be isolated from either fetal brain (embryonic NSPCs) or, as established more recently, from mature brain (in adults, both subventricular and subgranular zones in dentate gyrus of the hippocampus, forebrain, cerebellum and olfactory bulb) (Pagano et al., 2000; Roy et al., 2000; Klein et al., 2005; Behnan et al., 2017; Kempermann et al., 2018). Despite that adult stem cells bear expectations for an advanced translational approach (Kempermann et al., 2018), a limited number of rigorous evidence supports this research avenue at the moment.
Accumulating evidence for the properties of fetal NSPCs and phenotype specifications, confirmed by dedicated clinical trials, retains a focus on this stem cell type. Furthermore, embryonic NSPCs represent an essential tool for developmental biology research. Various methodological approaches were probed to isolate NSPCs from the fetal brains and to maintain cells in conditions determining their high viability, hence, progeny. Such conditions largely dictate phenotyping of differentiating progenitors to acquire the competitive NSPC-derived cells. The two main strategies for preserving the high viability of isolated NSPCs exist in i) cell culture in vitro and ii) grafting into a host tissue. A classical 2D culture, an adherently expanded monolayer of cells, has been routinely used for stem cell growth in vitro and/or for directed phenotypic differentiation (Conti et al., 2005; Pollard et al., 2006). In parallel, newer developed approach of generating 3D cultures, neuro-spheroids of an assembly of NSPCs at various phenotypic and developmental stages, has been emerged to represent more physiological conditions pertinent to neuronal differentiation rather than glial (Ignatova et al., 2002; Suslov et al., 2002; Makri et al., 2010). This approach can potentially enhance the therapeutic potential of NSPCs in vitro. Another alternative–grafting of viable NSPCs into brain tissue–fulfill a strategy for promoting the multi-lineage differentiation of progenitors within a host tissue, regulated by endogenous environment. Such an approach has recently been implemented in the treatment of the ischemia-damaged hippocampal tissue (Tsupykov et al., 2014; Kopach et al., 2018).
Here we provide the detailed protocol that we routinely use for isolation of viable NSPCs from the fetal mouse hippocampus. This protocol was also utilized for engraftment of undifferentiated cells to the hippocampal tissue for monitoring maturation of NSPC-derived cells within a host environment in our previous studies (Kopach et al., 2018). We describe all the details on how to test the viability of the obtained embryonic NSPCs and maintain the cells in multiple approaches for further proposed use.
Materials and Reagents
- Materials
- 15 ml and 50 ml centrifuge tubes (Corning, catalog numbers: 430790, 430828)
- 1.5 ml Eppendorf tubes (Sigma-Aldrich, catalog number: T9661)
- Glass Pasteur pipette (1 mm diameter) (Fisher Scientific, catalog number: 13-678-6A)
- 35 mm, 60 mm, and 100 mm tissue culture dishes (CELLSTAR, catalog numbers: 627160, 628160, 664160)
- Falcon® 40-µm cell strainer (Corning, Falcon®, catalog number: 352340)
- 0.45-µm sterile filter (Fisher Scientific, MerckTM, catalog number: 12279299)
- 6-well and 24-well tissue culture plates (Greiner Bio-One, catalog numbers: 657160, 662160)
- 13 mm glass coverslips (Henz Herenz, catalog number: 1051204)
- SuperFrost® glass slides (VWR, Thermo Fisher Scientific, catalog number: 631-0706)
- Plastic serological pipettes (5 ml, 10 ml, and 25 ml) (Corning® Costar® Stripette®, catalog numbers: 4487, 4488, 4489)
- Pipette tips (TipOne, STARLAB, catalog numbers: S1111-3700, S1111-1706, S1111-6701)
- Millipore® Millicell® cell culture plate inserts (Sigma-Aldrich, catalog number: Z353086)
- Whatman® qualitative filter paper (Sigma-Aldrich, catalog number: WHA1001110)
- Scalpel blades (Fisher Scientific, Swann-MortonTM, catalog number: 11772724)
- Scalpel with retractable blade (Eickemeyer®, catalog number: 100504)
- Parafilm® M (Sigma-Aldrich, catalog number: P7793)
- Animals
- Pregnant FVB-Cg-Tg(GFPU) 5Nagy/J GFP mice (obtained from the animal facilities of State Institute of Genetic and Regenerative Medicine)
- Embryos of FVB-Cg-Tg(GFPU) 5Nagy/J GFP mice at the developmental stage of E16-E17
- Pups of FVB mice (8 to 9-days old)
- Reagents
- HBBS without calcium and magnesium (Sigma-Aldrich, catalog number: 55021C)
- Paraformaldehyde (PFA, Sigma-Aldrich, catalog number: P6148)
- 0.25% trypsin-EDTA (Sigma-Aldrich, catalog number: T4049)
- 22% Percoll (Sigma-Aldrich, catalog number: 17-0891-01)
- Neurobasal® medium (Thermo Fisher Scientific, GibcoTM, catalog number: 21103049)
- B-27® supplement (50x) (Thermo Fisher Scientific, GibcoTM, catalog number: 17504044)
- GlutaMAXTM supplement (100x) (Thermo Fisher Scientific, GibcoTM, catalog number: 35050061)
- N-acetyl-L-cysteine (NAC) (Sigma-Aldrich, catalog number: A7250)
- Penicillin-streptomycin (P/S) solution (100,000 U/ml) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122)
- FGF-2 (Sigma-Aldrich, catalog number: SRP4037)
- Matrigel (BD Biosciences, catalog number: 354234)
- Triton® X-100 (Sigma-Aldrich, catalog number: T8787)
- Accutase® solution (Sigma-Aldrich, catalog number: A6964)
- 0.4% Trypan blue (Thermo Fisher Scientific, GibcoTM, catalog number: 15250061)
- Antibodies
- Primary
- Mouse anti-Nestin antibody (1:100) (Santa Cruz Biotechnology, catalog number: sc-23927)
- Rabbit anti-β-tubulin III (1:500) (Sigma-Aldrich, catalog number: T2200)
- Mouse anti-GFAP (1:200) (Sigma-Aldrich, catalog number: AMAB91033)
- Rabbit anti-olig-2 (1:200) (Abcam, catalog number: ab136253)
- Mouse anti-NeuN (1:100) (Millipore, catalog number: MAB377)
- Secondary
- Donkey anti-mouse Alexa Fluor 555 (1:1,000) (Thermo Fisher Scientific, catalog number: A-31570)
- Donkey anti-rabbit Alexa Fluor 647 (1:1,000) (Thermo Fisher Scientific, catalog number: A-31573)
- Nuclei tracer Hoechst 33342 (1:5,000) (Sigma-Aldrich, catalog number: 14533)
- Primary
- ImmunoHistoMountTM (Sigma-Aldrich, catalog number: I1161)
- 70% Ethanol (Sigma-Aldrich, catalog number: 459836)
- Sodium dihydrogen phosphate monobasic (NaH2PO4) (Sigma-Aldrich, catalog number: S0751)
- Disodium phosphate dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: S0876)
- Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: S8045)
- Minimum Essential Medium (MEM) (Sigma-Aldrich, catalog number: M2249)
- Tris (Sigma-Aldrich, catalog number: 252859)
- Sodium bicarbonate (Sigma-Aldrich, catalog number: 792519)
- HEPES (Thermo Fisher Scientific, catalog number: 15630080)
- D(+)-glucose (Sigma-Aldrich, catalog number: G8270)
- Horse serum (Sigma-Aldrich, catalog number: H1138)
- Phosphate buffered saline (PBS), pH 7.4 (Sigma-Aldrich, catalog number: P3813)
- BD Cell Wash buffer (BD Biosciences, catalog number: 349524)
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: 05470)
- 7-AAD (BD PharmingenTM, catalog number: 559925)
- 0.2 M Phosphate buffer (pH 7.4) (see Recipes)
- 4% Paraformaldehyde (PFA) in 0.1 M phosphate buffer (see Recipes)
- Growth medium (see Recipes)
- Tissue dissecting medium (pH 7.3) (see Recipes)
- Tissue culture medium (pH 7.2) (see Recipes)
- Antibody solution (see Recipes)
- Blocking solution (see Recipes)
Equipment
- Pipettes (Eppendorf, Research® Plus, catalog numbers: EP 3123000012; EP 3123000098; EP 3123000055; EP 3120000062)
- 250 ml Graduated cylinder (Karter Scientific, catalog number: 213I13)
- Dry heat sterilizing cabinet (Grande, model: GIO-072)
- Fine tools for dissection and manipulating embryonic brains
- Dissecting scissors (Dumont, catalog number: 14090-09)
- Small spatula (Dumont, catalog number: 10090-13)
- Forceps (Dumont, catalog number: 91197-00)
- Angled forceps (Dumont, catalog number: 00125-11)
- C-Chip disposable hemocytometer/Neubauer (Labtech International, Heathfield, UK, catalog number: DHC-N01)
- Refrigerated centrifuge (Thermo Fisher Scientific, model: IEC-CL30R)
- Water bath (Biosan, model: WB-4MS)
- Biosafety Cabinet Class II Type A2 (Labconco, model: Purifier Logic)
- CO2 incubator (Thermo Fisher Scientific, model: SteriCycle 371)
- Flow cytometer (Becton Dickinson, model: BD FACSAria сell sorter)
- Tissue chopper (McIlwain, model: MTC/2E)
- Alcohol burner (Sigma-Aldrich, catalog number: Z509604)
- Inverted microscope (Olympus, model: IX71, equipped with camera DP-20)
- Confocal microscope (Olympus, model: FV1000)
- Stereo microscope (Olympus, model: SZX7)
- 4 °C refrigerator (Haier Biomedical, model: HYC-940)
Software
- QuickPHOTO software (PROMICRA, s.r.o., Czech Republic)
- FV10-ASW software (Olympus, Japan)
- BD FACSDivaTM 6.1.2 software (BD Biosciences, USA)
Procedure
- Set-up (general preparation)
Note: All steps, including harvesting the embryos, are performed in a flow hood.- Sterilization of the working area
Expose the biosafety cabinet hood to UV light for 1 h before starting to use it. Wipe all working surfaces and the tools with 70% ethanol. - Prepare the Matrigel-coated coverslips for cell cultures
Dissolve Matrigel in a cold Neurobasal medium (10 mg/ml) by pipetting up and down to mix the Matrigel and medium very well. Put a sterile glass coverslip at each well of a 24-well plate. Add the prepared mixture onto each coverslip in a volume of approximately 0.5 ml per coverslip, making sure the surface of each coverslip is well covered. Keep coverslips for 40 min at room temperature to let them be coated with Matrigel. Afterward, remove Matrigel and wash the coverslips with Neurobasal medium 3 times. Avoid letting the Matrigel-coated coverslips become completely dry after washing.
Note: For monolayer cell cultures, prepare the Matrigel-coated coverslips at a 24-well plate. The number of coverslips/wells plated depends on experimental design and density of desired cultures and may need to be adjusted accordingly. Seeding cells onto glass coverslips is also preferable for the purpose of further immunostaining. Glass coverslips may be prepared in advance. In such a case, seal the 24-well plate containing the Matrigel-coated coverslips very well with parafilm. The Matrigel-coated coverslips should not be kept longer than for 24 h. - Polishing and sterilizing glass Pasteur pipettes
Prepare the glass Pasteur pipettes by polishing the pipette tips over flame of an alcohol burner. To obtain pipettes with tips of various sizes, rotate each pipette slowly over flame from few to ten seconds until the glass of pipette tip’s edge become polished that can be visually seen (Figure 1). The smallest-sized tips have an internal diameter of approximately 0.2-0.3 mm, the medium-sized–approximately 0.5-0.6 mm, and the large-sized–about 0.9 mm (Figure 1B). Before use, sterilize the flame-polished pipettes at 180 °C for 1-2 h.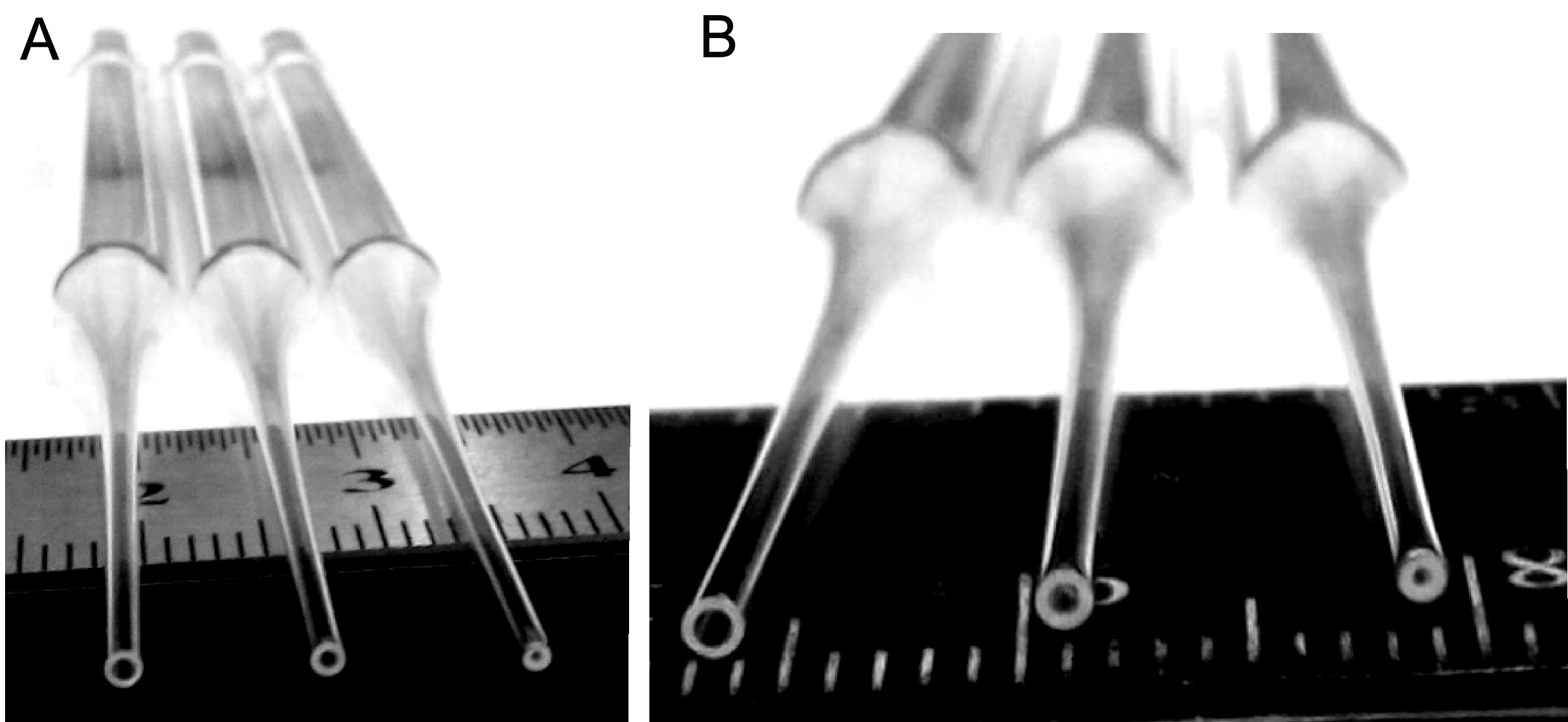
Figure 1. Snapshots of the glass Pasteur pipettes with flame-polished tips of different sizes. The pipettes are used for mechanical dissociation of the cells from the fetal mouse hippocampi, starting using the pipette with the largest-sized internal tip diameter (from left to right on A and B).
- Sterilization of the working area
- Harvesting embryos and dissecting the fetal hippocampi
- Sacrifice a pregnant female mouse at the desired stage (we use E16-E17) according to the procedures approved by your Institute.
- Wipe the abdominal skin with 70% ethanol and make a longitudinal incision to open the abdominal wall. Use another pair of sterile tools (forceps and scissors) for opening.
- Cut the uterus and carefully take the embryos out using a stereo microscope to visually control the procedure. If needed, use PBS to wash out the tissue. Place the embryos in a sterile 100 mm Petri dish containing a cold PBS (approximately 10 ml).
- Dissect out the brains from all embryos and carefully transfer to a 35 mm Petri dish containing 1 ml of HBBS. The dish should be kept on ice to ensure well-chilled medium.
- Very carefully dissect both hippocampi out of the brain by dissecting first the olfactory bulbs and the cerebellum and splitting then the hemispheres either. For each of the hemisphere, remove the thalamus and very gently pull the hippocampus out of the brain with the help of a spatula.
- Isolating NSPCs
- Take 0.5 ml of 0.25% trypsin-EDTA to a 15 ml Falcon tube and warm it in a CO2 incubator (35 °C, for 20-30 min).
- Enzymatic dissociation of hippocampal cells
Transfer the isolated hippocampi into a warm 0.25% trypsin-EDTA. Incubate in a CO2 incubator for 5-7 min. After incubation, stop the enzymatic digestion by adding Neurobasal medium in a volume of 1 ml (room temperature). Give a gentle flick to the tube containing the tissue to mix it up.
Note: Each hippocampus may be cut to several parts using a sterile scalpel in order to facilitate enzymatic digestion of the tissue. Also, we recommend shaking the tube over tissue incubation period (2-3 times each minute). - Mechanical dissociation of the cells
Pipette the hippocampal tissue up and down using the flame-polished glass Pasteur pipettes. Start to pipette the tissue using the pipette with the largest-sized internal tip diameter first, followed by the medium-sized one. Finally, pipette the dissociated cells using the pipette with the smallest-sized tip. All steps are performed at room temperature.
Note: Use each pipette for up and down pipetting not more than 10 times in order to increase the cell viability upon mechanical dissociation of the cells. Gently apply very small pressure to minimize cell damage while breaking cell aggregates. Avoid bubbles at any step of cell isolation. - Take 5 ml of Neurobasal medium and add to the obtained cell suspension, gently mixing while adding the medium.
- Let the mixture of isolated cells in Neurobasal medium go through a 40-µm cell strainer into a 50 ml centrifuge tube.
- Centrifuge the obtained suspension at 200 x g for 10 min (room temperature).
- Aspirate the supernatant, leaving the cell pellet intact. Add 10 ml of Neurobasal medium to the pellet and gently resuspend it.
- Centrifuge cells at 200 x g for 10 min, as before (Figure 2).
- Aspirate the supernatant, leaving the cell pellet intact. Add 1 ml of PBS to the pellet and gently re-suspend the cell pellet.
- Centrifugation in a density gradient
- Take 1 ml of the obtained cell suspension and transfer to the surface of 22% Percoll solution (total volume: 10 ml).
- Centrifuge the mixture at 450 x g for 10 min (room temperature). Remove the supernatant, leaving the cell pellet intact (Figure 2).
- Add 10 ml of Neurobasal medium to the pellet and gently re-suspend it.
- Centrifuge the obtained NSPCs at 200 x g for 10 min (room temperature). Remove the supernatant, add 1 ml of growth medium to the pellet and gently resuspend it.
Note: Use a 1 ml pipette for mixing and resuspending the cell suspension. This minimizes cell damage and increases the viability of isolated NSPCs.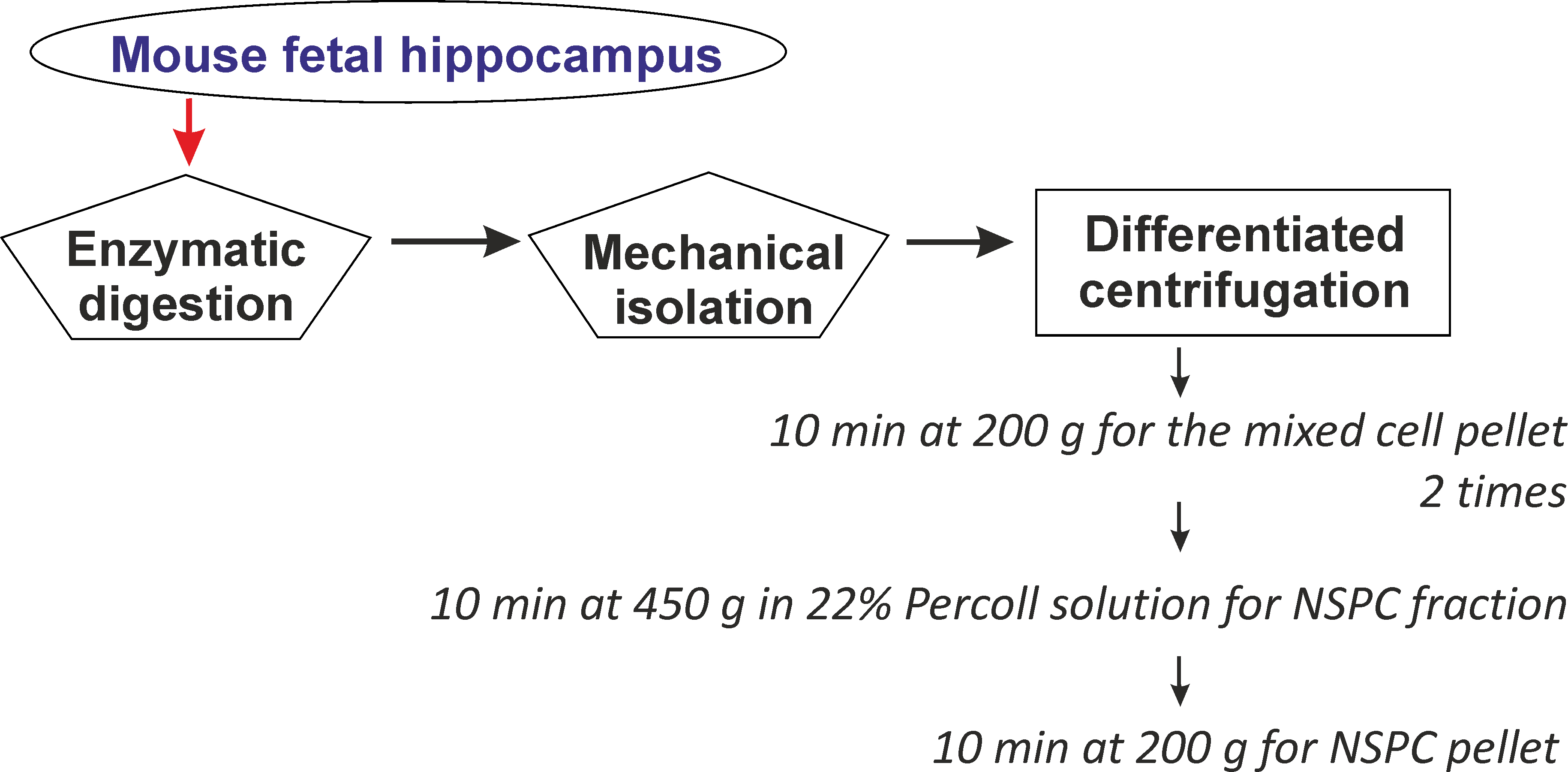
Figure 2. Schematic illustration of the main steps for NSPC isolation from the fetal mouse hippocampus through controlled enzymatic digestion and subsequent mechanical dissociation of the cells, followed by a number of centrifugation/resuspension steps
- Testing the viability of isolated stem cells
- Trypan blue staining
Once the suspension of NSPCs has been obtained, perform testing the cell viability using trypan blue. For this, take 20 µl of cell suspension and add 20 µl of 0.4% trypan blue solution to a droplet. Mix the sample well. Load the mixture into a glass hemocytometer very gently. Let the cells fill the chamber of hemocytometer by capillary action. Count the viable cells versus damaged cells (considering those brightly stained with trypan blue). It is advised that the proportion of viable cells shall to get over at least 75%.
Note: For the assessment of cell viability, another dye can be else used. We probed propidium iodide, a commonly used red-fluorescent DNA counterstain, to mark dead cells in a population. This approach requires equipment for detection of the propidium iodide-mediated fluorescence. - FACS-based analysis
Take a suspension of the obtained NSPCs (approximately 5 x 105 cells) and resuspend it in 100 µl of BD Cell Wash buffer. Transfer the mixture to a flow cytometer and add 7-AAD (5 µl). Mix carefully. Incubate the cells for 10 min at room temperature. After incubation, perform analysis (we use BD FACSDivaTM 6.1.2 software). Typically, viable cells count up to ~88% (see Figure 3). This confirms the high viability of embryonic NSPCs obtained from mouse hippocampi using the described protocol.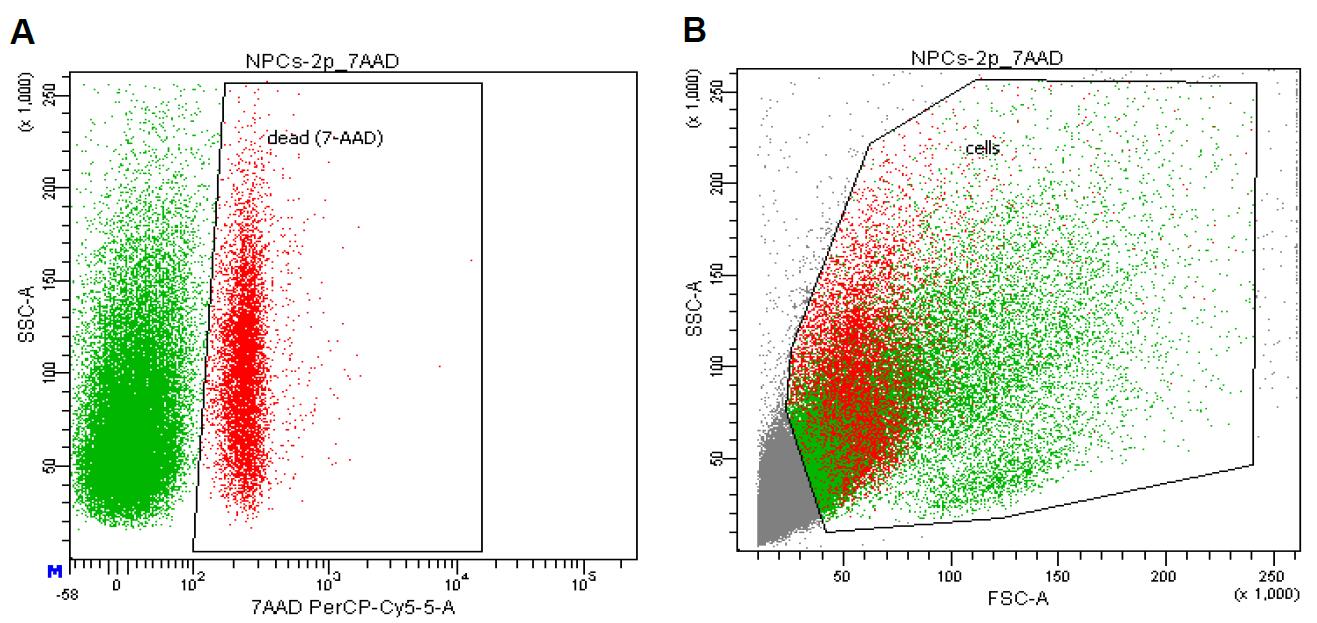
Figure 3. FACS-based analysis of embryonic NSPCs, freshly isolated from the fetal mouse hippocampi. A. Dot plot with gate of 7-AAD-positive non-viable (red) cells to show the proportion of dead cells after isolation. B. Distribution of viable (green) and non-viable (red) cells by morphology on the dot plot FSC vs. SSC. At least 5 x 104 cells were analyzed for each sample using BD FACSDivaTM software.
- Trypan blue staining
- Growing embryonic NSPCs in a 2D culture
- Take the obtained NSPC suspension (1 x 105 cells/ml) and add 250 µl of FGF-2 (20 ng/ml) to the cells. We seed the cells on the Matrigel-coated coverslips at a 24-well plate by diluting the mixture to get the total volume of 50 ml.
Note: The total volume depends on the desired cell density (number of coverslips) to grow. - To seed NSPCs in a 2D culture, take 0.5 ml of the mixture and transfer the cells onto the Matrigel-coated coverslips at a 24-well plate.
Note: For a 2D culture, seed NSPCs by distributing cells evenly across the coverslip surface, since cell aggregates dramatically influence growth and subsequent differentiation of NSPCs in a monolayer cell culture. - Place the plate in a CO2 incubator (35 °C, 5% CO2) and incubate for the next 24 h.
- Next day, check the cell survival using an inverted microscope. At this point, change the growth medium (0.5 ml/well) (see Recipes) by gently aspirating the entire medium from each well containing coverslips with the cells.
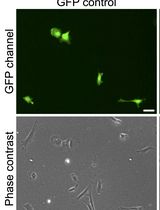
- Change the growth medium every two days. While performing the changes, visually assess cultures for cell growth. We present an example of embryonic stem cells shortly after plating in a 2D culture (see Figure 4A). Undifferentiated cells can be confirmed by immunostaining for nestin, a specific marker of undifferentiated neural stem cells, showing typically up to 97% nestin-positive staining in 2D cultures (see Figure 4B).
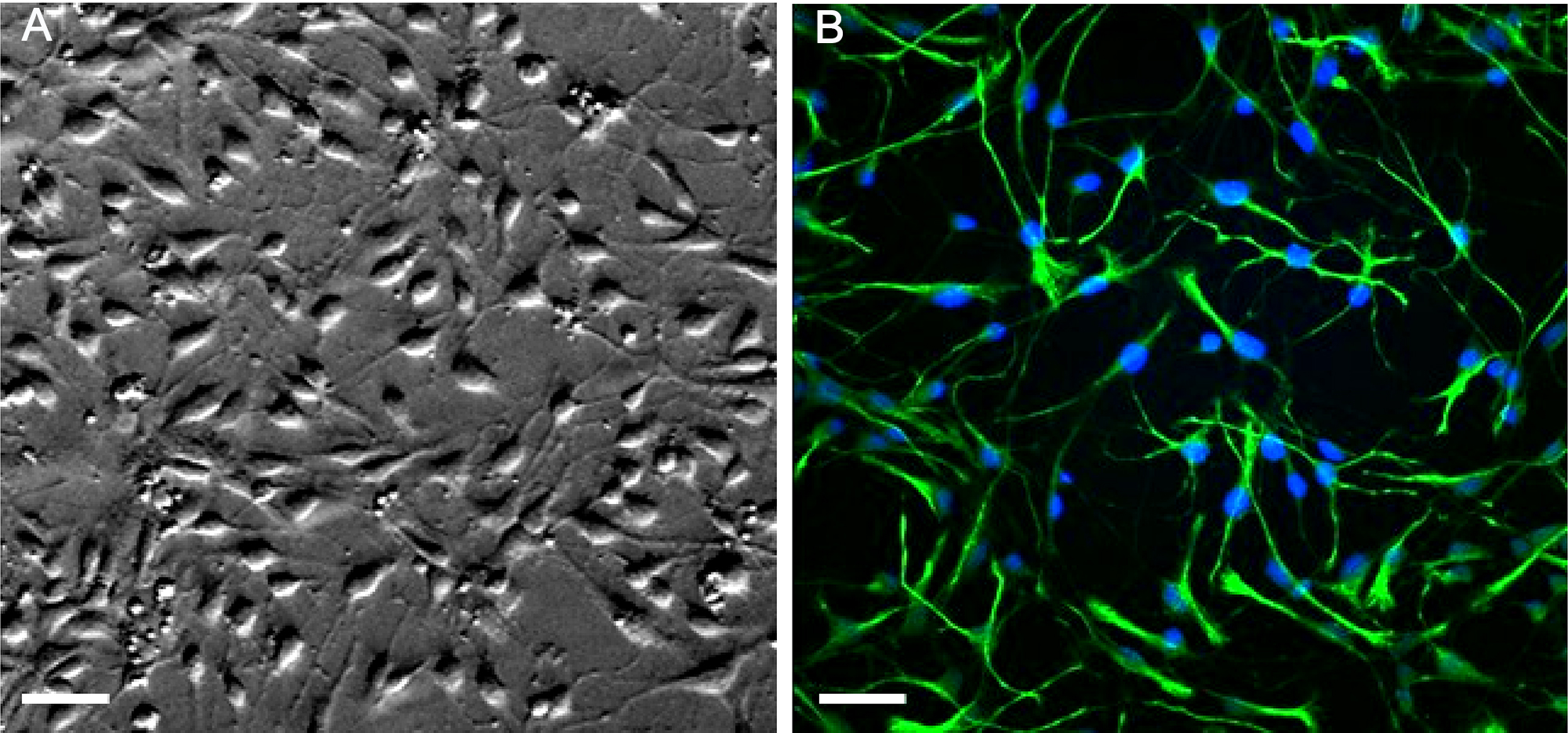
Figure 4. Example images of embryonic NSPCs in a 2D culture. A. Transmitted light image of undifferentiated NSPCs isolated from the fetal mouse hippocampi shortly after plating in an adherent 2D culture (snapshot taken on Day 1 in vitro). B. Confocal image of immunostained NSPCs for nestin (green) and Hoechst 33342 (blue) staining. Scale bars, 30 µm.
- Take the obtained NSPC suspension (1 x 105 cells/ml) and add 250 µl of FGF-2 (20 ng/ml) to the cells. We seed the cells on the Matrigel-coated coverslips at a 24-well plate by diluting the mixture to get the total volume of 50 ml.
- Passaging NSPCs in a 2D culture
- Passage the cells once NSPCs reach about 80% confluency.
- Aspirate all media from each well containing the cells at a 24-well plate. Gently wash the attached cells with a warm PBS (see Recipes).
- Take 200 µl Accutase (1x solution, thawed in advance at room temperature) and add it to the cells (each well of the plate). Incubate the cells with Accutase in a CO2 incubator (at 35 °C) for 5 min.
- Check under a microscope that cells are detached and then add 1 ml of growth medium to the cells (to dilute/deactivate Accutase).
Note: If cells are not detached after 5 min of incubation with Accutase, return the 24-well plate into a CO2 incubator (up to 5 min), by checking every minute or two if the cells have detached. - Collect the detached cells to a 1.5 ml Eppendorf tube and centrifuge the suspension at 200 x g for 10 min (room temperature). Discard the supernatant and re-suspend the pellet in 1 ml of growth medium.
Note: Perform a check of the NSPC viability for each of the obtained passage (we use trypan blue staining, but another approach can also be used). Also, estimate the density of the obtained suspension before seeding the cells. If the density is higher than 1 x 105 cells/ml, dilute the suspension accordingly (typically, 1:2). - Take 0.5 ml of the obtained suspension and transfer the cells onto each of the Matrigel-coated coverslips in a 24-well plate for seeding the next passage of NSPCs.
Note: We recommend seeding NSPCs in a 2D culture in the density at least 3 x 104 cells per coverslip, as we have established experimentally.
- Generating 3D neuro-spheroids by embryonic NSPCs
- Formation of the adherent neuro-spheroids (neurospheres)
For the formation of 3D neurospheres take a suspension of NSPCs (8 x 104 cells per coverslip) and resuspend the cells in the culture medium (without supplementing FGF-2). As we have observed routinely, removing FGF-2 from the culture medium boosts spontaneous differentiation of embryonic NSPCs and facilitates the formation of the adherent neuro-spheroids. - Transfer cells onto the Matrigel-coated coverslips at a 24-well plate in a volume of 0.5 ml per coverslip. Place the plate in a CO2 incubator (35 °C, 5% CO2).
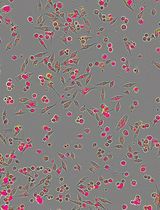
Note: For the formation of adherent neuro-spheroids, coverslips need to be coated with Matrigel. There is no requirement to coat coverslips if experimental design includes formation of floating neuro-spheroids. - Neurospheres will become forming within first 24-48 h after plating the cells. There is no need to change the culture medium during that period of time in order to not disrupt the formations. Neurospheres progressively grow over the time in a 3D culture. After 6-7 days in a 3D culture, embryonic NSPCs generate stable neuro-spheroids, which can develop up to 150-200 µm (see Figure 5).

Figure 5. Typical neuro-spheroids generated by embryonic mouse NSPCs after 5 days in an adherent 3D culture. Scale bar: 50 µm.
Note: We recommend not growing embryonic NSPCs in a 3D culture for over 9-10 days since 3D neurospheres facilitate spontaneous cell differentiation. At that time-point, 3D neuro-spheroids can reach above 300 µm that could promote cell death within the formation core. Therefore, it is strongly advised passaging NSPCs before 3D neuro-spheroids reach overgrowth. - Formation of the adherent neuro-spheroids (neurospheres)
- Immunostaining of NSPCs in 2D or 3D cultures
- Aspirate all the media from 2D or 3D cultures of embryonic NSPCs. Wash the cells with PBS (pre-warmed to room temperature) to remove dead cells and any debris.
- Add 500 µl of 4% PFA in 0.1 M phosphate buffer to each well containing the cells. Incubate for 30 min at room temperature.
- Wash with PBS at least 2 times.
Note: We recommend using ~0.5 ml of the buffer to ensure sufficient washing. At this step, cells may be stored until proceeding with immunostaining (up to a week time). In such a case, seal the plate containing coverslips with the cells hermetically with parafilm to avoid drying and keep it at 4 °C. - For blocking unspecific staining incubate the cells (or neuro-spheroids) in blocking solution containing 0.3% Triton X-100 and 0.5% BSA (see Recipes) for 1.5 h at room temperature.
- Incubate with primary antibodies in antibody solution containing 0.3% Triton X-100 and 0.5% BSA (see Recipes) for overnight at 4 °C. We use 200 µl of antibody solution per well.
Note: Shaking the plate in a slow rate is useful (but not required) during incubation in blocking solution or with antibodies for facilitating the treatment. - Wash the cells/neurospheres 3 times in PBS, each for 15 min.
- Incubate with secondary antibody(ies) in antibody solution containing 0.5% BSA (without Triton X-100) for 1 h at room temperature in the dark. We use 200 µl of the solution per well.
- Wash 3 times in PBS for 15 min.
- Incubate with Hoechst 33342 (1:5,000 in PBS) for 3-5 min at room temperature in the dark.
- Wash 3 times in PBS. Then wash in ddH2O to rinse out all salts. The cells/spheroids are ready for imaging.
- Imaging
We carry out imaging of the immunostained cells on a glass slide. For this, mount the coverslips containing a 2D culture (or 3D neuro-spheroids) onto the SuperFrost® glass slides, using a mounting medium (ImmunoHistoMount). Then let the slides air-dry for overnight at room temperature. Keep the samples at 4 °C. With such an approach, the immunostained cell preparations can be stored as long as needed. We present the example images for immunostaining of i) the adherent neuro-spheroids generated by embryonic NSPCs for both nestin and β-tubulin III staining (see Figure 6) and ii) an adherent monolayer of embryonic NSPCs for both nestin and Hoechst staining in a 2D culture (see Figure 4B). Staining for other proteins can also be performed using this method.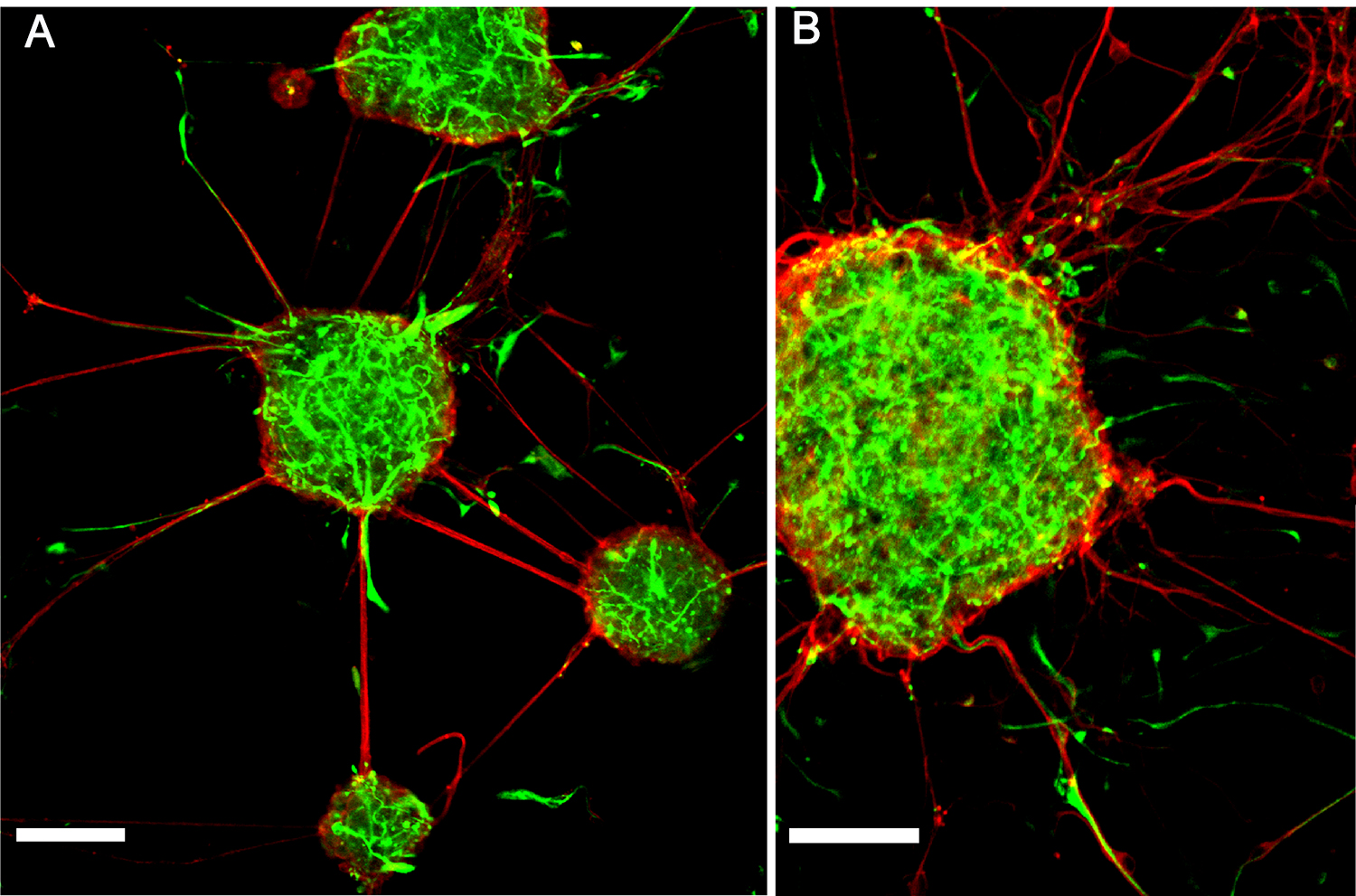
Figure 6. Immunofluorescent staining of neuro-spheroids generated from mouse embryonic NSPCs in a 3D culture. A and B. Confocal images of neuro-spheroids generated by embryonic NSPCs merged for nestin (green) and β-tubulin III (red) staining on Day 5 in a 3D culture. Scale bars, 50 µm (A) and 40 µm (B).
- Engraftment of mouse embryonic NSPCs onto organotypic hippocampal tissue
- Preparation of the hippocampal slices
Sacrifice pups of FVB mice at P8-P9 according to the procedures approved by your Institute. Decapitate the animals, dissect out the brains and remove both hippocampi into a cold dissecting medium (see Recipes), as we have described above and previously (Rybachuk et al., 2017). Place each hippocampus on a tissue chopper disc, mounted on a tissue chopper (McIlwain), and initiate chopping to cut the tissue to the transverse slices of 350-μm thick. Flash hippocampal slices into a Petri dish containing a cold dissecting medium and turn slices over to make their medial side faces up. Collect only hippocampal slices those have a well-recognized hippocampal-like morphology.
Note: For ‘good’ slicing the tissue, we recommend to put a filter paper above the chopper disc and place the hippocampus on it to avoid slipping and wobbling the tissue by the blade. Also, remove excess medium around the tissue before initiating chopping. - Organotypic hippocampal tissue cultures
Transfer slices using a glass Pasteur pipette with the flame-polished large-sized tip (inner diameter of near 3 mm) onto a surface of 0.4-μm membrane inserts placed at a 6-well plate. We typically plate four slices on a 0.4-μm membrane insert. Remove the spare medium using a glass Pasteur pipette of a small-sized tip (inner diameter of around 0.8 mm). Add 1 ml of culture medium (see Recipes) to each well containing slices on membrane insert. Place the plate in a CO2 incubator (35°C, 5% CO2). Change the culture medium on Day 2 after plating. Afterward, change the media every two days.
Note: A number of membrane inserts (wells at a tissue culture plate) depend on the amount of slices cut ‘good’ and may be adjusted from a 6-well plate to a 24-well plate. - Engraftment of embryonic NSPCs
Take 50 µl of a suspension of freshly isolated NSPCs and place the cells onto the surface of organotypic hippocampal tissue using a standard pipette (Eppendorf, Research® Plus). We grafted embryonic NSPCs onto hippocampal tissue at 7 days post-plating, but any time-point can be used. Any timing needs to be adjusted accordingly to experimental design. Transfer NSPCs by very carefully distributing the cells evenly across the tissue surface, not forming cell aggregates. Also, make sure that NSPCs settle exclusively on the tissue surface. Do not move the plate sharply to prevent the freshly engrafted cells drop down on membrane inserts. At Day 2 after engraftment, change the culture medium to wash out NSPCs not attached to a host tissue.
Note: We recommend engraftment of NSPCs at the density of 0.25 x 105 cells per slice. NSPCs from 2D or 3D cultures may also be engrafted after careful dissociation of the cells to ensure homogeneous distribution across the surface of a host tissue. - Growth of embryonic stem cells within a host organotypic tissue.
Maintain organotypic hippocampal tissue with NSPC grafts in a CO2 incubator (35 °C, 5% CO2) until use. Change the culture medium every two days.
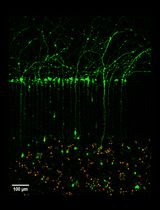
Note: We recommend changing the culture medium by aspirating it under membrane inserts. - Check the growth of differentiating NSPCs within a host tissue. This can be performed at any time-point due to simple tracing of the GFP-labeled NSPCs within the tissue. After 3 days, the grafted NSPCs began to incorporate into a host tissue (see Figure 7). Immunostaining in organotypic hippocampal tissue is performed similarly to the procedure described for cell cultures above and in our previous works (Rybachuk et al., 2017; Kopach et al., 2018).
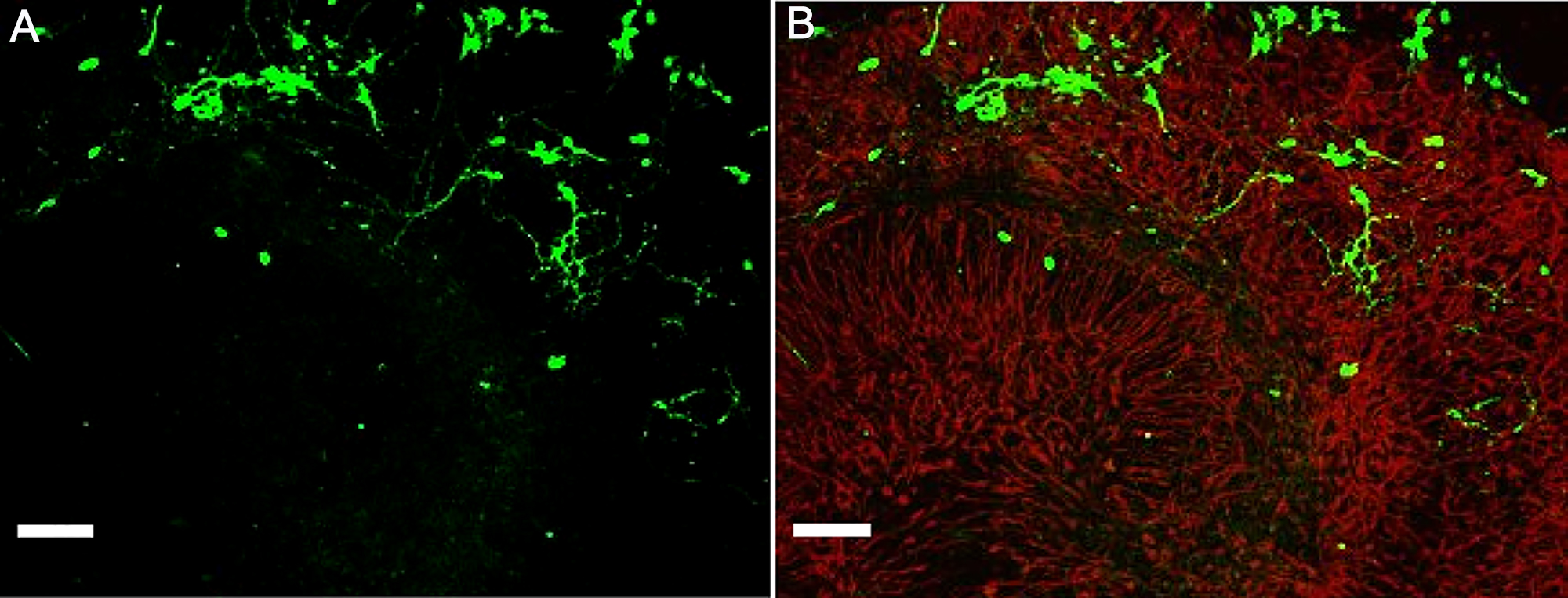
Figure 7. Example of embryonic mouse NSPCs grafted onto a host hippocampal tissue. A and B. Confocal images of immunostained organotypic hippocampal tissue with GFP-labeled NSPCs (green, A) and merge image of GFP-labeled NSPCs (green) and GFAP (red) staining (B) on Day 3 after engraftment. Scale bars, 100 µm.
- Preparation of the hippocampal slices
Data analysis
The obtained suspension of embryonic NSPCs can be used to grow and/or differentiation of the cells in vitro or within endogenous environment of a host tissue.
- In a 2D culture, the growth of the cells can be monitored visually and confirmed through immunostaining for nestin (red) and Hoechst 33342 (blue), as shown in Figure 4. See also Kopach et al. (2018), Figure 1B, for immunostaining images of NSPC fraction at 1 d in vitro and also Figure 6A for statistical analysis of the proportion of nestin-positive NSPCs (undifferentiated progenitors) in different experimental conditions.
- In 3D cultures, NSPCs can follow a robust multi-lineage differentiation. NSPCs isolated from the fetal mouse hippocampi can generate neuro-spheroids, the formation that enhances cell growth and differentiation and facilitates maintaining the high viability of differentiating progenitors. For the assessment whether neurons or glia are generated from embryonic NSPCs in a 3D culture, use of the cell type-specific markers is required. For visualization of phenotypic distribution within neuro-spheroids, we carried out immunostaining of differentiating progenitors for astrocytic marker GFAP together with neuronal marker NeuN and oligodendrocyte-specific marker olig-2. The example shown are images of phenotypic differentiation of embryonic NSPCs taken on Day 5 of 3D culture (see Figure 8).
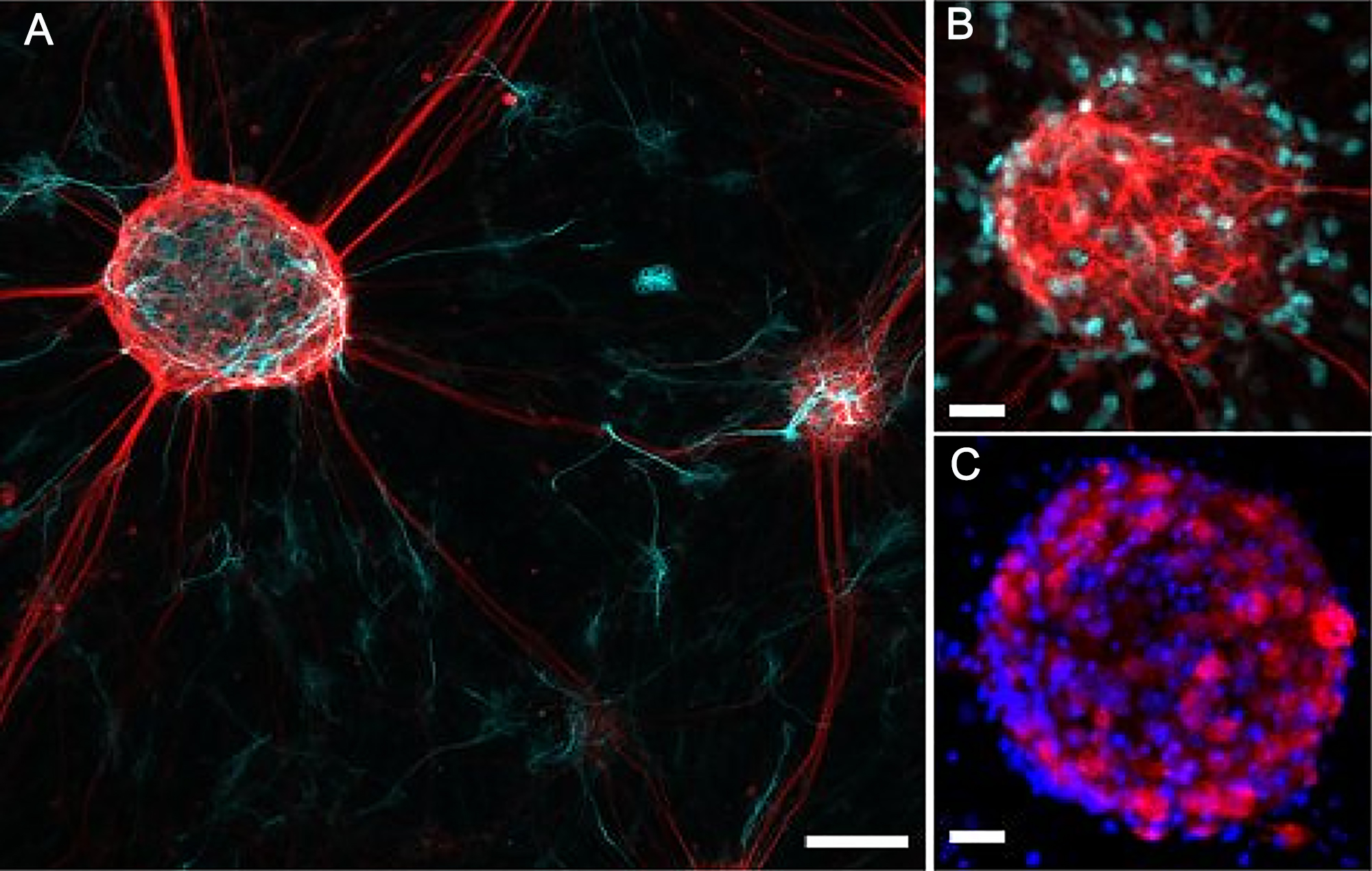
Figure 8. Immunofluorescent staining of 3D neuro-spheroids generated by embryonic mouse NSPCs. A. Confocal image of immunostained neuro-spheroids of embryonic hippocampal NSPCs for GFAP (cyan) and β-tubulin III (red) staining on Day 5 of 3D culture. B. Confocal image of neuro-spheroid generated by embryonic NSPCs for GFAP (red) and olig-2 (cyan) staining. C. Confocal image of the NSPC-generating neuro-spheroid for Hoechst 33342 (blue) and NeuN (red) staining. Scale bars, 50 µm (A) and 20 µm (B and C). - In a host brain tissue, organotypic hippocampal slices where both morphological layer architecture and signaling pathway assemblies remained preserved for the required period of time (weeks of tissue maintenance), embryonic NSPCs displayed a prompt multi-lineage neurogenesis. Using organotypic hippocampal tissue, we enabled to trace the time-dependent maturation of NSPC-derived hippocampal neurons through monitoring neuronal excitability with electrophysiological approaches. In particular, we recorded the passive membrane properties from differentiating progenitors [see Kopach et al. (2018), Figure 1], neuronal firing [see Kopach et al. (2018), Figure 3] and synaptic events spontaneously evoked in NSPC-derived neurons [see Kopach et al. (2018), Figure 2]. Organotypic hippocampal tissue represents a useful tool to feasibly assess maturation of neurophysiological properties of differentiating NSPCs within endogenous host environment at various time-points after engraftment. Moreover, it is implementable to carry out the direct quantitative comparisons between the functional properties of maturing progenitors versus endogenous principal neurons. Glial lineage of differentiating NSPCs can be further confirmed using immunostaining for the cell type-specific markers (see the description of the protocol in Kopach et al. [2018] and Figure 6F, for images of NSPC-derived oligodendrocytes or astroglia at 2 or 3 weeks post-grafting).
Recipes
- 0.2 M Phosphate buffer (total volume 250 ml; pH 7.4)
- Take 1.56 g sodium dihydrogen phosphate (NaH2PO4) and add 50 ml ddH2O
- Take 5.68 g disodium phosphate (Na2HPO4) and add 200 ml ddH2O
- Mix 200 ml diluted Na2HPO4 with 50 ml diluted NaH2PO4
- Store 0.2 M phosphate buffer (pH 7.4) at 4 °C
- 4% Paraformaldehyde (PFA) in 0.1 M phosphate buffer (total volume 100 ml)
- Take 4 g PFA and add it into 50 ml ddH2O while stirring at 60 °C
- To facilitate dilution of PFA in ddH2O, add 20 μl NaOH to the mixture while stirring
- Stir the mixture for approximately 30 min
- Add 50 ml of 0.2 M phosphate buffer to the mixture
- Filter the mixture using a 0.45-μm filter
- Store 4% PFA at -20 °C if not use it over the next days
- Growth medium (total volume 50 ml)
- Take 48.5 ml Neurobasal medium and add it to a 50 ml Falcon tube
- Add 1 ml B-27® (50x)
- Add 0.5 ml GlutaMAXTM (100x)
- Add 50 µl N-acetyl-L-cysteine (NAC) and 0.25 ml P/S
- Store the medium at 4 °C if not use immediately
- Add 20 ng/ml FGF-2 to the medium just before use it
- Tissue dissecting medium (total volume 100 ml; pH 7.3)
- Take 50 ml MEM and add it to a Falcon tube
- Add 25 ml HBSS
- Add 60 µg Tris to get its final concentration of 5 mM
- Add 17.5 µg NaHCO3 (final concentration 2 mM)
- Add 1.26 ml HEPES (final concentration 12.5 mM)
- Add 276.5 µg glucose (final concentration 15 mM)
- Add 1 ml P/S
- Dilute in ddH2O to get the total volume of 100 ml
- Filter the prepared medium using a 0.45-µm sterile filter
- Store the medium at 4 °C (or freeze if not use it over the next days)
- Tissue culture medium (total volume 100 ml; pH 7.2)
- Take 50 ml MEM and add it to a Falcon tube
- Add 25 ml HBSS
- Add 30 µg Tris (final concentration 2.5 mM)
- Add 17.5 µg NaHCO3 (2 mM)
- Add 1.26 ml HEPES (12.5 mM)
- Add 276.5 µg glucose (15 mM)
- Add 1 ml P/S
- Filter the medium through a 0.45-µm sterile filter and store it at 4 °C (or freeze it if not use)
- Add 250 µl/ml horse serum to the medium right before using
- Add 20 µl/ml B-27® supplement (50x) to the medium before using
- Antibody solution
Dissolve BSA (0.5%) in PBS just before use it - Blocking solution
Dissolve BSA (0.5%) and Triton X-100 (0.3%) in PBS before use
Acknowledgments
The work was supported by grants from the National Academy of Sciences of Ukraine.
Competing interests
Authors declare that there are no conflicts of interest or competing interests.
Ethics
All procedures were used in accordance with the protocols approved by the Animal Care and Use Committee at Bogomoletz Institute of Physiology and State Institute of Genetic and Regenerative Medicine (Kyiv, Ukraine) and were within the European Commission Directive (86/609/EEC) guidelines.
References
- Behnan, J., Stangeland, B., Langella, T., Finocchiaro, G., Tringali, G., Meling, T. R. and Murrell, W. (2017). Identification and characterization of a new source of adult human neural progenitors. Cell Death Dis 8(8): e2991.
- Conti, L., Pollard, S. M., Gorba, T., Reitano, E., Toselli, M., Biella, G., Sun, Y., Sanzone, S., Ying, Q. L., Cattaneo, E. and Smith, A. (2005). Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol 3(9): e283.
- Ignatova, T. N., Kukekov, V. G., Laywell, E. D., Suslov, O. N., Vrionis, F. D. and Steindler, D. A. (2002). Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 39(3): 193-206.
- Kempermann, G., Gage, F. H., Aigner, L., Song, H., Curtis, M. A., Thuret, S., Kuhn, H. G., Jessberger, S., Frankland, P. W., Cameron, H. A., Gould, E., Hen, R., Abrous, D. N., Toni, N., Schinder, A. F., Zhao, X., Lucassen, P. J. and Frisen, J. (2018). Human adult neurogenesis: evidence and remaining questions. Cell Stem Cell 23(1): 25-30.
- Klein, C., Butt, S. J., Machold, R. P., Johnson, J. E. and Fishell, G. (2005). Cerebellum- and forebrain-derived stem cells possess intrinsic regional character. Development 132(20): 4497-4508.
- Kopach, O. and Pivneva, T. (2018). Cell-based therapies for neural replacement strategies in stroke-related neurodegeneration: neurophysiological insights into stem progenitor cell neurogenesis within a host environment. Neural Regen Res 13(8): 1350-1351.
- Kopach, O., Rybachuk, O., Krotov, V., Kyryk, V., Voitenko, N. and Pivneva, T. (2018). Maturation of neural stem cells and integration into hippocampal circuits - a functional study in an in situ model of cerebral ischemia. J Cell Sci 131(4) pii: jcs210989.
- Makri, G., Lavdas, A. A., Katsimpardi, L., Charneau, P., Thomaidou, D. and Matsas, R. (2010). Transplantation of embryonic neural stem/precursor cells overexpressing BM88/Cend1 enhances the generation of neuronal cells in the injured mouse cortex. Stem Cells 28(1): 127-139.
- Pagano, S. F., Impagnatiello, F., Girelli, M., Cova, L., Grioni, E., Onofri, M., Cavallaro, M., Etteri, S., Vitello, F., Giombini, S., Solero, C. L. and Parati, E. A. (2000). Isolation and characterization of neural stem cells from the adult human olfactory bulb. Stem Cells 18(4): 295-300.
- Pollard, S. M., Conti, L., Sun, Y., Goffredo, D. and Smith, A. (2006). Adherent neural stem (NS) cells from fetal and adult forebrain. Cereb Cortex 16 Suppl 1: i112-120.
- Roy, N. S., Wang, S., Jiang, L., Kang, J., Benraiss, A., Harrison-Restelli, C., Fraser, R. A., Couldwell, W. T., Kawaguchi, A., Okano, H., Nedergaard, M. and Goldman, S. A. (2000). In vitro neurogenesis by progenitor cells isolated from the adult human hippocampus. Nat Med 6(3): 271-277.
- Rybachuk, O., Kopach, O., Krotov, V., Voitenko, N. and Pivneva, T. (2017). Optimized model of cerebral ischemia in situ for the long-lasting assessment of hippocampal cell death. Front Neurosci 11: 388.
- Suslov, O. N., Kukekov, V. G., Ignatova, T. N. and Steindler, D. A. (2002). Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc Natl Acad Sci U S A 99(22): 14506-14511.
- Tsupykov, O., Kyryk, V., Smozhanik, E., Rybachuk, O., Butenko, G., Pivneva, T. and Skibo, G. (2014). Long-term fate of grafted hippocampal neural progenitor cells following ischemic injury. J Neurosci Res 92(8): 964-974.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Rybachuk, O., Kopach, O., Pivneva, T. and Kyryk, V. (2019). Isolation of Neural Stem Cells from the Embryonic Mouse Hippocampus for in vitro Growth or Engraftment into a Host Tissue. Bio-protocol 9(4): e3165. DOI: 10.21769/BioProtoc.3165.
Category
Stem Cell > Embryonic stem cell > Maintenance and differentiation
Neuroscience > Cellular mechanisms > Tissue isolation and culture
Cell Biology > Cell viability > Cell death
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.