- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Imaging Higher-order Chromatin Structures in Single Cells Using Stochastic Optical Reconstruction Microscopy
Published: Vol 9, Iss 3, Feb 5, 2019 DOI: 10.21769/BioProtoc.3160 Views: 7606
Reviewed by: Zinan ZhouMichael Nguyen TrinhVishal S Parekh
Original research article
The authors used this protocol in:

Jul 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
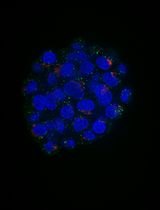

Abstract
Higher-order chromatin organization shaped by epigenetic modifications influence the chromatin environment and subsequently regulate gene expression. Direct visualization of the higher-order chromatin structure at their epigenomic states is of great importance for understanding chromatin compaction and its subsequent effect on gene expression and various cellular processes. With the recent advances in super-resolution microscopy, the higher-order chromatin structure can now be directly visualized in situ down to the scale of ~30 nm. This protocol provides detailed description of super-resolution imaging of higher-order chromatin structure using stochastic optical reconstruction microscopy (STORM). We discussed fluorescence staining methods of DNA and histone proteins and crucial technical factors to obtain high-quality super-resolution images.
Keywords: Higher-order chromatinBackground
Recent advances in super-resolution imaging technique provide new potentials to observe the biological structures at the molecular scale. In particular, single-molecule localization based super-resolution technique such as stochastic optical reconstruction microscopy (STORM) has become a valuable tool to directly observe higher-order chromatin structure in situ down to ~20-30 nm resolution (Xu et al., 2018). Although many detailed protocols have been devoted for STORM imaging in general, few have been focused on imaging higher-order chromatin structures. Chromatin has densely packed structure, and the nucleus of mammalian cells tends to be thicker than other membrane-based proteins. Such samples tend to give higher background and present more overlapping fluorescent emitters. Therefore, the proper optimization of nuclear staining for STORM imaging is critical to obtain high-quality super-resolution images of chromatin structure. A compromise in any of staining steps may lead to significant image artifacts and degradation in image resolution. Here we provide detailed protocols for STORM-based super-resolution imaging of higher-order chromatin structure marked by either DNA and histone proteins. We also provide detailed procedures on the conjugation of the fluorophores with secondary antibodies, for single-color and two-color STORM imaging. The protocol presented here is optimized for STORM imaging, we believe that this protocol can also be extended to most high-resolution fluorescence imaging of chromatin structures or other proteins of interest.
Materials and Reagents
- Microcentrifuge tube
- Glass-bottom dish (World Precision Instruments, catalog number: FD3510)
- NAP-5 size-exclusion columns (GE Healthcare, catalog number: 17-0853-02)
- MCF-10A cells
- DMEM/F12 (Invitrogen, catalog number: 11039-021)
- Horse serum (Invitrogen, catalog number: 16050-122)
- Pen/Strep (100x solution) (Invitrogen, catalog number: 15070-063)
- EGF (Peprotech, 1 mg) (Resuspend at 100 μg/ml in sterile ddH2O. Store aliquots at -20 °C)
- Hydrocortisone (Sigma-Aldrich, catalog number: H0888) (Resuspend at 1 mg/ml in 200-proof ethanol and store aliquots at -20 °C)
- Cholera toxin (Sigma-Aldrich, catalog number: C-8052) (Resuspend at 1 mg/ml in sterile ddH2O and allow to reconstitute for about 10 min. Store aliquots at 4 °C)
- Insulin (Sigma-Aldrich, catalog number: I-1882) (Resuspend at 10 mg/ml in sterile ddH2O containing 1% glacial acetic acid. Shake solution and allow 10-15 min to reconstitute. Store aliquots at -20 °C)
- FluoSpheresTM Carboxylate-Modified Microspheres, 0.1 μm, yellow-green fluorescent (505/515) (Thermo Fisher Scientific, catalog number: F8803)
- TetraSpeckTM Microspheres, 0.1 μm, fluorescent blue/green/orange/dark red (Thermo Fisher Scientific, catalog number: T7279)
- Phosphate buffered saline (PBS) (Lonza)
- Triton X-100 (Sigma-Aldrich, catalog number: T8787)
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A9647)
- Paraformaldehyde (Sigma-Aldrich, catalog number: P6148)
- Primary antibody
Rabbit anti H3K4me3 antibody (EMD Millipore, catalog number: 07-473)
Mouse anti H3K9ac antibody (Abcam, catalog number: ab12179) - Secondary antibodies
Donkey anti-rabbit antibody (Jackson ImmunoResearch, catalog number: 711-005-152)
Donkey anti-mouse antibody (Jackson ImmunoResearch, catalog number: 715-005-151) - Alexa 405 carboxylic acid succinimidyl ester (Thermo Fisher Scientific, catalog number: A30000)
- Alexa 647 carboxylic acid succinimidyl ester (Thermo Fisher Scientific, catalog number: A20006)
- Cy2 and Cy3B reactive dye (GE Healthcare, catalog numbers: PA22000, PA63101)
- Dimethylsulfoxide (DMSO; anhydrous)
- Click-iTTM EdU Alexa FluorTM 647 Imaging Kit (Thermo Fisher Scientific, catalog number: C10340)
- Glucose (Sigma-Aldrich, catalog number: G7021)
- Glucose oxidase from Aspergillus niger-Type VII (Sigma-Aldrich, catalog number: G2133)
- Catalase from bovine liver-lyophilized powder (Sigma-Aldrich, catalog number: C40)
- 1 M Tris, pH 8.0
- NaCl (Sigma-Aldrich, catalog number: S9888)
- 2-mercaptoethanol (β-ME) (Sigma-Aldrich, catalog number: 63698)
- Cysteamine (MEA) (Sigma-Aldrich, catalog number: 30070)
- NaHCO3 (Sigma-Aldrich, catalog number: S5761)
- Culture medium for MCF-10A (see Recipes)
- Washing buffer (see Recipes)
- Blocking buffer (see Recipes)
- STORM Imaging buffer (see Recipes)
Equipment
- Olympus IX71 inverted microscope frame with an oil-immersion objective (100x, NA = 1.4, UPLSAPO 100XO; Olympus, model: IX71)
- sCMOS camera (pco.edge 4.2, PCO-TECH)
Two-color dSTORM images were acquired on a custom system built upon an Olympus IX71 inverted microscope frame with an oil-immersion objective and an sCMOS camera attached on the side camera port. The 0.5x adaptor was used, such that each pixel on the camera corresponds to 130 nm on the sample plane (the pixel size of approximately 80-160 nm is acceptable). - Objective nanopositioner (Mad City Labs, model: Nano-F100S)
- N-STORM system (Nikon Instruments)
- NanoDrop 2000 microspectrophotometer (Thermo Fisher Scientific, model: NanoDropTM 2000)
- Rocking platform
Software
- ImageJ (National Institutes of Health, https://imagej.nih.gov/ij/download.html)
- ThunderSTORM plug in Ovesný et al. (2014) (downloadable at https://github.com/zitmen/thunderstorm)
- Labview (National Instrument)
Procedure
- Conjugation of photo-switchable fluorophores to secondary antibody
- Dissolve Alexa 405 (1.0 mg tube) and Alexa 647 (tube) in 100 μl DMSO, divide into 50 aliquots containing 0.02 mg for each. Dissolve Cy2 (1 pack) and Cy3B (1 pack) in 20 μl DMSO and divide into 10 aliquots. For long-term storage, lyophilize and store at -20 °C under dry conditions.
- Take one tube of each dye above (lyophilized powder), add 40 μl DMSO to Alexa 647 tube (0.5 μg/μl), add 10 μl DMSO to Alexa 405 (2 μg/μl), Cy2 and Cy3B tubes (10 μg/μl), dissolve properly.
- Prepare fresh 0.5 M NaHCO3 solution by dissolving 0.021 g NaHCO3 into 5 ml H2O.
- To conjugate secondary antibodies with single fluorophore Alexa 647 or Cy3B for dSTORM imaging, thoroughly mix 40 μl donkey anti-rabbit/mouse antibody, 10 μl of 0.5 M NaHCO3, and 2 μl Alexa 647 dilution from Step A2 and incubate for 30 min at room temperature, protected from light. Similarly, mix 40 μl donkey anti-rabbit/mouse antibody, 10 μl NaHCO3, and 1 μl Cy3B dilution from Step A2, and incubate under the same conditions.
- To conjugate secondary antibodies with dye pairs: Mix 40 μl donkey anti-rabbit/mouse antibody, 10 μl NaHCO3, 5 μl Cy2 dilution from Step A2, and 1 μl Alexa 647 dilution from Step A2, and incubate at room temperature for 30 min, protected from light. Similarly, mix 40 μl donkey anti-rabbit/mouse antibody, 10 μl NaHCO3, 5 μl Alexa 405 dilution from Step A2, and 1 μl Alexa 647 dilution from Step A2, and incubate under the same conditions.
- During the reaction, wash the NAP-5 size-exclusion columns (see Materials and Reagents) three times by 1,000 μl PBS. When the reaction is complete, add 150 μl PBS to bring the reaction volume to 200 μl.
- Add the entire reaction solution to the columns and allow the liquid to be completely absorbed into the column. Add 550 μl PBS to wash, then add another 300 μl PBS and collect the fluorophore-conjugated antibody in a microcentrifuge tube.
- Measure the absorbance of the fluorophore-conjugated secondary antibody using a NanoDrop 2000 Spectrophotometer to calculate the conjugation efficiency. Measure the absorbance of antibody at 280 nm, Alexa 405 at 401 nm, Cy2 at 489 nm, Cy3B at 559 nm, and Alexa 647 at 650 nm. Store the labeled antibody fraction at 4 °C. For long-term storage, aliquot and store at -20 °C.
- Two-color staining of DNA and histone modifications (see Figure 1. Procedure takes example of DNA and H3K4me3)
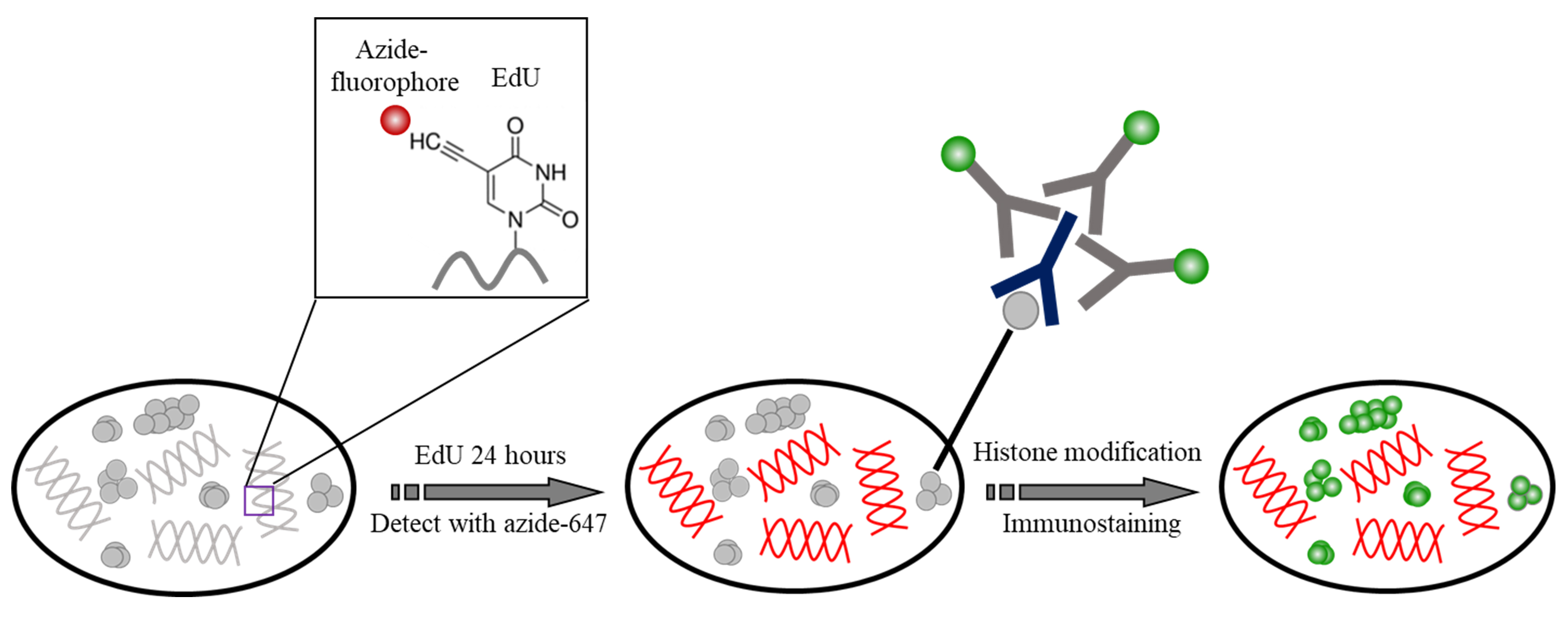
Figure 1. The schematic of the staining of DNA and histone modifications. DNA was incorporated with EdU and detected with Azide Alexa 647. Histone modification was then immunolabeled with Cy3B.- Plate MCF-10A cells on glass bottom dish at confluency 50%-70%, for the WPI (FD3510) dishes used in this protocol, place 200 μl cell suspension (~50 cells/μl) in the well. Incubate overnight.
- Add EdU to the cells at final concentration 1 μM, total volume 200 μl, incubate for 24 h.
Note: EdU concentration needs to be optimized for different samples or experimental conditions. We fully labeled DNA, by incubation throughout a complete cell cycle. For STORM imaging, we suggest using lower concentration (0.5-2 μM) compared to the manufacturer’s recommendation of 10 μM. Over-labeling may cause extremely high blinking density and high background at the STORM imaging condition, and result in poor image resolution and artifacts. - Fix cells with 4% PFA for 15 min, wash 3 times with PBS.
Note: Other commonly used alcohol fixative can also be used, such as methanol/ethanol (1:1) or methanol/acetone (1:1). Our test showed comparable higher-order chromatin structures compared to those from 4% PFA (Similar cluster size and density. Please refer to Xu et al., 2018). - Permeabilize with 0.2% Triton X-100 in PBS for 10 min, wash 3 times with PBS. This step is not required if alcohol fixative was used.
- Prepare Click-iT Plus reaction cocktails as follows. For a total volume of 500 μl, cocktails contain 440 μl 1x Click-iT reaction buffer, 10 μl copper protectant, 1.2 μl Alexa 647 picolyl azide, and 50 μl reaction buffer additive. All components were provided by the manufacturer’s imaging kits.
Note: Similar to EdU-based labeling, the concentration of Alexa 647 picolyl azide may also need adjustment to achieve the optimized labeling density for dSTORM imaging. - Add Click-iT Plus reaction cocktails to cells and incubate for 30 min, protect from light. Wash 3 times with washing buffer.
- Incubate cells with 150 μl rabbit anti-H3K4me3 primary antibody diluted in blocking buffer (1:600) at 4 °С overnight.
- Wash cells 3 times with washing buffer, 5 min per wash.
- Incubate with Cy3B conjugated donkey anti-rabbit secondary antibody diluted in blocking buffer for 2 h at room temperature, protected from light.
Note: Two-color dSTORM imaging needs simultaneous labeling of two fluorophores at two distinct wavelengths. Alexa 647 and Cy5 are often the first choice as they perform the best blinking properties in the red/near-infrared channel; for the second channel, based on our experience, Cy3B and CF568 perform better blinking behavior compared to other dyes with the similar wavelength range. - Wash cells 3 times with washing buffer, 5 min per wash.
- Postfix with 4% PFA for 10 min, wash with PBS.
- Deposit FluoSpheresTM Carboxylate-Modified Microspheres (Materials and Reagents #12) to the culture dish for drift correction. Dilute fluorescent beads at 1:500,000 in PBS, add 200 μl beads solution to the dish, deposit for 30 min, then switch to PBS. Samples are ready for imaging.
Note: Concentration and deposition time of the beads are adjustable, to ensure at least 5 fluorescent beads in the field of view for drift correction (Principles were discussed previously, Ma et al., [2017]).
- Two-color staining of different histone modifications (H3K9ac and H3K4me3)
- Plate MCF-10A cells on glass bottom dish at confluency 50%, incubate overnight.
- Fix cells with 4% PFA for 15 mi, wash 3 times with PBS.
- Permeabilize with 0.2% Triton X-100 for 10 min, wash 3 times with blocking buffer.
- Incubate cells with two primary antibodies (Mouse anti H3K9ac antibody, rabbit anti H3K4me3 antibody) simultaneously diluted in blocking buffer at 4 °С overnight.
Note: High efficiency and specificity of the primary antibody are critical for high-quality super-resolution imaging. We recommend testing ChIP grade or Knockout (KO) validated antibodies for high specificity (low background) and sufficient localization number (~ > 20 localizations per cluster), prior to biological experiments. - Wash cells 3 times with washing buffer, 5 min per wash.
- Incubate with secondary antibodies (dye pair Alexa 405-647 conjugated donkey anti-mouse secondary antibody, Cy2-Alexa 647 conjugated donkey anti-rabbit secondary antibody) diluted in blocking buffer for 2 h at room temperature; Protect from light.
- Wash cells 3 times with washing buffer, 5 min per wash.
- Postfix with 4% PFA for 10 min, wash with PBS.
- Deposit fluorescent beads to the culture dish for drift correction. Dilute fluorescent beads at 1:500,000, deposit for 30 min, then switch to PBS. The samples are ready for imaging.
Note: If a commercial N-STORM system is used (e.g., N-STORM, Nikon), it is equipped with the focus lock system for drift correction, this step may not be necessary.
- STORM imaging
- Prepare STORM imaging buffer freshly.
Note: For dSTORM imaging, β-ME based buffer is preferred; for two-color STORM using dye pairs, MEA based buffer is preferred. See Recipes section below. - Add STORM imaging buffer to the sample and put the sample on the microscope stage.
- Use lower laser power (~1 mW) to identify the imaging objects, adjust focal plane to find the clearest nuclear periphery.
- Increase the laser power to maximum (~2-10 kW•cm-2 power density) to turn “off” fluorescent molecules and trigger photo-switching (see Video 1).
Note: If the blinking is too dense (significant overlapping emitters and high background), bleach the sample for 30 sec-3 min before data acquisition. If after bleaching for several minutes, the blinking is still too dense, sample re-preparation with lower labeling density may be needed. See Figure 2. Video 1. Imaging set up and real-time data acquisition of STORM imaging
Video 1. Imaging set up and real-time data acquisition of STORM imaging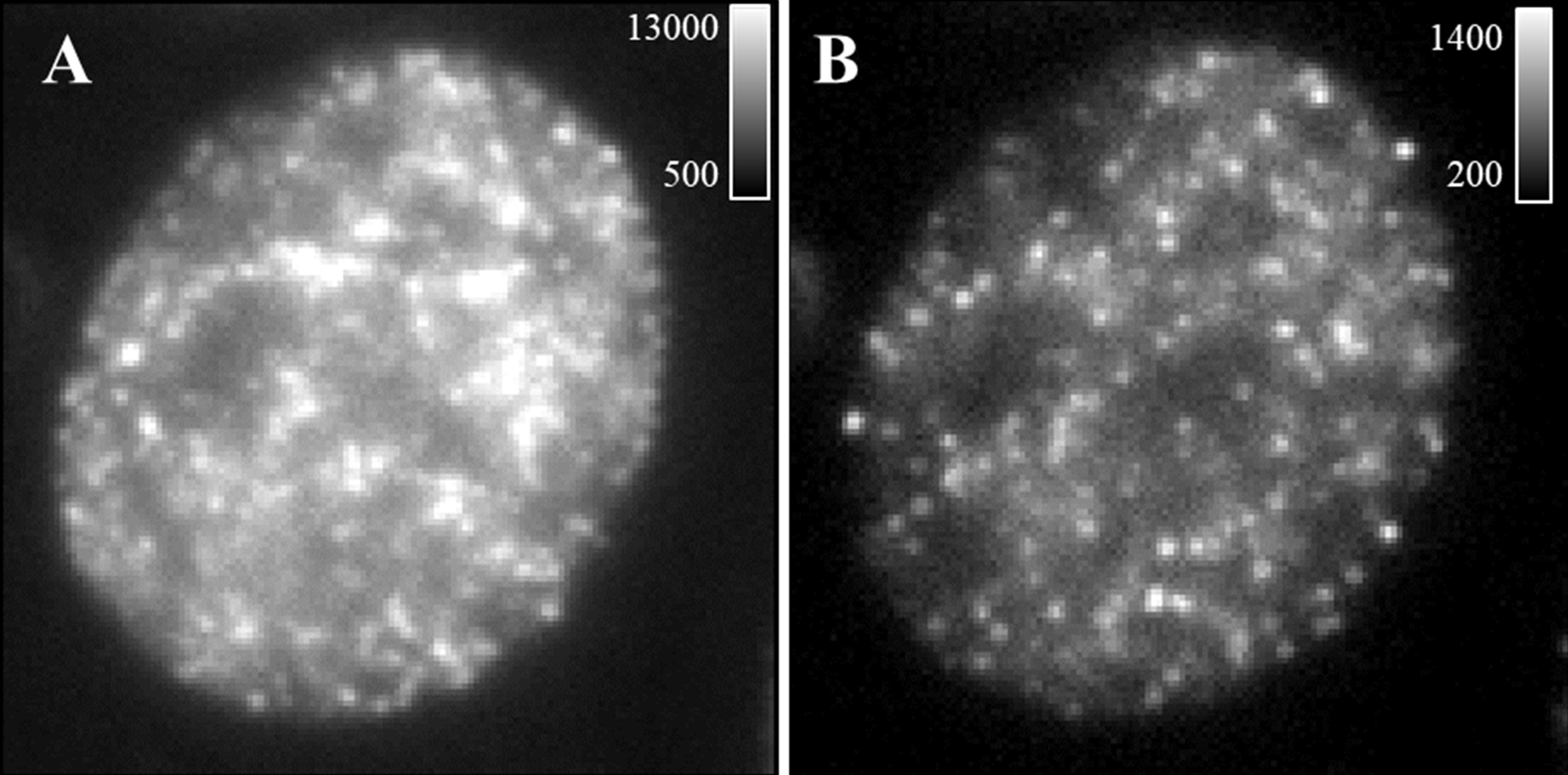
Figure 2. Representative single frame of raw data. A. An example of a single-frame image of the raw data with insufficient bleaching (dense emitters and high background). B. An example of a single frame of the raw data after bleaching for 2 min with acceptable emitter density and background. Calibration bar shows photon counts. - Data acquisition
- Two-color dSTORM imaging
Continuous illumination with 642 nm and 561 nm lasers is used in the two-color dSTORM imaging. The two channels are imaged sequentially at an exposure time of 20 ms for 30,000 frames using 642 nm excitation, followed by 30,000 frames using 561 nm excitation. One hundred nm yellow-green fluorescent beads are used as fiduciary markers on the coverslip to correct for 3D system drift every 200 frames as previously described (Ma et al., 2017; Xu et al., 2017). Low power (1 to 100 μW) of 405 nm can be added during data acquisition to facilitate the blinking if the “on” molecule in each frame becomes very sparse.
Notes:- For drift correction, at the beginning of the experiment, move the position of the objective nanopositioner to the focal plane where at least 4-5 fiducial markers (e.g., fluorescent beads) are visible; then record a set of 2D images of the fiducial markers at different axial positions around the focal planes set by the objective nanopositioner control software written in Labview; the initial position of fiducial markers is determined using the method described in our previous publication (Ma et al., 2017). During data acquisition, for every 200 frames (~4 s), the axial position of the objective was moved to the initial position of the fiducial markers (with excitation wavelength of 488 nm) determined at the beginning of the experiment via objective nanopositioner, and take a 2D image of the fiducial markers and the patterns of point spread function from the individual fiducial markers are used to track the axial position of the samples; and adjust the focal plane of the samples accordingly as described in detail in our previous publication (Ma et al., 2017). The entire process is controlled using the software written in Labview.
- If a commercial STORM system is used (e.g., N-STORM, Nikon), it should be equipped with the focus lock for drift correction, based on the reflection between coverslip and mounting media. If there is a large mismatch of the refractive index between the coverslip (n = 1.515) and the mounting media (if aqueous medium, n = ~1.34), it will work well.
- For two-color dSTORM imaging using Alexa 647 and Cy3B, it is preferred to image the Alexa 647 channel first, since the 561-nm laser will strongly bleach the Alexa 647 dye if Cy3B is imaged first.
- Two-color STORM imaging based on dye pairs
For two-color STORM imaging, 405-nm and 488-nm lasers are used as activation lasers, and 647-nm laser is used as imaging laser. The sample is periodically illuminated using a sequence of activation-imaging laser pulse cycles. In each cycle, one of the activation lasers is turned on for one frame (activation frames), followed by three continuous frames of 647-nm imaging laser (imaging frames), acquire ~10,000 cycles for each color (a total of 40,000 frames). The imaging frame immediately after the activation pulse (imaging frame 1) is recognized as a controlled activation event, and used for the image reconstruction.
Take an example of two-color STORM imaging using dye pairs Alexa 405-647 and Cy2-Alexa 647. Two activator lasers (405 nm and 488 nm) are pulsed on: when the 405-nm laser is pulsed on (at low power) to turn on the Alexa 405-647 channel, the imaging laser (647 nm) is turned on (at maximum power) for image acquisition in Color 1 channel; when the 488-nm laser is pulsed on (at low power) to turn on the Cy2-Alexa 647 channel, the 647-nm imaging laser is turned on for image acquisition in Color 2 channel.
Note: It is important to adjust the power of activator laser (405 nm and 488nm) to optimize the “on-off” switch and emitter density of the imaging frames. When the activator laser is on, appropriate density of blinking should be seen; when the activator laser is off, very few molecules should be at the “on” state. See Figure 3B.
- Two-color dSTORM imaging
- Prepare STORM imaging buffer freshly.
Data analysis
- Image reconstruction of dSTORM
A final STORM image is reconstructed from the accumulated positions by Gaussian fitting of single fluorescent emitters (“on” molecules) recorded at each frame. Image reconstruction software is generally provided by the commercial STORM system (e.g., N-STORM). A lot of open-source image reconstruction software is available (Henriques et al., 2010; Wolter et al., 2012; Min et al., 2014; Ovesný et al., 2014). We recommend ThunderSTORM (Ovesný et al., 2014). Briefly as follows:- Import the entire image sequence of raw data into ImageJ.
- Set camera parameters (e.g., pixel size, photoelectrons per A/D count and base level).
- Open “Run Analysis” in ThunderSTORM plugin and set corresponding parameters (e.g., Image filtering, localization methods) and the suggestions on how to set parameters have been described in detail in Xu et al. (2017).
- After all the parameters are set up, click “OK” to start the image reconstruction (see Video 2 using 100 frames as an example. The actual image reconstruction often needs 10,000-40,000 frames).
Video 2. STORM image reconstruction by ThunderSTORM ImageJ plugin - Image reconstruction of STORM imaging based on dye pairs
If dye pairs are used in STORM imaging, for each channel, only the imaging frames immediately after each activation pulse (imaging frame 1 in Figure 3B) are used for the image reconstruction following the same approach as dSTORM described above. The second and third frames (imaging frames 1 and 2 in Figure 3B) following each activation pulses were considered as non-specific activation signals, which are moved by a cross-talk subtraction algorithm described in Bates et al., 2007.
Note: For two-color STORM imaging using dye pairs, chromatic correction is not needed since the same imaging fluorophore was used for two color channels. As the correction for chromatic aberration is rarely perfect, in cases where the accurate localization of two targets is crucial to answering the biological question, dye-pair based two-color STORM free of chromatic aberration can be advantageous.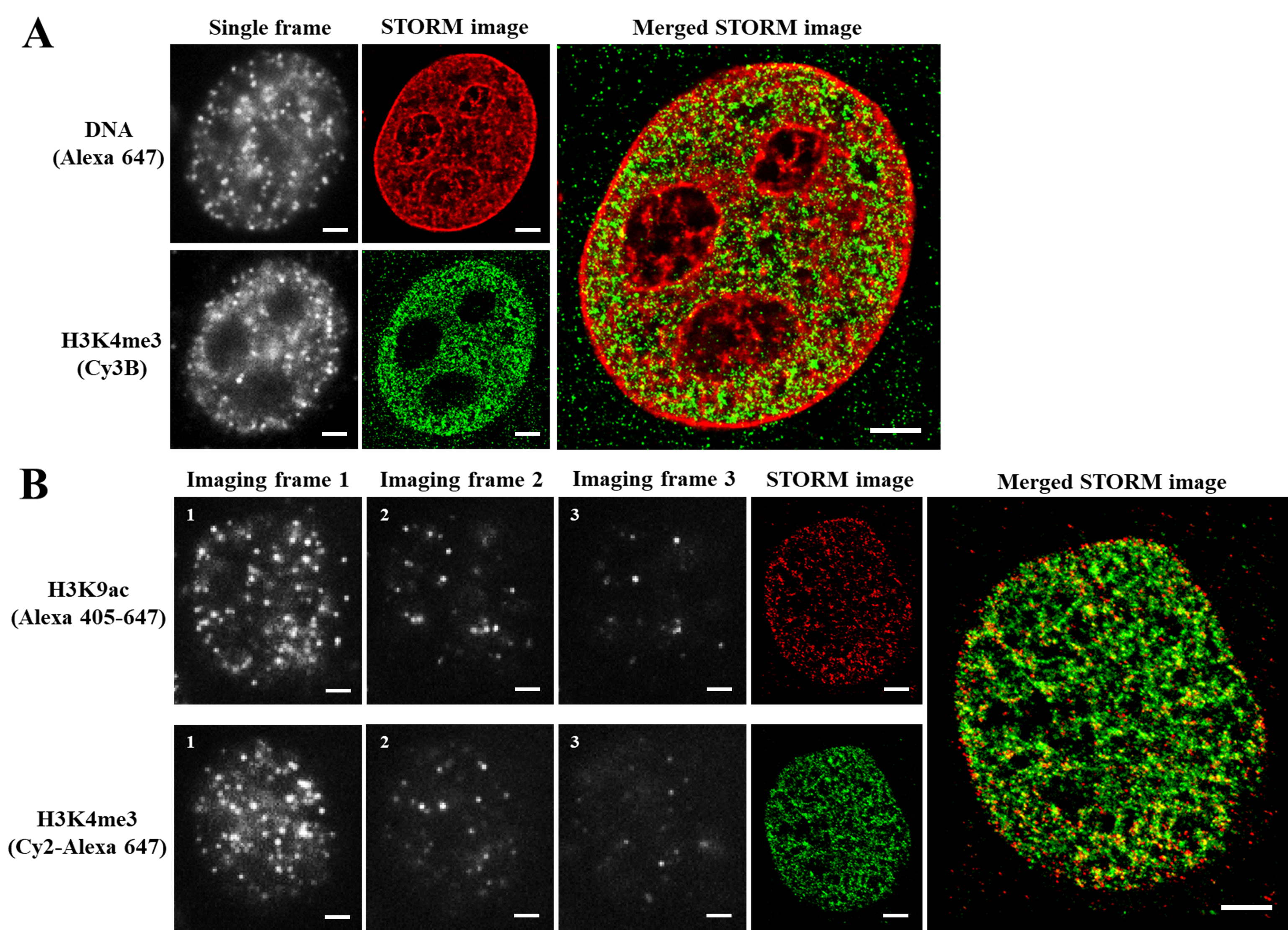
Figure 3. Representative two-color STORM imaging (raw data and STORM) based on dSTORM or dye pair. A. Representative raw images and two-color dSTORM images of DNA (EdU) and H3K4me3 labeled with Alexa 647 and Cy3B, respectively. B. Representative raw images (imaging frames 1-3 for one activation-imaging cycle) and two-color STORM images of H3K9ac and H3K4me3 based on dye-pairs. H3K9ac was labeled with Alexa 405-647 and H3K4me3 were labeled with Cy2-Alexa 647. Scale bar: 2 μm.
Recipes
- Culture medium for MCF-10A
DMEM/F12
5% horse serum
10 mg/ml insulin
20 ng/ml EGF
0.5 mg/ml hydrocortisone
100 ng/ml cholera toxin - Washing buffer
0.2% BSA + 0.05% Triton X-100 in PBS - Blocking buffer
3% BSA + 0.05% Trion X-100 in PBS - STORM Imaging buffer
10% (w/v) glucose
0.56 mg/ml glucose oxidase
0.17 mg/ml catalase
10 mM NaCl
50 mM Tris (pH 8.0)
For dSTORM imaging, use 0.14 M 2-mercaptoethanol; for two-color imaging with dye pair, use 0.1 M mercaptoethylamine (MEA)
Acknowledgments
We acknowledge the funding support from National Institute of Health Grant Number R01CA185363 and R33CA225494. Our protocols were adapted based on the previously published protocols (Bates et al., 2007 and 2013; van de Linde et al., 2011).
Competing interests
The authors declare no competing interests.
References
- Bates, M., Huang, B., Dempsey, G. T. and Zhuang, X. (2007). Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science 317(5845): 1749-1753.
- Bates, M., Jones, S. A. and Zhuang, X. (2013). Preparation of photoswitchable labeled antibodies for STORM imaging. Cold Spring Harb Protoc 2013(6): 540-541.
- Henriques, R., Lelek, M., Fornasiero, E. F., Valtorta, F., Zimmer, C. and Mhlanga, M. M. (2010). QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat Methods 7(5): 339-340.
- Ma, H., Xu, J., Jin, J., Huang, Y. and Liu, Y. (2017). A simple marker-assisted 3D nanometer drift correction method for superresolution microscopy. Biophys J 112(10): 2196-2208.
- Min, J., Vonesch, C., Kirshner, H., Carlini, L., Olivier, N., Holden, S., Manley, S., Ye, J. C. and Unser, M. (2014). FALCON: fast and unbiased reconstruction of high-density super-resolution microscopy data. Sci Rep 4: 4577.
- Ovesný, M., Křížek, P., Borkovec, J., Švindrych, Z. and Hagen, G. M. (2014). ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 30(16): 2389-2390.
- Sigal, Y. M., Speer, C. M., Babcock, H. P. and Zhuang, X. (2015). Mapping synaptic input fields of neurons with super-resolution imaging. Cell 163(2): 493-505.
- van de Linde, S., Loschberger, A., Klein, T., Heidbreder, M., Wolter, S., Heilemann, M. and Sauer, M. (2011). Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat Protoc 6(7): 991-1009.
- Wolter, S., Loschberger, A., Holm, T., Aufmkolk, S., Dabauvalle, M. C., van de Linde, S. and Sauer, M. (2012). rapidSTORM: accurate, fast open-source software for localization microscopy. Nat Methods 9(11): 1040-1041.
- Xu, J., Ma, H. and Liu, Y. (2017). Stochastic Optical Reconstruction Microscopy (STORM). Curr Protoc Cytom 81(1): 12.46.11-12.46.27.
- Xu, J., Ma, H., Jin, J., Uttam, S., Fu, R., Huang, Y. and Liu, Y. (2018). Super-resolution imaging of higher-order chromatin structures at different epigenomic states in single mammalian cells. Cell Rep 24(4): 873-882.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Xu, J. and Liu, Y. (2019). Imaging Higher-order Chromatin Structures in Single Cells Using Stochastic Optical Reconstruction Microscopy. Bio-protocol 9(3): e3160. DOI: 10.21769/BioProtoc.3160.
Category
Cell Biology > Cell imaging > Fluorescence
Molecular Biology > DNA > DNA structure
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.