- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Measuring Homologous Recombination Rates between Chromosomal Locations in Salmonella

Published: Vol 9, Iss 3, Feb 5, 2019 DOI: 10.21769/BioProtoc.3159 Views: 7587
Reviewed by: Valentine V TrotterSneha JaniAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Homologous recombination between two similar DNA molecules, plays an important role in the repair of double-stranded DNA breaks. Recombination can occur between two sister chromosomes, or between two locations of similar sequence identity within the same chromosome. The assay described here is designed to measure the rate of homologous recombination between two locations with sequence similarity within the same bacterial chromosome. For this purpose, a selectable/counter-selectable genetic cassette is inserted into one of the locations and homologous recombination repair rates are measured as a function of recombinational removal of the inserted cassette. This recombinational repair process is called gene conversion, non-reciprocal recombination. We used this method to measure the recombination rates between genes within gene families and to study the stability of mobile genetic elements inserted into members of gene families.
Keywords: Homologous recombinationBackground
Homologous recombination can occur between two strands of DNA that share a sufficient amount of sequence identity. Homologous recombination can result in either reciprocal exchanges (for example, during meiosis in diploid eukaryotes, when genetic information is exchanged between two homologous parental chromosomes to create novel gametic chromosomes), or non-reciprocal exchanges (such as during mating-type switching in Saccharomyces cerevisiae, when genetic information is copied from one location on a chromosome to another homologous location on the same chromosome, resulting in gene conversion). In reciprocal recombination no genetic information is lost, it is simply exchanged between two homologous chromosomes. In contrast, in a non-reciprocal recombination event, genetic information from one location is copied into another homologous location, and replaces the original information, resulting in gene conversion. In haploid bacteria there is no meiosis, but homologous recombination can take place between the bacterial chromosome and a foreign fragment of DNA, leading to a horizontal gene transfer event (Thomas and Nielsen, 2005) or between two homologous locations within the same chromosome, which can lead to gene conversion (Anderson and Roth, 1977; Abdulkarim and Hughes, 1996; Hughes, 2000).
To measure homologous recombination rates within bacterial chromosomes requires the ability to select for successful recombinant cells. Traditionally, this was achieved by the use of selectable markers. These were inserted in a mutated, inactive state into two locations of the chromosome and could recombine to form an active, selectable gene (Segall and Roth, 1989). The drawback of this method is that recombination occurs between the two inserted sequences and does not provide any information about the homologous recombination rates between native sites within the chromosome. We have developed a method that overcomes this drawback by the use of a counter-selectable marker (Brandis et al., 2018). A selectable/counter-selectable cat-sacB-yfp cassette (cat: selectable for chloramphenicol resistance, sacB: counter-selectable on sucrose (Gay et al., 1985); yfp: yellow fluorescence protein for screening) is inserted into a chromosomal location (selecting for chloramphenicol resistance) (Figures 1A and 1B) and homologous recombination rates are measured as a function of recombinational removal of the inserted cassette (selecting for loss of sacB, growth in the presence of sucrose) (Figures 1C and 1D).
This method enables the measurement of recombination rates between any native homologous sites within the bacterial chromosome. 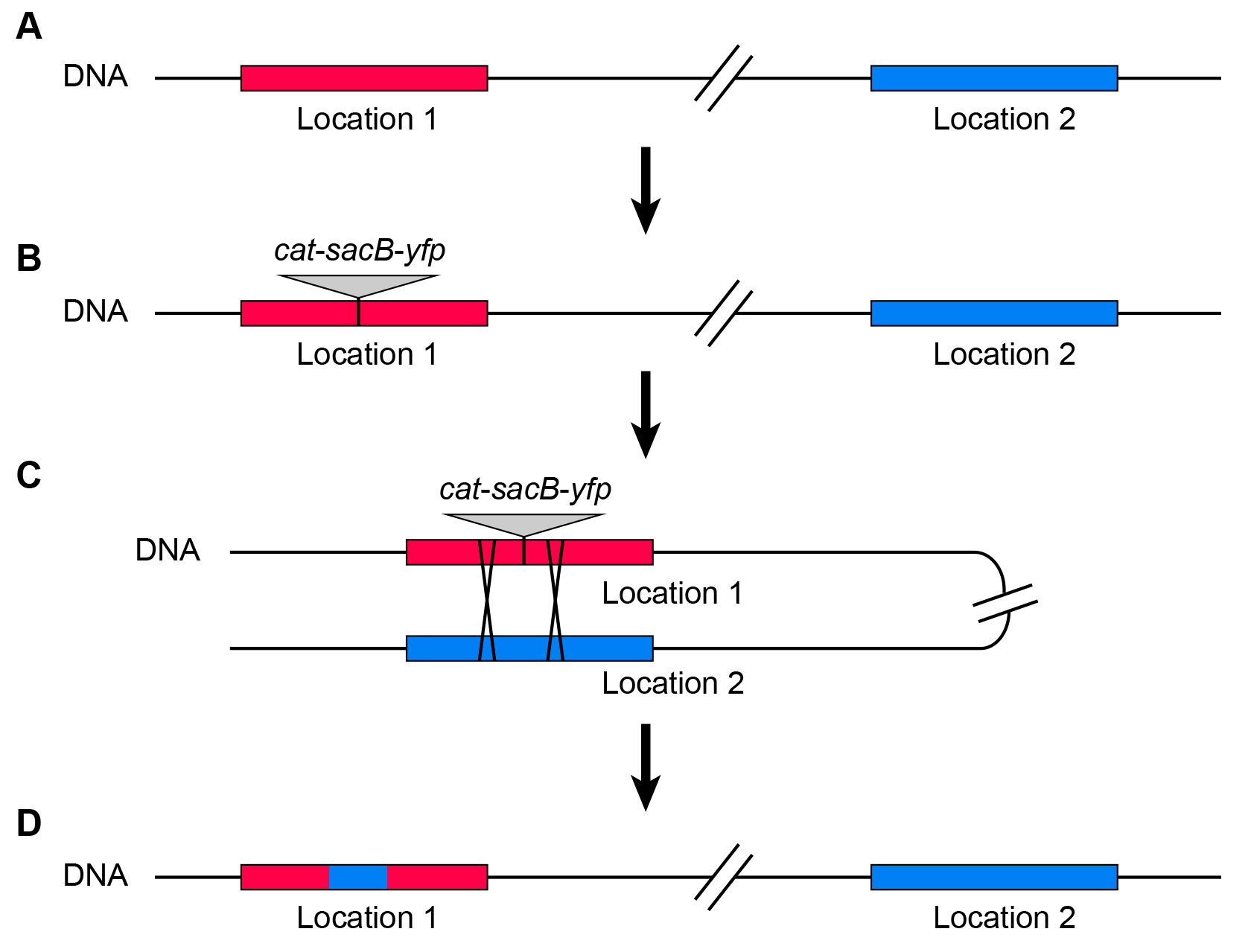
Figure 1. Principle of the assay. A. The assay measures recombination rates between two homologous locations within the chromosome. B. Initially, a selectable/counter-selectable marker is inserted into one of the homologous locations (not part of this protocol). C and D. Spontaneous homologous recombination between the two locations (C) can lead to the removal of the inserted cassette (gene conversion) and the resulting strains (D) can be isolated due to the loss of the counter-selectable marker gene.
Materials and Reagents
- PCR tubes
- 1.5 ml Eppendorf tubes
- 15 ml test tubes
- Plastic Petri dishes (9 cm diameter) for bacterial growth
- Sterile loops (1 µl)
- Pipette tips
- 250 ml E-flask
- 50 ml plastic tubes
- Electroporation cuvettes, 1 mm gap (BTX, product number: 45-0124)
- Salmonella enterica serovar Typhimurium strain LT2 (Biosafety level 2)
- PCR primers that bind outside of the cassette insertion site
- Ampicillin (Sigma-Aldrich, catalog number: A9518)
- Chloramphenicol (Sigma-Aldrich, catalog number: C0378)
- Glucose (Sigma-Aldrich, catalog number: G7021)
- Bacteriological agar (Oxoid, catalog number: LP0011)
- Ethanol (95%)
- PCR Master Mix (2x) (Thermo Fisher, catalog number: K0171)
- Sodium chloride (Sigma-Aldrich, catalog number: S3014)
- Sucrose (Sigma-Aldrich, catalog number: 84100)
- Tryptone (Oxoid, catalog number: LP0042)
- Yeast extract (Oxoid, catalog number: LP0021)
- LB medium (see Recipes)
- LA plates (see Recipes)
- Salt-free LA + 5% sucrose (see Recipes)
Equipment
- Test tube racks
- Autoclave
- Incubator at 37 °C
- PCR machine
- Pipettes (10 µl, 20 µl, 100 µl and 1,000 µl)
- Shaking incubator at 37 °C
- UV Transilluminator (e.g., 365 nm)
- Shaking incubator at 30 °C
- Shaking water bath at 43 °C
- Centrifuge (temperature controlled) for 50 ml tubes (Thermo Scientific, model: Haraeus Megafuge 16R)
- Centrifuge (temperature controlled) for Eppendorf tubes (Eppendorf, model: Centrifuge 5424R)
- Electroporator (Bio-Rad, model: Gene Pulser Xcell)
Procedure
- Insert the genetic cassette into the chromosomal location for which recombination rates should be determined
Note: This step is specific for each location at which recombination rates will be measured. The details of the procedure will also vary depending on the Lambda-red recombination system that is used. For detailed instructions on Lambda-red recombineering see Yu et al., 2000. The following steps are a summary for pSIM6-based recombineering.- Amplify the cat-sacB-yfp cassette by PCR and purify the product.
Note: Primer design and amplification conditions are specific for the used selectable/counter-selectable cassette and insertion location and therefore not further described in this protocol. - Start an overnight culture: Inoculate 2 ml sterile LB containing 100 mg/L ampicillin in a 15 ml test tube with cells from a single colony.
- Incubate in an incubator at 30 °C, shaking at 200 rpm for ~20 h.
- Mix 50 ml salt-free LB containing 100 mg/L ampicillin and 0.2% glucose with 500 µl o/n culture in a 250 ml E-flask.
- Incubate in an incubator at 30 °C, shaking at 200 rpm for ~2 h.
- To induce red expression, move the flask to a 43 °C shaking water bath for 15 min.
- Cool in an ice-water bath for at least 10 min. Swirl carefully to cool cells.
- Transfer the culture to a cold 50 ml plastic tube and pellet the cells (3,500 x g, 8 min, 4 °C).
- Remove as much as possible of the supernatant and resuspend the cells in 20 ml ice-cold sterile water. Do not vortex! Pellet the cells at 3,500 x g, 8 min, 4 °C.
- Resuspend the cells in 1 ml ice-cold water and transfer into a cold Eppendorf tube. Pellet the cells at 16,000 x g, 3 min, 4 °C and remove as much as possible of the supernatant.
- Resupend cells in 100 µl ice-cold water.
- Mix 50 µl cells with 5 µl purified PCR product, incubate on ice for 5 min and transfer into a cold electroporation cuvette.
- Electroporate (1.8 kV, 200 Ω, 25 µF). Immediately, resuspend the cells in 1 ml pre-warmed (37 °C) LB and transfer back into the Eppendorf tube.
- Let the cells recover for at least 3 h or overnight at 37 °C.
- Plate 100 µl recovered culture on an LA plate containing 25 mg/L chloramphenicol, incubate the plate o/n at 37 °C.
- Restreak colonies on LA plates containing 25 mg/L chloramphenicol, incubate the plates o/n at 37 °C.
- Confirm the correct insertion by PCR size verification and test the phenotype of the recombinant cells (YFP fluorescence and sensitivity to growth on salt-free LA plates containing 5% sucrose).
- Amplify the cat-sacB-yfp cassette by PCR and purify the product.
- Determine the number of colony-forming units per ml of overnight cultures
- Start three independent overnight cultures for each strain to be measured: Inoculate 2 ml sterile LB in a 15 ml test tube with cells from a single colony.
- Incubate in an incubator at 37 °C, shaking at 200 rpm for ~20 h.
- Make a 10-fold dilution series in a sterile 0.9% NaCl solution to a dilution factor of 10-7.
- Plate 100 µl of dilutions 10-5, 10-6 and 10-7 on LA plates.
Note: Under these incubation conditions Salmonella cultures usually grow to density of approximately 4 x 109 cfu/ml. Change the plated dilution factors for strains or conditions that form cultures with a different density. - Incubate the LA plates in an incubator at 37 °C for ~20 h.
- Count the colonies per plate. Choose a dilution with 30-500 colonies per plate to calculate the original culture density according to formula 1 (see Data analysis).
- Determine the volume of overnight culture to be plated for assay
Note: Steps B1-B3 are identical to “Procedure A” and the same dilutions can be used for both parts.- Start three independent overnight cultures for each strain to be measured: Inoculate 2 ml sterile LB in a 15 ml test tube with cells from a single colony.
- Incubate in an incubator at 37 °C, shaking at 200 rpm for ~20 h.
- Make a 10-fold dilution series in a sterile 0.9% NaCl solution to a dilution factor of 10-5.
- Plate 100 µl of each dilution (100 to 10-5) on salt-free LA plates containing 5% sucrose.
- Incubate in an incubator at 37 °C for ~20 h.
- Assess colonies under UV light for fluorescence. Re-streak non-fluorescent colonies onto salt-free LA plates containing 5% sucrose.
Note: Use UV light with a wavelength that does not cause DNA damage (e.g., 365 nm), re-streak two colonies per colony morphology (different colony shapes or sizes) if more than 5 colonies are non-fluorescent. - Incubate in an incubator at 37 °C for ~20 h.
- Confirm recombinational removal of the cat-sacB-yfp cassette by PCR size verification followed by sequencing of the insertion site.
- Estimate the dilution factor where approximately half of the cultures would yield a colony with recombinational removal of the cat-sacB-yfp cassette.
Note: The estimated dilution does not need to be part of the 10-fold dilution series but can be any dilution.
- Measure recombination rate
- Start twenty independent overnight cultures for each strain to be measured: Inoculate 2 ml sterile LB in a 15 ml test tube with cells from a single colony.
- Incubate in an incubator at 37 °C, shaking at 200 rpm for ~20 h.
- Make appropriate dilutions, as determined in “Procedure B”, in a sterile 0.9% NaCl solution.
- Plate 100 µl of each dilution on salt-free LA plates containing 5% sucrose.
- Incubate in an incubator at 37 °C for ~20 h.
- Assess colonies under UV light for fluorescence. Re-streak non-fluorescent colonies onto salt-free LA plates containing 5% sucrose.
Note: Use UV light with a wavelength that does not cause DNA damage (e.g., 365 nm). Re-streak two colonies per colony morphology (different colony shapes or sizes) if more than 5 colonies are non-fluorescent. - Incubate in an incubator at 37 °C for ~20 h.
- Confirm recombinational removal of the cat-sacB-yfp cassette by PCR size verification followed by sequencing of the insertion site.
- Calculate the recombination rate according to the formula 2 (see Data analysis).
Data analysis
- The culture density is calculated according to the formula:
P = N/V*D (1)
where P is the population density in cfu/ml, N is the number of colonies on the plate, V is the volume plated in ml and D is the dilution factor. - The recombination rate is calculated according to the formula:
µ = -ln(P0)/N (2)
where µ is the mutation rate, P0 is the proportion of cultures without recombinational removal of the cat-sacB-yfp cassette and N is the number of cells plated for the assay.
Notes
- This protocol can be performed in any bacterial strain in which lambda-red recombineering can be used.
- This protocol is written for the use of a cat-sacB-yfp cassette (available on request), but any selectable/counter-selectable cassette and screening marker can be used.
Recipes
- LB medium
Mix and autoclave:
10 g Tryptone
5 g Yeast extract
10 g Sodium chloride (omit in salt-free LB)
1 L water - LA plates
Mix and autoclave:
10 g Tryptone
5 g Yeast extract
10 g Sodium chloride
15 g Agar
1 L water
Pour media into plastic Petri dishes and let solidify - Salt-free LA + 5% sucrose
- Mix and autoclave
10 g Tryptone
5 g Yeast extract
15 g Agar
700 ml water - Mix and autoclave
50 g Sucrose
300 ml water - Mix both solutions, pour media into plastic Petri dishes and let solidify
- Mix and autoclave
Acknowledgments
This work was supported by grants to DH from the Swedish Science Research Council (Vetenskapsrådet, grant numbers 2016-04449, 2017-03953) and the Carl Trygger Foundation, (grant numbers CTS16:194, CTS17:204).
Competing interests
The authors have no conflict of interest.
References
- Abdulkarim, F. and Hughes, D. (1996). Homologous recombination between the tuf genes of Salmonella typhimurium. J Mol Biol 260(4): 506-522.
- Anderson, R. P. and Roth, J. R. (1977). Tandem genetic duplications in phage and bacteria. Annu Rev Microbiol 31: 473-505.
- Brandis, G., Cao, S. and Hughes, D. (2018). Co-evolution with recombination affects the stability of mobile genetic element insertions within gene families of Salmonella. Mol Microbiol 108(6): 697-710.
- Gay, P., Le Coq, D., Steinmetz, M., Berkelman, T. and Kado, C. I. (1985). Positive selection procedure for entrapment of insertion sequence elements in gram-negative bacteria. J Bacteriol 164(2): 918-921.
- Hughes, D. (2000). Co-evolution of the tuf genes links gene conversion with the generation of chromosomal inversions. J Mol Biol 297(2): 355-364.
- Segall, A.M. and Roth J.R. (1989). Recombination between homologies in direct and inverse orientation in the chromosome of Salmonella: intervals which are nonpermissive for inversion formation. Genetics 122(4): 737-747.
- Thomas, C. M. and Nielsen, K. M. (2005). Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat Rev Microbiol 3(9): 711-721.
- Yu, D., Ellis, H. M., Lee, E. C., Jenkins, N. A., Copeland, N. G. and Court, D. L. (2000). An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A 97(11): 5978-5983.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Brandis, G., Cao, S. and Hughes, D. (2019). Measuring Homologous Recombination Rates between Chromosomal Locations in Salmonella. Bio-protocol 9(3): e3159. DOI: 10.21769/BioProtoc.3159.
Category
Microbiology > Microbial genetics > Recombination
Molecular Biology > DNA > DNA recombination
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.