- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

pNP Transgenic RNAi System Manual in Drosophila

Published: Vol 9, Iss 3, Feb 5, 2019 DOI: 10.21769/BioProtoc.3158 Views: 7345
Reviewed by: Gal HaimovichPradeep Kumar BhaskarAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Much of our knowledge about the mechanisms underlying biological processes relies on genetic approaches, whereby gene activity is reduced and the phenotypic consequences of perturbation are analyzed in detail. For functional genomic studies, a specific, systematic, and cost-effective manner is critical. Transgenic RNAi system is the top priority choice to study gene functions due to its simple and practical characteristics in Drosophila. We established a novel system that works well in both soma and germ cells which is efficient and specific. With this system, we can precisely and efficiently modulate highly expressed genes, and simultaneously knock down multiple genes in one step. In this study, we provide a detailed protocol of the pNP system, which replaces other transgenic systems, and expect it can provide some help to researchers who are using this system.
Keywords: DrosophilaBackground
Drosophila melanogaster is easy to culture, and has a short life cycle, clear genetic background, and abundant research tools. More importantly, most genes in Drosophila are highly homologous to human genes, which over the past century made Drosophila a powerful model to study the genes in vivo.
RNAi technology can specifically reduce gene expression, which is efficient and easy to operate, so it has been widely used in different model organisms to study gene functions (Dietzl et al., 2007; Ni et al., 2008, 2009 and 2011; Perrimon et al., 2010). In Drosophila, transgenic RNAi approach is a binary system, where UAS-hairpin is controlled by GAL4 to achieve tissue- or developmental stage-specific gene knockout (Brand and Perrimon, 1993). Without Gal4, the hairpin downstream of the UAS will not be transcribed, thus transgenic hairpin lines behave similar to wild type. That is why we can keep thousands of transgenic RNAi lines. However, current RNAi systems exhibit many limitations, such as off-target effects, leaky expression of the basal promoter, poor efficiency to highly expressed genes and the unsolved challenge of simultaneously targeting multiple genes.
We developed a new transgenic RNAi system based on pNP vector, which has an intergenic linker between miR-2a-1 and miR-2b-2 and a modified promoter (Qiao et al., 2018). The pNP transgenic RNAi system can efficiently target highly expressed genes and produce expected severe phenotypes, which overcomes the poor efficiency of current RNAi systems (Ma et al., 2006; Markstein et al., 2008). Furthermore, the pNP transgenic RNAi system can innovatively knock down multiple genes simultaneously in one step, for example simultaneously depleting all the core components of PRC1 in the eye using the pNP system led to smaller eyes (Qiao et al., 2018). The efficiency of multiple mRNA depletion was further confirmed by qRT-PCR, which showed reduced expression levels of these targeted genes. Moreover, the loss-of-function phenotypes produced by targeting multiple genes simultaneously are more robust than the effect of sequentially depleting the genes. The following protocol details the steps from vector construction to transgenic RNAi flies’ screening, which gives us the approach to study the autonomous or non-autonomous roles of genes, including protein complexes or redundant genes.
Materials and Reagents
- 1.5 ml MaxyClear Microtubes (Axygen, catalog number: MCT-150-C)
- 0.2 ml Polypropylene PCR Tube (Axygen, catalog number: PCR-0208-C)
- Pipette tips (Corning, Axygen®)
- 0.22 μm Millipore filters (Biosharp, BS-QT-011)
- Petri dishes
- Drosophila lines (Tinghua Fly Center, http://fly.redbux.cn/)
- y[1] sc[1] v[1] p{y[+t7.7]}=nos-phiC31\int.NLS}X; P{y[+t7.7]=CaryP}attP40 (TB00016)
- y[1] sc[1] v[1] p{y[+t7.7]}=nos-phiC31\int.NLS}X; P{y[+t7.7]=CaryP}attP2 (TB00018)
- y[1] sc[1] v[1]; wg[Gla-1] Bc[1]/CyO (TB00023)
- y[1] sc[1] v[1]; Dr[1] e[1]/TM3, Sb[1] (TB00139)
- y[1] sc[1] v[1] (TB00077)
- pNP vector (Ampicillin resistant, plasmid map is shown in Figure 1. This vector is not commercial, but can be provided for free. If you want, please email your need to nijq@mail.tsinghua.edu.cn)
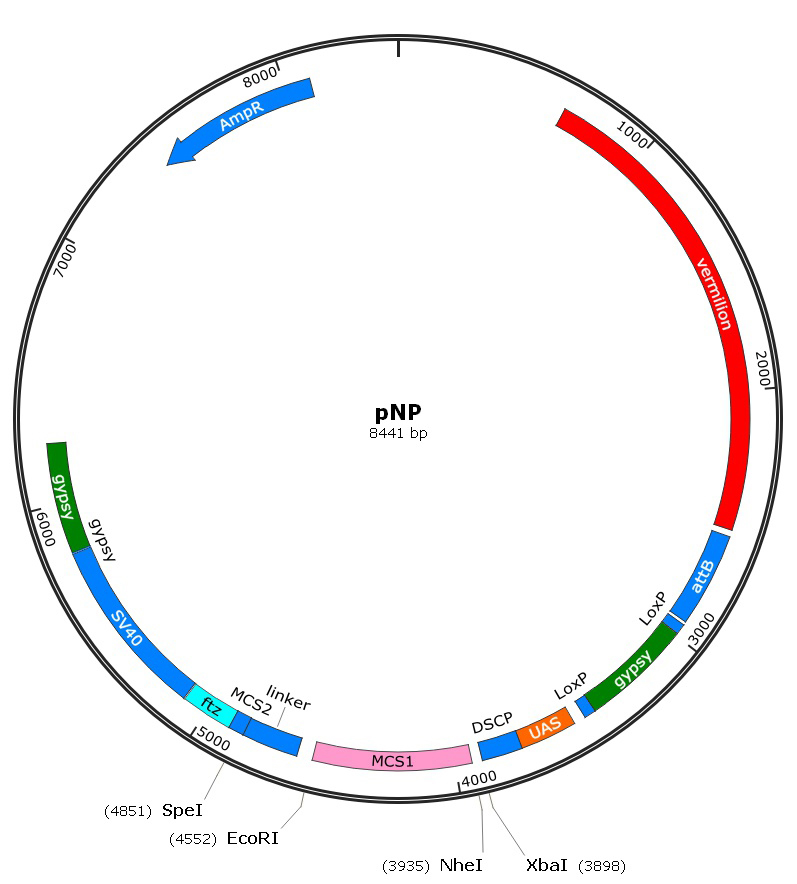
Figure 1. The map of pNP construct. Vermilion is a selectable marker. attB sequence is used for phiC31 targeted integration at genomic attP landing sites. MCS1 allows a single shRNA to be cloned in both orientations, while with the help of MCS2 and the linker it could simultaneously generate multiple shRNAs once. - EcoRI-HF (NEB, catalog number: R3101L)
- NheI-HF (NEB, catalog number: R3131L)
- SpeI (NEB, catalog number: R3133L)
- XbaI (NEB, catalog number: R0145L)
- Alkaline Phosphatase, Calf Intestinal (CIP, NEB, catalog number: M0290S)
- AxyPrepTM DNA Gel Extraction Kit (Corning, Axygen, catalog number: AP-GX-250)
- T4 DNA ligase (NEB, catalog number: M0202L)
- Trans5α Chemically Competent Cell (Transgen Biotech, catalog number: CD201-01)
- Go Tag Green Master Mix, 2x (Promega, catalog number: 000179370)
- AxyPrepTM Plasmid Miniprep Kit (Corning, Axygen, catalog number: AP-MN-P-250)
- PurePlasmid Mini Kit (CWBiotech, catalog number: CW0500M)
- Primers sequences:
U-F: 5’-GCTGAGAGCATCAGTTGTGA-3’; Ftz: 5’-TAATCGTGTGTGATGCCTACC-3’ - Buffer PB (QIAGEN, catalog number: 154051779)
- LB Broth, Miller (Luria Bertani) (Becton Dickinson)
- Agar (Beyotime)
- Ampicillin (Solarbio, catalog number: A8180)
- Tris-HCl (AMRESCO, catalog number: 1185-53-1)
- EDTA (AMRESCO, catalog number: 60-00-4)
- NaCl (AMRESCO, catalog number: 7647-14-5)
- KCl (AMRESCO, catalog number: 7447-40-7)
- Ethidium bromide (Sigma, catalog number: E8751)
- 50x TAE (Double Helix, catalog number: P0309A)
- Sodium phosphate buffer stock solution (pH 6.8) (see Recipes)
- 10x Annealing Buffer (see Recipes)
- Injection Buffer (see Recipes)
- LB (Luria Bertani) medium (see Recipes)
- LB/ampicillin plates (see Recipes)
- 1x TAE (see Recipes)
- 1.5% agarose gel (see Recipes)
Equipment
- Pipette (Eppendorf)
- Microwave (SANYO, model: EM-2509EB1)
- Autoclave (SANYO, model: MLS-3780)
- PCR Thermal Cycler (Eppendorf, Mastercycler nexus GSX1)
- Refrigerated centrifuge (Eppendorf, model: 5417R)
- Stereo Microscope (Olympus Corporation)
- NanoDrop 2000 (Thermo Scientific)
Software
- DSIR website: http://biodev.cea.fr/DSIR/DSIR.html
- FlyBase website: http://flybase.org/
- EMBOSS Needle: https://www.ebi.ac.uk/Tools/psa/emboss_needle/nucleotide.html
- Tsinghua Fly Center: http://fly.redbux.cn
Procedure
- Vector construction
- Single short hairpin construction
- Short hairpins design
Design primers for short hairpins (21 nt) on http://biodev.cea.fr/DSIR/DSIR.html website: first, copy the sequence of exon within the gene, and then paste the sequence to predict the siRNA with this web tool. Based on the parameters we defined (Figure 2), the siRNA with the highest score will be selected. To avoid the off-target effect, use BLAST to compare the siRNA against the fly genome of FlyBase website (http://flybase.org/) to ensure the 21 nt sequence has more than 5 nt that mismatch to other genes. “As re-com” sequence is sense sequence of the short hairpin. “As” sequence is the antisense sequence of the short hairpin.
Primer-F: ctagcagt “As re-com” tagttatattcaagcata “As” gcg
Primer-R: aattcgc “As re-com” tatgcttgaatataacta “As” actg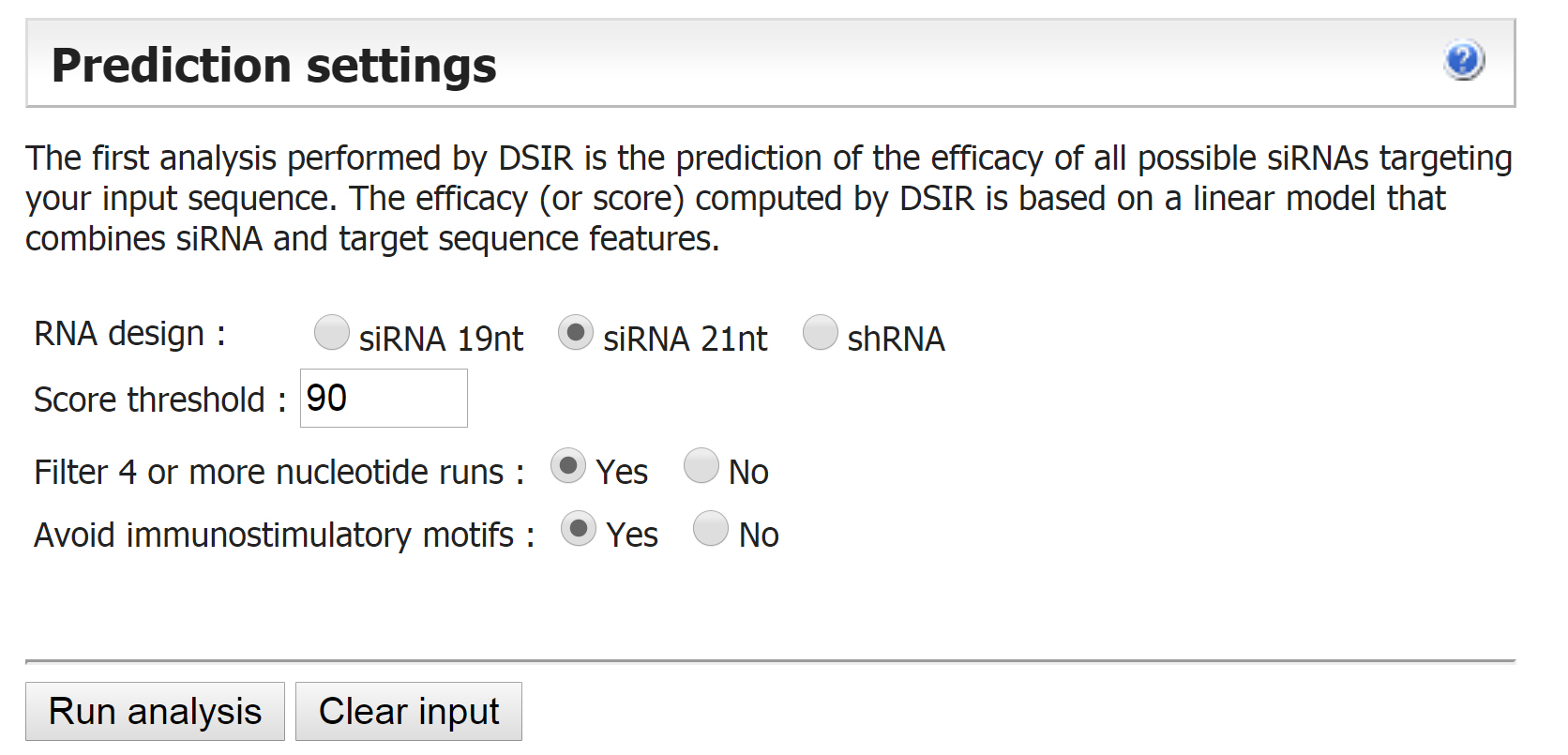
Figure 2. Parameters for short hairpins design - Annealing
Perform the annealing reaction as follows, and then put the Eppendorf tube into PCR equipment. Denature the primers at 95 °C for 5 min, and then turn off the power of PCR equipment to let it cool down slowly to room temperature.
- Digestion
Use EcoRI/NheI restriction endonucleases for pNP vector (Figure 1) digestion, and then separate the fragments with 1.5% agarose gels. Cut and purify the larger DNA fragment around 8,000 bp with AxyPrepTM DNA Gel Extraction Kit. Perform the digestion reaction as follows: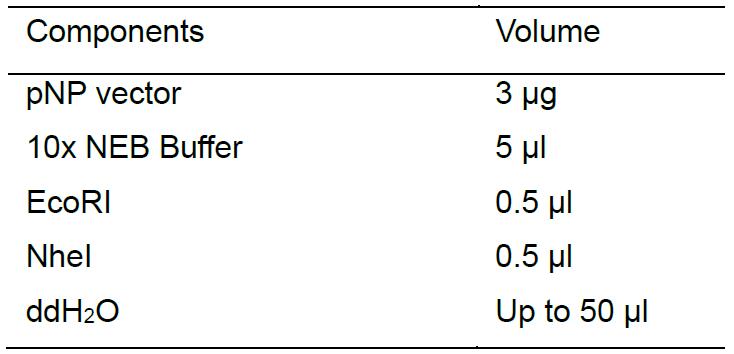
- Ligation
Ligate the linearized pNP vector with the annealing product using T4 DNA ligase. Perform the ligation reaction as follows: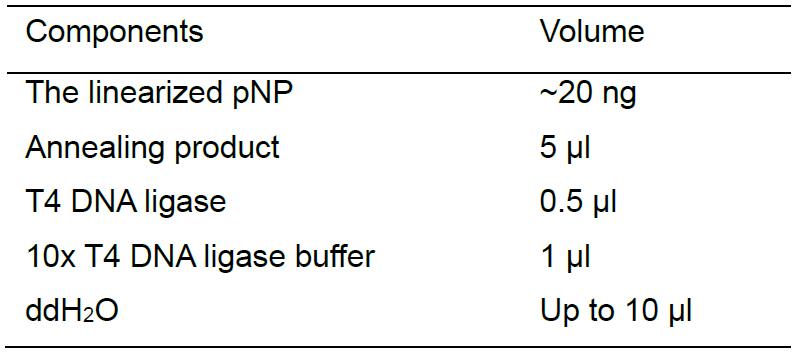
Mix and incubate at 25 °C for 20 min, and then put 2-5 µl ligation product into 10 µl Trans5α Chemically Competent Cell following the standard protocol, and spread on an LB-agar plate containing 100 µg/ml ampicillin. Incubate the plate overnight at 37 °C, upside down. - Select correct clones
- Mark and pick clones to the PCR mix. Primers sequences are U-F: 5’-GCTGAGAGCATCAGTTGTGA-3’, Ftz: 5’-TAATCGTGTGTGATGCCTACC-3’.

- Set up and run the PCR program as follows:
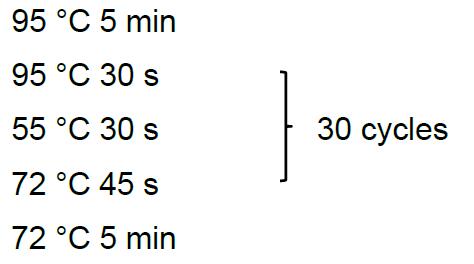
- Identify the correct clones based on the size of the PCR product. By gel electrophoresis, the correct clones give around 750 bp bands, and the wrong clones appear either 1,000 bp or no band.
- Amplify the correct clone: Pick up the correct clone into 5 ml of LB medium containing 100 µg/ml ampicillin and incubate overnight at 37 °C at 250 rpm.
- Extract plasmid using AxyPrepTM Plasmid Miniprep Kit, and further confirm the right clone by DNA sequencing with the primer (U-F: 5’-GCTGAGAGCATCAGTTGTGA-3’).
- Mark and pick clones to the PCR mix. Primers sequences are U-F: 5’-GCTGAGAGCATCAGTTGTGA-3’, Ftz: 5’-TAATCGTGTGTGATGCCTACC-3’.
- Short hairpins design
- Construct multiple shRNAs in one vector
- For the two or more short hairpins in one vector, we first clone all short hairpins into the pNP vector respectively as mentioned above.
- Linearize vector pNP-shRNA1 using SpeI at 37 °C for 90 min as follows:

Before gel electrophoresis, use CIP to treat the cutting product to dephosphorylating at 37 °C for 10 min as follows:
Purify the product using AxyPrepTM DNA Gel Extraction Kit. - Digest another hairpin vector using SpeI and XbaI to release the fragment (300 bp) containing a different short hairpin as follows:
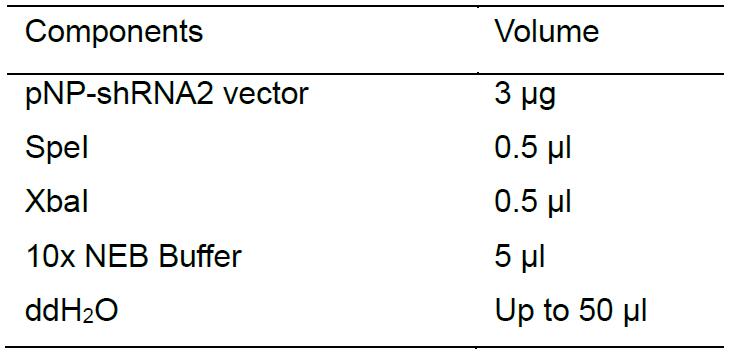
Separate the digested products on an agarose gel and extract smaller DNA fragment around 300 bp from the agarose gel with AxyPrepTM DNA Gel Extraction Kit. Finally, using T4 DNA ligase ligate the linearized pNP-shRNA1 with the released fragment (shRNA2). - Following transformation, use primers (U-F: 5’-GCTGAGAGCATCAGTTGTGA-3’, Ftz: 5’-TAATCGTGTGTGATGCCTACC-3’) to identify the correct clones by PCR, which give around 1,000 bp. Then further confirm these correct plasmids by DNA sequencing.
- To clone the third shRNA, the two shRNA containing vector is digested with SpeI again and then ligated with another short hairpin fragment released from hairpin vector by SpeI and XbaI digestion.
- Single short hairpin construction
- Purify and inject the correct pNP-shRNA vector
- Purification of the plasmid DNA
Extract plasmid DNA (~10 mg) using PurePlasmid Mini Kit as per the manufacturer’s instructions. Elute the plasmid DNA with 70 μl of 1x Injection Buffer. The appropriate concentration of each sample is 100-200 mg/ml, which is determined by using NanoDrop 2000. - Injection of the plasmid DNA to flies
Inject the purified plasmid DNA to flies (Chromosome II TB00016; Chromosome III TB00018) following the standard protocol (Ni et al., 2011). For knocking down a gene on the second chromosome, choose TB00016 to microinject, while a gene on the third chromosome to choose TB00018. Keep the injected embryos at 25 °C and 60% humidity to adult (G0). - Selection protocols after G0 eclosion (Figures 3 and 4 are overviews of screening procedure)
- Once the G0 flies begin to hatch into adults, cross them to the TB00077 (vermillion eye color).
Note: For injections into y,v stocks, G0s should all have vermillion eyes.
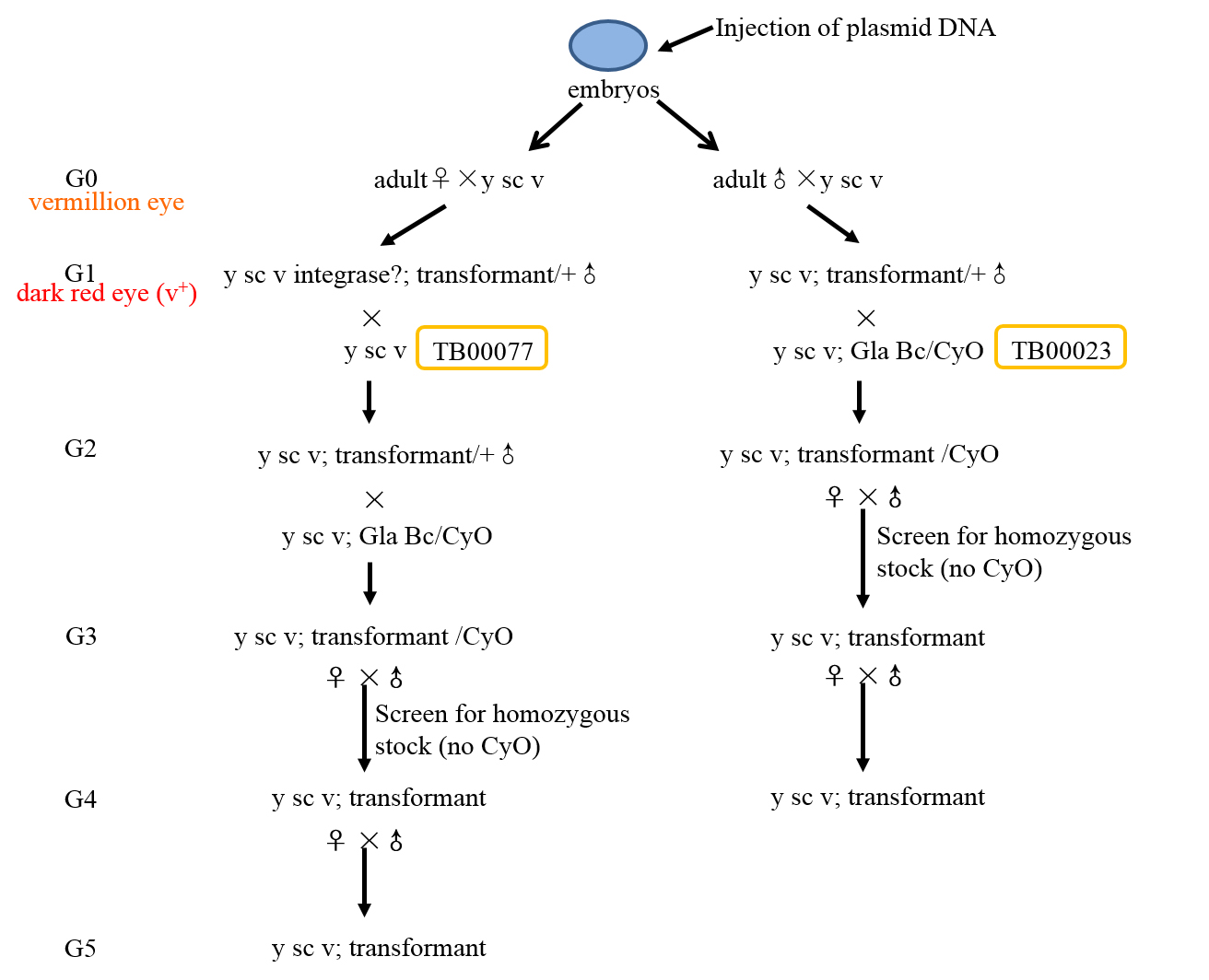
Figure 3. Overview of the selection procedures for generating Chromosome II transgenic flies- When adult G1 flies begin to eclose, screen G1 transformants. Remember that the pNP vector carry a copy of vermillion (v+), so G1 transformants will have a dark red wild type (v+) eye color.
- Cross the appropriate balancer stocks to ♂ G1 transformants from ♂ G0 parents (For Chromosome II integration into an attP40 site, G1 flies were crossed with TB00023. For Chromosome III integration into an attP2 site, G1 flies were crossed with TB00139). When G2 flies begin to eclose, collect ♂ and virgin ♀ siblings that have v+ (wild type) eye color and CyO (for Chromosome II insertions) or TM3 (for Chromosome III insertions), and cross siblings to each other to screen for homozygous transgenic flies. Collect ♂ G3 and virgin ♀ G3 siblings, and cross siblings to each other to make sure that the homozygous transgenic flies were fertile.
- ♂ transformants from ♀ G0 parents need to backcross TB00077 again to remove phiC31 integrase. Collect wild type (v+) eye color ♂ G2 transformants to cross the appropriate balancer stocks. The following sequential steps are consistent with the Step B3c.
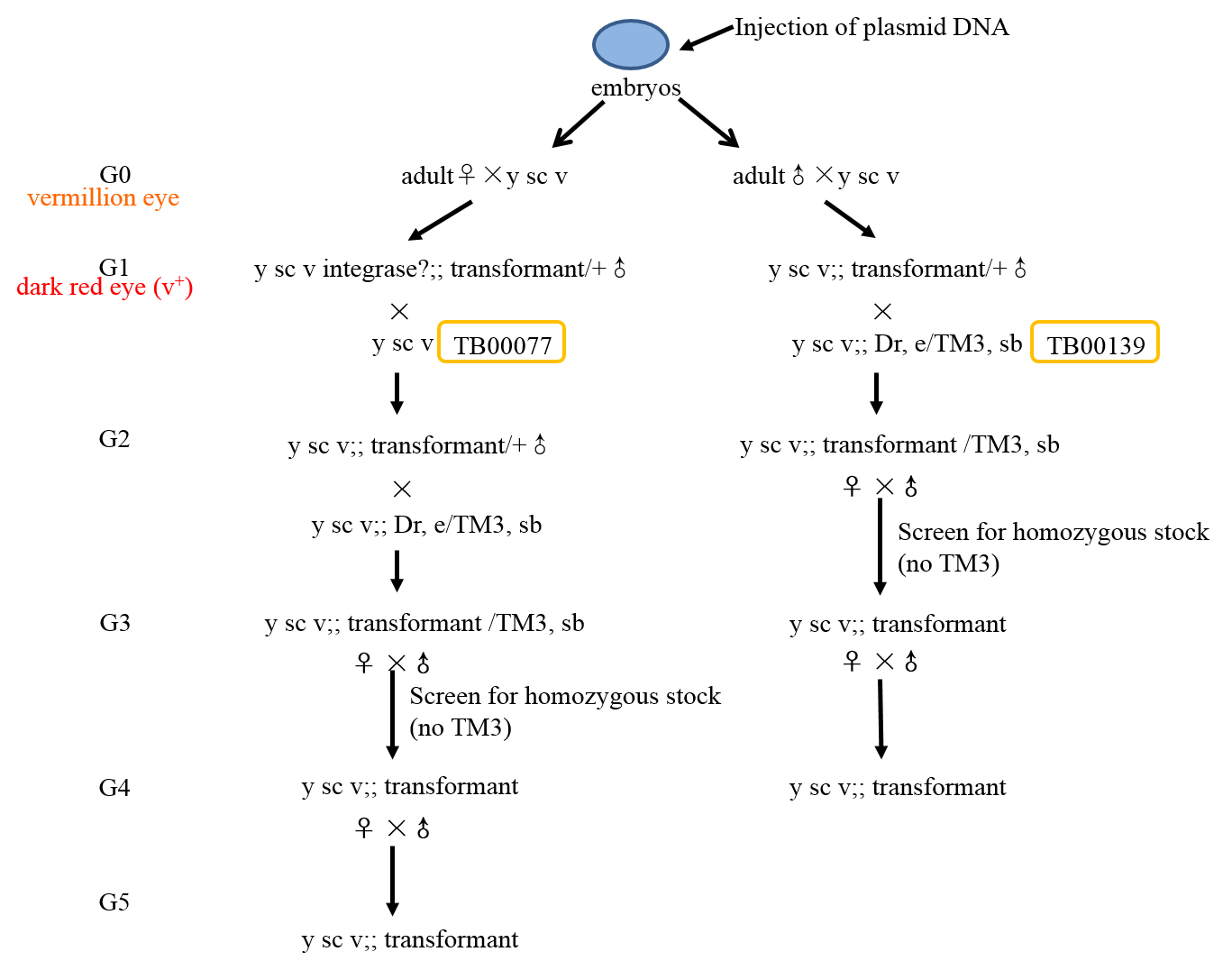
Figure 4. Overview of the selection procedures for generating Chromosome III transgenic flies - Once the G0 flies begin to hatch into adults, cross them to the TB00077 (vermillion eye color).
- Purification of the plasmid DNA
Data analysis
EMBOSS Needle (https://www.ebi.ac.uk/Tools/psa/emboss_needle/nucleotide.html) is used for sequence alignment. Detailed information on the data of this manual is available from the corresponding author upon reasonable request.
Recipes
- 10x Annealing Buffer (store at RT)
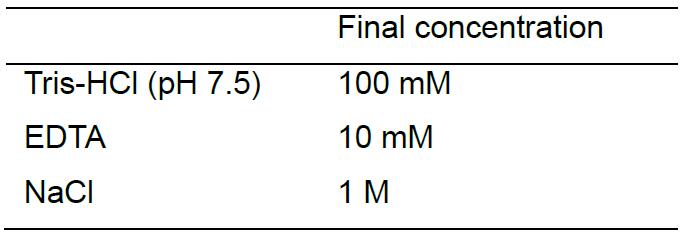
- Injection Buffer
Prepare 1 mM sodium phosphate Buffer (pH 6.8) and 50 mM KCl in sterile bi-distilled water, and then filter through 0.22 μm Millipore filters, store at -20 °C - Sodium phosphate buffer (pH 6.8) (stock solution)
51 ml 0.2 M NaH2PO4
49 ml 0.2 M Na2HPO4
Store at room temperature- 0.2 M Na2HPO4
Dissolve 71.6 g Na2HPO4•12H2O in 1 L of ddH2O, mix thoroughly
Store at room temperature - 0.2 M NaH2PO4
Dissolve 31.2 g Na2HPO4•2H2O in 1 L of ddH2O, mix thoroughly
Store at room temperature
- 0.2 M Na2HPO4
- LB (Luria Bertani) medium
Dissolve 25 g of the powder in 1 L of ddH2O, mix thoroughly
Autoclave at 121 °C for 15 min - LB/ampicillin plates
Add 25 g of LB media and 7 g Agar in 1 L of ddH2O
Autoclave at 121 °C for 15 min
After sterilization, allow the medium to cool before adding 100 µg/ml of ampicillin, pour 15 ml of medium into each Petri dish - 1x TAE
20 ml 50x TAE
980 ml ddH2O
Store at room temperature - 1.5% agarose gel
1.5 g agarose
100 ml 1x TAE
Heat solution to dissolve agarose in a microwave
Add ethidium bromide to a final concentration of 0.2 μg/ml
Acknowledgments
This work was supported by the National Key Technology Research and Development Program of the Ministry of Science and Technology of the People’s Republic of China (2016YFE0113700), and the National Natural Science Foundation of China (31571320, 91729301, 81630103), and the National Science Fund for Distinguished Young Scholars (31725023).
Competing interests
The authors declare no competing financial interests.
References
- Brand, A. H. and Perrimon, N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118(2): 401-415.
- Dietzl, G., Chen, D., Schnorrer, F., Su, K. C., Barinova, Y., Fellner, M., Gasser, B., Kinsey, K., Oppel, S., Scheiblauer, S., Couto, A., Marra, V., Keleman, K. and Dickson, B. J. (2007). A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448(7150): 151-156.
- Ni, J. Q., Liu, L. P., Binari, R., Hardy, R., Shim, H. S., Cavallaro, A., Booker, M., Pfeiffer, B. D., Markstein, M., Wang, H., Villalta, C., Laverty, T. R., Perkins, L. A. and Perrimon, N. (2009). A Drosophila resource of transgenic RNAi lines for neurogenetics. Genetics 182(4): 1089-1100.
- Ni, J. Q., Markstein, M., Binari, R., Pfeiffer, B., Liu, L. P., Villalta, C., Booker, M., Perkins, L. and Perrimon, N. (2008). Vector and parameters for targeted transgenic RNA interference in Drosophila melanogaster. Nat Methods 5(1): 49-51.
- Ni, J. Q., Zhou, R., Czech, B., Liu, L. P., Holderbaum, L., Yang-Zhou, D., Shim, H. S., Tao, R., Handler, D., Karpowicz, P., Binari, R., Booker, M., Brennecke, J., Perkins, L. A., Hannon, G. J. and Perrimon, N. (2011). A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat Methods 8(5): 405-407.
- Perrimon, N., Ni, J. Q. and Perkins, L. (2010). In vivo RNAi: today and tomorrow. Cold Spring Harb Perspect Biol 2(8): a003640.
- Qiao, H. H., Wang, F., Xu, R. G., Sun, J., Zhu, R., Mao, D., Ren, X., Wang, X., Jia, Y., Peng, P., Shen, D., Liu, L. P., Chang, Z., Wang, G., Li, S., Ji, J. Y., Liu, Q. and Ni, J. Q. (2018). An efficient and multiple target transgenic RNAi technique with low toxicity in Drosophila. Nat Commun 9(1): 4160.
- Ma, Y., Creanga, A., Lum, L., Beachy, P. A. (2006). Prevalence of off-target effects in Drosophila RNA interference screens. Nature 443(7109): 359-63.
- Markstein, M., Pitsouli, C., Villalta, C., Celniker, S. E., Perrimon, N. (2008). Exploiting position effects and the gypsy retrovirus insulator to engineer precisely expressed transgenes. Nat Genet 40(4): 476-483.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wang, F., Qiao, H., Xu, R., Sun, J., Zhu, R., Mao, D. and Ni, J. (2019). pNP Transgenic RNAi System Manual in Drosophila. Bio-protocol 9(3): e3158. DOI: 10.21769/BioProtoc.3158.
Category
Molecular Biology > RNA > RNA interference
Molecular Biology > DNA > Gene expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.