- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Protein-DNA Binding Assay (AlphaScreen® Technology)
Published: Vol 9, Iss 3, Feb 5, 2019 DOI: 10.21769/BioProtoc.3155 Views: 10241
Reviewed by: Rajeevkumar SarmaNeha AgarwalAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Identification of specific DNA binding sites of transcription factors is important in understanding their functions. Recent techniques allow us to investigate genome-wide in vivo binding positions by chromatin immunoprecipitation combined with high-throughput sequencing. However, to further explore the binding motifs of transcription factors, in-depth biochemical analysis is required. Here, we describe an efficient protocol of protein-DNA interactions based on a combination of our in vitro transcription/translation system and AlphaScreen® technology. The in vitro transcription/translation system supports an efficient and quick way of protein synthesis by alleviating cumbersome cloning steps. In addition, AlphaScreen® system provides a highly sensitive, quick, and easy handling platform to investigate the protein-DNA interactions in vitro. Thus, our method largely contributes to comprehensive analysis of the biochemical properties of transcription factors.
Keywords: Transcription factorBackground
Upon exposure to abiotic and biotic stresses, major transcriptional changes are induced in plants to help them adapt to these environmental stimuli. Transcriptional regulation is important not only for stress responses but also for plant development. In general, one transcription factor regulates an array of target genes by recognizing its corresponding cis-regulatory elements. Thus, to study the physiological role of transcription factors, identification of their specific binding sequences is a major challenge. Recently, chromatin immunoprecipitation combined with high-throughput sequencing (ChIP-seq) has enabled researchers to determine the genome-wide locations of a transcription factor-DNA interaction. However, the precise binding capability should be further confirmed by in-depth biochemical experiments.
For the past several decades, electrophoretic mobility shift assay (EMSA) and transient reporter assay using viable cells have been widely used to determine protein-DNA interactions. For EMSA, a protein-DNA complex is subjected to electrophoresis followed by the autoradiographic or immunodetection of the band shift. For transient assay, protoplasts or tissues are transformed with a plasmid that consists of a promoter sequence harboring candidate cis-regulatory elements fused with a reporter gene encoding such as luciferase or green fluorescent protein (Chalfie et al., 1994). Although these are powerful methods to investigate the direct binding site of a transcription factor and its transcriptional activity, they are time-consuming and thus cannot be applicable to a comprehensive analysis.
On the other hand, Amplified Luminescence Proximity Homogenous Assay (Alpha) technology is an emerging alternative method to effectively detect protein-DNA interaction (AlphaScreen®). AlphaScreen assay uses two types of small beads (250 nm in diameter), Acceptor and Donor beads, which are specifically designed to associate with either a target protein fused with a protein tag or a biotinylated cis-regulatory element. When the protein-DNA interaction occurs, illumination at 680 nm releases singlet oxygen from donor beads, which subsequently transfers energy to acceptor beads to generate light at 520-620 nm. Since the half-life of singlet oxygen is only 4 µsec, the interaction is not detected without close proximity (see also web site: www.perkinelmer.com/alphascreen). Since AlphaScreen® shows high sensitivity and low background in a microplate format, it can adapt to a high-throughput screening platform.
Compared to EMSA and transient assay, AlphaScreen® is considerably easier to handle and quicker for the detection of protein-DNA interaction. Moreover, we have developed a highly efficient way to synthesize proteins in vitro (Nomoto and Tada, 2018a and 2018b). This method enables us to synthesize proteins that are difficult to obtain in cell-based production systems such as E. coli. Furthermore, this method is highly time efficient because the DNA template for in vitro transcription can be made by the second round of PCR using any type of cloning vectors. To take these advantages, we describe a protocol from protein synthesis to AlphaScreen® assay for detecting protein-DNA interactions. We confirm that our method contributes to the understanding of biological roles of transcription factors function in diverse signaling pathways.
Materials and Reagents
- Aluminum foil
- 1.5 ml microcentrifuge tubes
- 0.2 ml 8 strip PCR tube and cap strips
- 5’-biotinylated and non-biotinylated 50 base single-strand DNA and complementary unmodified 50 base single-strand DNA (Eurofins, Japan)
- Plasmid containing cDNA region of the gene of interest
- Gene-specific primers for 1st and 2nd PCR (see Table 1)
- Ultrapure water (Milli-Q water)
- KOD-Plus-Neo (including 10x PCR buffer, 2 mM dNTPs, 25 mM MgSO4 and KOD-Plus-Neo DNA polymerase) (TOYOBO, catalog number: KOD-401)
- Agarose (VWR, catalog number: 0710-500g)
- Quick-Load Purple 1 kb Plus DNA ladder (0.1-10.0 kb) (New England BioLabs, catalog number N0550S)
- Ethidium bromide (Wako Pure Chemical, catalog number: 547-00101)
- RNase-free water
- 4 M ammonium acetate diluted with RNase-free water
- 99.5% ethanol (Wako Pure Chemical, special grade, catalog number: 057-00451)
- In Vitro Transcription/Translation Kit (NUProtein, catalog number: PSS3050)
- Polyacrylamide gel
- AlphaScreen FLAG® (M2) Detection kit (PerkinElmer, catalog number: 6760613C)
- AlphaPlate-384, 384-well plate (PerkinElmer, catalog number: 6005350)
- Tween® 20 (TCI, catalog number: T0543)
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A2058-5G)
- Polydeoxyadenylic acid-Polythymidylic acid (dAdT) (Sigma-Aldrich, catalog number: P9764-5UN)
- DL-dithiothreitol (DTT) (Wako Pure Chemical, catalog number: 047-08973)
- Tris (hydroxymethyl) aminomethane (Wako Pure Chemical, catalog number: 207-06275)
- Glacial acetic acid (Wako Pure Chemical, catalog number: 518-33985)
- Ethylenediaminetetraacetic acid (EDTA) (DOJINDO, catalog number: 345-01865)
- Glycerol (Wako Pure Chemical, catalog number: 075-00616)
- Sodium dodecyl sulfate (SDS) (Wako Pure Chemical, catalog number: 191-07145)
- Bromophenol blue (Wako Pure Chemical, catalog number: 021-02911)
- Xylene cyanolFF (Wako Pure Chemical, catalog number: 244-00461)
- 100x TAE buffer (see Recipes)
- 10x loading dye (see Recipes)
For SDS-PAGE and Western blotting
- Nitrocellulose membrane (GE Healthcare, catalog number: 10600007)
- WhatmanTM 3 MM Chr Chromatography Paper (GE Healthcare, catalog number: 05-714-5)
- Hybridization bag
- MXJB III Film (IBI, catalog number: 6567291)
- Precision Plus ProteinTM Dual color standards (Bio-Rad Laboratories, catalog number: 1610374)
- SDS polyacrylamide gel
- Methanol (Wako pure chemical, special grade, catalog number: 131-01826)
- Anti-DYKDDDDK tag antibody (diluted 1:2,000 in blocking buffer) (Wako Pure Chemical, catalog number: 014-22383)
- Goat anti-mouse IgG-HRP (diluted 1:1,000 in blocking buffer) (Cosmo Bio, catalog number: 1030-05)
- SuperSignalTM West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific, catalog number: 34577)
- Tris (hydroxymethyl) aminomethane (Wako Pure Chemical, catalog number: 207-06275)
- SDS (Wako Pure Chemical, catalog number: 191-07145)
- Bromophenol blue (Wako Pure Chemical, catalog number: 021-02911)
- Xylene cyano lFF (Wako Pure Chemical, catalog number: 244-00461)
- NaCl (Wako Pure Chemical, catalog number: 191-01665)
- Na2HPO4 (Wako Pure Chemical, catalog number: 197-02865)
- KCl (Wako Pure Chemical, catalog number: 163-03545)
- KH2PO4 (Wako Pure Chemical, catalog number: 169-04245)
- Skimmed milk powder (Wako Pure Chemical, catalog number: 190-12865)
- 10x running buffer (see Recipes)
- 4x SDS sample buffer (see Recipes)
- Transfer buffer (see Recipes)
- 10x PBS (see Recipes)
- Blocking buffer (see Recipes)
Equipment
- Pipettes
- PCR thermal cycler (Thermo Fisher Scientific, model: Veriti 200)
- Refrigerated centrifuge (Hitachi, model: himac CF 15R)
- Vortex mixer
- Block incubator (You can use a water incubator instead of block incubator.)
- Wonder Shaker (NISSIN, model: NA-4X)
- Multi Shaker (Tokyo Rikakikai Co., Ltd., model: MMS-120H)
- Heat Sealer (Taiyo Electric Co., Ltd., model: HS-400)
- Trans-Blot® SD Semi-Dry Electrophoretic Transfer Cell (Bio-Rad Laboratories, catalog number: 170-3940)
- Fuji Medical Film Processor (FUJIFILM Medical, model: FPM100)
- X-ray film cassette
- EnSpireTM Alpha Plate Reader (PerkinElmer, model: 2390-00000)
Note: Equipment #5 to #11 are shown for your reference for SDS-PAGE and Western blotting.
Procedure
- In vitro transcription/translation of a transcription factor using AlphaScreen®
- Preparation of DNA template for in vitro transcription/translation
To attach a protein tag, in this case, “FLAG®-tag” to N-terminus of a target transcription factor, perform the two-step PCR method. All primers for making transcription templates are shown in Table 1.
Note: For details, please refer to articles published in Genes Cells (Nomoto and Tada, 2018a) and Methods Mol Biol (Nomoto and Tada, 2018b) which include all the procedures for in vitro transcription/translation.
Table 1. Primer sequences for transcription template PCR
- Prepare the following 1st PCR mix (total volume: 50 µl/sample) on ice:
5 µl of 10x PCR buffer for KOD-Plus-Neo
5 µl of 2 mM dNTPs
3 µl of 25 mM MgSO4
1 µl of KOD-Plus-Neo
1 µl of plasmid (1 ng/µl)
1 µl of 10 µM 1st-gene-specific-NF primer
1 µl of 10 µM 1st-gene-specific-NR primer
Add 33 µl of ultrapure water to make up the volume of the reaction to 50 µl
Note: You can use any plasmid containing cDNA region of the gene of interest. In this case, cDNA region cloned into pENTR/D-TOPO was used as a 1st PCR template. - Run the following 1st PCR
Step 1: 94 °C for 5 min
Step 2: 30 cycles of
a) 98 °C for 10 s
b) 55 °C for 30 s
c) 68 °C for 3 min (30-60 s/kb)
Step 3: 72 °C for 2 min
Step 4: 4 °C until agarose gel electrophoresis
Note: Temperature in this step (hybridization step) depends on primer sequences. You can change the temperature for this step according to normal PCR methods. It should be between 55 °C and 65 °C. - Run 2 µl of the 1st PCR sample on agarose gel (1.5% [w/v] in 1x TAE buffer) electrophoresis to check the quantity and size of PCR amplicon (Figure 1).
Note: If multiple PCR amplicons are detected in single PCR, gel extraction of a target band is recommended. - Prepare the following 2nd PCR mix (total volume: 50 µl/sample) on ice:
5 µl of 10x PCR buffer for KOD-Plus-Neo
5 µl of 2 mM dNTPs
3 µl of 25 mM MgSO4
1 µl of KOD-Plus-Neo
1 µl of 1st PCR sample
1 µl of 10 µM 2nd-T7-NF2 primer
1 µl of 100 nM 2nd-protein tag(FLAG)-NF1 primer
1 µl of 10 µM 2nd-3’ SU-NCR2 primer
1 µl of 100 nM 2nd-3’ SU-NR1 primer
Add 31 µl of ultrapure water to make up the volume of the reaction to 50 µl - Run the following 2nd PCR
Step 1: 98 °C for 1 min
Step 2: 10 cycles of
a) 98 °C for 10 s
b) 60 °C for 1 min
c) 68 °C for 3 min (30-60 s/kb)
Step 3: 30 cycles of
a) 98 °C for 10 s
b) 60 °C for 15 s
c) 68 °C for 3 min (30-60 s/kb)
Step 4: 72 °C for 2 min
Step 5: 4 °C until agarose gel electrophoresis - Run 2 µl of the 2nd PCR sample on agarose gel (1.5% [w/v] in 1x TAE buffer) electrophoresis to check the quantity and size of PCR amplicon (Figure 1).
Note: If multiple PCR amplicons are detected in single PCR, gel extraction of a target band is recommended. One transcription reaction requires 25 µl of purified 2nd PCR amplicon containing 300 to 600 ng DNA.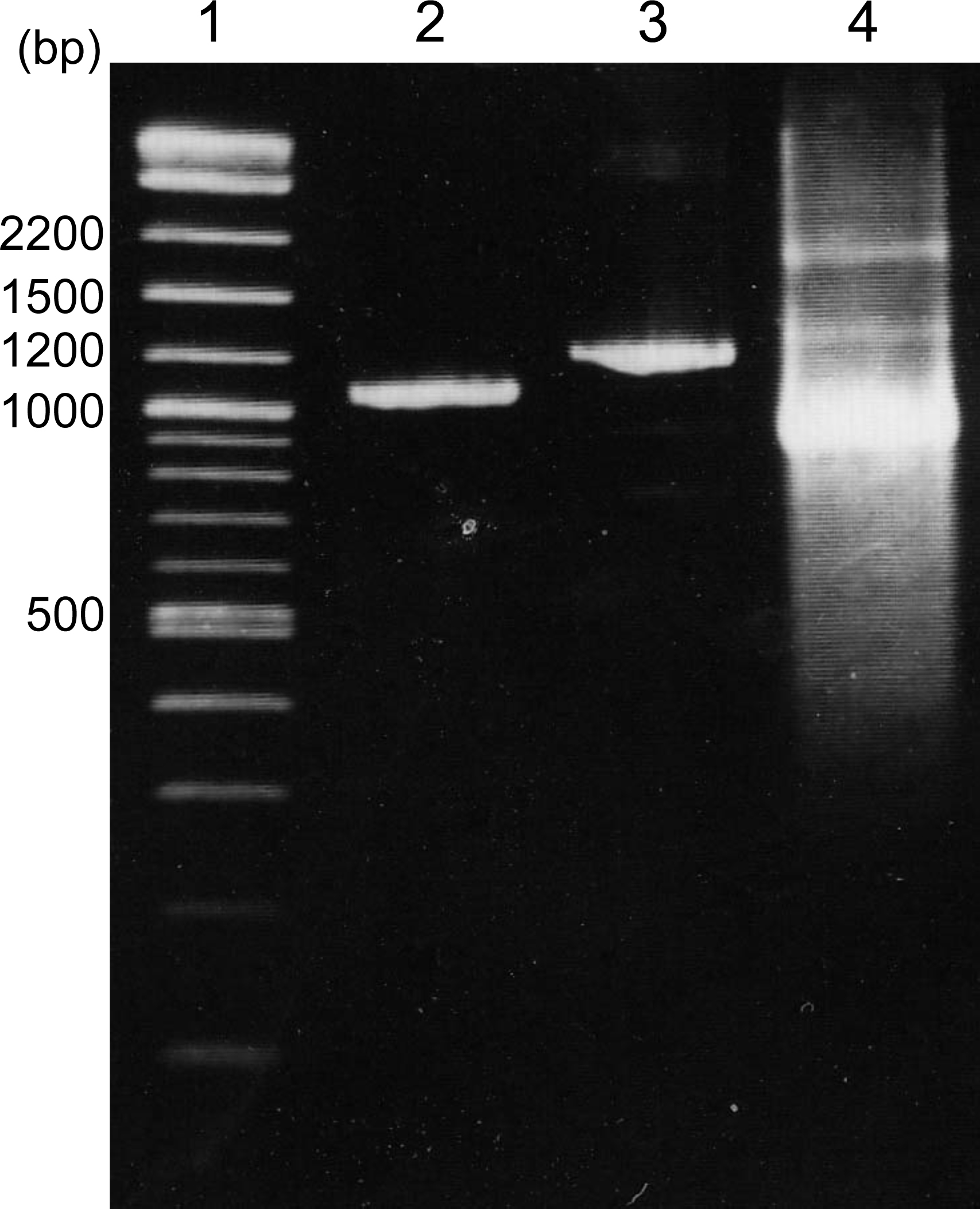
Figure 1. Agarose gel electrophoresis of products from the 1st PCR, 2nd PCR, and transcription products. Lane 1: DNA marker (2-log ladder), lane 2: 1st PCR product, lane 3: 2nd PCR product, lane 4: transcription reaction product. MYB30 coding region cloned into pENTR/D-TOPO vector is used as the template for 1st PCR. MYB30 coding region is 969 base pairs.
- Prepare the following 1st PCR mix (total volume: 50 µl/sample) on ice:
- In vitro transcription
Perform In vitro transcription/translation reaction using NUProtein kit following the manufacturer’s protocol.- Prepare the following transcriptional master mix (for one sample) on ice:
2.5 µl of 10x Transcription Buffer (provided in the kit)
1.25 µl of 0.1 M DTT (provided in the kit)
2.5 µl of 25 mM NTPs (provided in the kit)
1 µl of T7 RNA polymerase (provided in the kit)
Add 15.25 µl of RNase-free water
Note: If RNA degradation occurs, an RNase inhibitor should be added to the transcription mix. - Dispense 22.5 µl of the above transcription mix to each new tube (RNase-free tube recommended) and add 2.5 µl of 2nd PCR product carefully (Purification of 2nd PCR products is not necessarily required).
Note: NUProtein kit contains positive control DNA (2nd PCR amplicon) which can be used as a positive control for the in vitro transcription reaction. - Incubate the sample at 37 °C for 3 h in a block incubator.
Note: Incubation time depends on the type and length of mRNA, and may vary from 30 min to 3 h. Water incubator can also be used instead of block incubator. - Run 1 µl of the transcription reaction sample can be loaded on agarose gel (1.5% [w/v] in 1x TAE buffer) electrophoresis to check the quantity and quality of mRNA (Figure 1).
- Add 10 µl of 4 M ammonium acetate and 100 µl of 99.5 % ethanol per sample (total volume: 124 µl/sample.
- Mix the sample by gentle vortexing or tapping and incubate at -20 °C for 20 min after centrifugation for a few seconds.
- Centrifuge the sample at 17,700 x g for 20 min at 4 °C and discard the supernatant carefully.
- Repeat centrifugation for few seconds to remove the residual solution completely using a pipette.
- Dry the pellet by keeping the tube lid open for about 10 min.
- Add 70 µl of RNase-free water per sample and incubate at RT for about 15 min.
- Dissolve the pellet well by gentle tapping and centrifuging for a few seconds.
- Prepare the following transcriptional master mix (for one sample) on ice:
- In vitro protein synthesis
- Prepare the following translation reaction master mix (for one sample) into 1.5 ml tube on ice:
20 µl of wheat germ extract (WGE) (provided in the kit)
20 µl of amino acid mixture (AAM) (provided in the kit) - Mix the translation reaction master mix gently, and incubate at RT for 15 min.
Note: Preincubation of WGE and AAM may enhance the translation efficiency. - Add 40 µl of the translation reaction mix to 70 µl of prepared mRNA carefully (total volume: 110 µl/sample).
Note: Do not make bubbles during additions. Make AlphaScreen® negative control by adding 40 µl of the translation reaction mix to 70 µl of RNase-free water. - Incubate the sample at 16 °C for 10 h without agitation.
- Centrifuge the sample at 17,700 x g for 10 min at 4 °C and transfer the supernatant (approximately 118 µl) to a new tube.
- Mix 1 µl of the in vitro-synthesized protein, 4 µl of 4x SDS sample buffer, 1 µl of 2 M DTT, and 6 µl of water in a new tube.
Note: Remaining protein sample can be stored at -80 °C. We recommend flash-freezing the sample using liquid N2. Repeated freeze-thaw cycles decrease the activity of synthesized proteins. When using frozen protein for any assay, quickly thaw the sample by hand. - Heat protein sample at 70 °C for 20 min and perform SDS-PAGE in a 10% (w/v) polyacrylamide gel and confirm the synthesized target protein by western blotting with anti-DYKDDDDK tag antibody.
- Prepare the following translation reaction master mix (for one sample) into 1.5 ml tube on ice:
- Preparation of DNA template for in vitro transcription/translation
- AlphaScreen® assay
- Preparation of double-stranded DNA probes by annealing
For AlphaScreen®, detecting the interaction between protein and DNA, prepare biotinylated (target sample) and non-biotinylated (control for the detection) 50 bp DNA probes. For obtaining stable AlphaScreen® results, we recommend designing the DNA probes that contain putative DNA binding sequence of the target transcription factor in the central region (11 bp to 40 bp from 5’ end of the probe [Figure 2A]).- Mix 20 µl of 20 µM biotinylated 50 bp single-strand DNA (or 20 µl of 20 µM non-biotinylated 50 bp single strand DNA) and 20 µl of 20 µM complementary unmodified 50 bp single-strand DNA in 1.5 ml tube (total volume: 40 µl).
- Incubate the DNA mixture at 95 °C for 10 min, let it cool down until it reaches RT.
Note: Double-strand DNA probes can be stored at -20 °C. - Check annealed double-stranded DNA probes by polyacrylamide gel (12% w/v) electrophoresis (Figure 2B).
Note: At the same time prepare DNA probe for the negative control, which does not contain any putative DNA binding sequence of the target transcription factor. If required, also prepare the DNA probes that contain mutation in the target sequence.
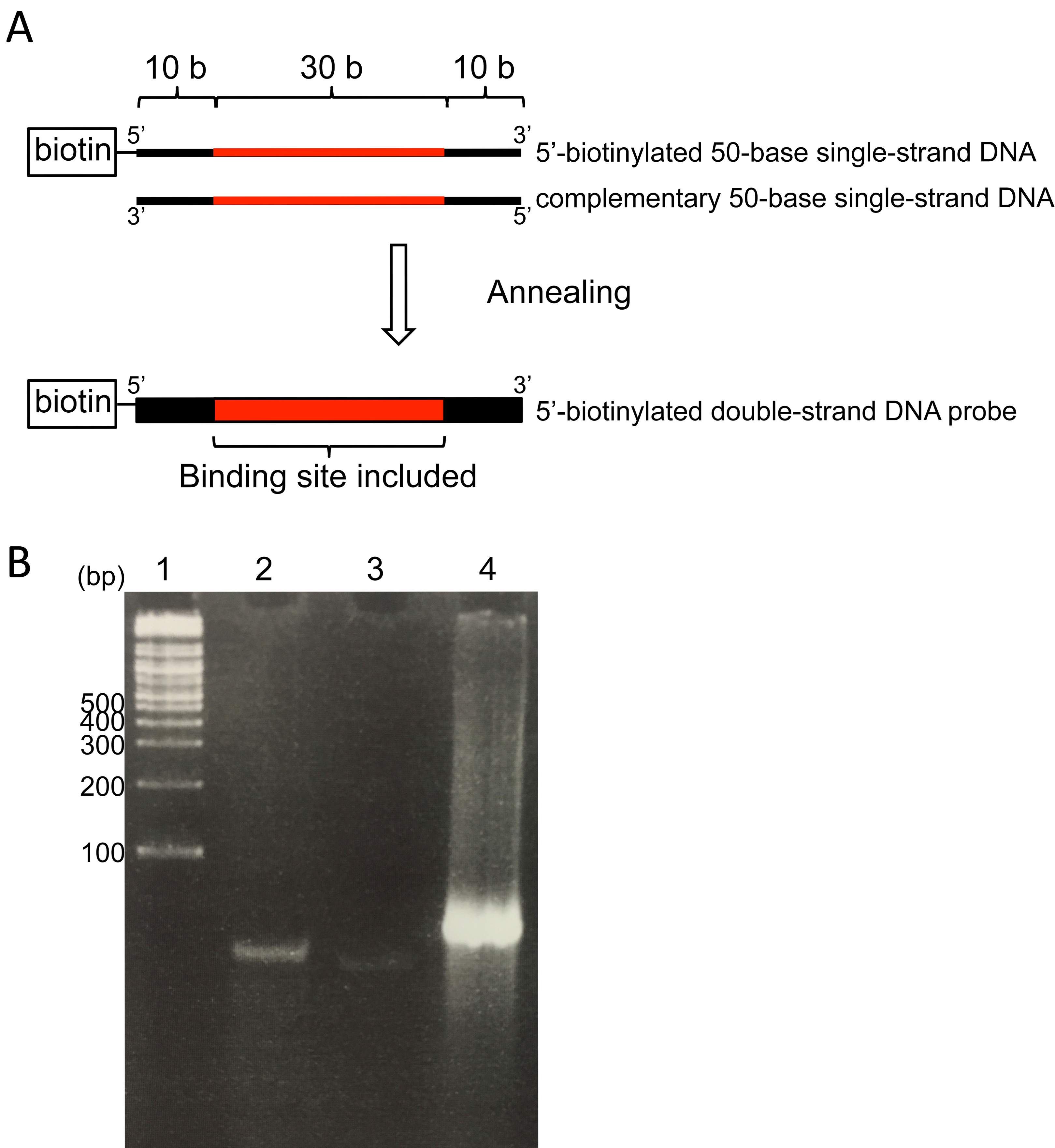
Figure 2. Schematic diagram of DNA probe for AlphaScreen ®. A. Schematic diagram of annealed double-strand DNA probe. Black lines and box indicate scaffold region (10 bases in length). Red lines and box indicate the region that contains putative DNA binding sequence of the target transcription factor. B. Polyacrylamide gel (10%) electrophoresis of DNA probes. Lane 1: DNA marker (2-log ladder), lane 2: 5’-biotinylated 50-base single-strand DNA, lane 3: Complementary 50-base single-strand DNA, lane 4: Annealed double-strand DNA. The polyacrylamide gel was stained with ethidium bromide after electrophoresis. - AlphaScreen® assay with FLAG (M2) Detection kit
- Prepare the following protein-DNA binding mix (total volume: 13 µl/sample):
2.5 µl of 10x control buffer [provided in FLAG (M2) Detection kit]
2.5 µl of 0.1% (w/v) Tween® 20
2.5 µl of 1% (w/v) BSA
1 µl of 1 ng/µl dAdT
Add 4.5 µl of ultrapure water.
Note: Depending on the target transcription factor, dGdC should be used instead of dAdT. - Dispense 13.0 µl of the protein-DNA binding mix to the 384-well plate and add 2 µl of 625 nM of double-stranded DNA probe and 4-fold diluted FLAG-tagged target transcription factor protein.
Note: Also add 2 µl of 4-fold diluted negative control protein. - Incubate protein-DNA binding mix at RT for 1 h.
Note: Dilution range of synthesized protein depends on the feature of target transcription factor. Two to ten times dilution can be used for this assay. Incubation under dark condition is not necessary for this step. - Add 4 µl of 40-fold diluted (with ultrapure water) acceptor beads and incubate at RT for 1 h.
Note: Incubation under dark condition is not necessary for this step. - Add 4 µl of 40-fold diluted (with ultrapure water) donor beads and incubate at RT for 1 h to 12 h in the dark (total volume: 25 µl).
Note: To ensure that the plate is in the dark, wrap it with aluminum foil and remove it before keeping it in the Alpha plate reader. - Detect signal by EnSpireTM Alpha Plate Reader. After the excitation at 680 nm, the emission wavelengths between 520 and 620 nm are measured as AlphaScreen® unit.
- Prepare the following protein-DNA binding mix (total volume: 13 µl/sample):
- Preparation of double-stranded DNA probes by annealing
Data analysis
- Divide the signal intensity from the mix of biotinylated DNA probe by the signal intensity from the mix of non-biotinylated DNA probe. This signal can be used as “Relative signal intensity” for plotting a graph.
Note: The signal intensity from negative controls (without target protein) usually shows very low signal. - Data from 3 technical replicates are combined for the statistical analysis, and the same experiment using different batches of proteins is repeated at least 3 times with similar results.
Note: One example of AlphaScreen® result can be found in Figures 4B and 4C in Mabuchi et al., 2018. As an alternative way of representing data, you may also be able to show all the signals, including one from negative controls.
Notes
- In this protocol, we used protein that is synthesized by our in vitro transcription/translation system. However, one may use protein that is synthesized by other lab protocols (e.g., using bacteria, yeast, and any other systems).
- In this protocol, we use FLAG®-tagged protein. However, one may use other protein tags, (e.g., GST, 6-Histidine, c-myc, and HA) as long as detection kits for “AlphaScreen® Fusion Tag” are available from PerkinElmer (see also web site: www.perkinelmer.com/alphascreen).
Recipes
- 100x TAE buffer
400 mM Tris-HCl
400 mM glacial acetic aid
10 mM EDTA, pH 8.0 - 10x loading dye
50% (w/v) glycerol
0.9% (w/v) SDS
10 mM EDTA, pH 8.0
0.05% (w/v) bromophenol blue
0.05% (w/v) xylene cyanol - 4x SDS sample buffer
200 mM Tris-HCl, pH 6.8
40% (w/v) glycerol
8% (w/v) SDS
0.08% (w/v) bromophenol blue - 10x running buffer
250 mM Tris
1.92 M glycine
1% (w/v) SDS - Transfer buffer
192 mM glycine
20% (w/v) methanol
25 mM Tris - 10x PBS
1.37 M NaCl
81 mM Na2HPO4
26.8 mM KCl
14.7 mM KH2PO4 - Blocking buffer
1x PBS
1% (w/v) non-fat skimmed milk
0.1% (w/v) Tween® 20
Acknowledgments
This work was supported by Ministry of Education, Culture, Sports, Science, and Technology Grant-in-Aid for Scientific Research on Innovative Areas 26113508 to H.T., JSPS KAKENHI (No. 13J10800) and Program for Leading Graduate Schools “Integrative Graduate Education and Research in Green Natural Sciences”, MEXT, Japan to M.N., Grant-in-Aid for Scientific Research on Innovative Areas (No. 23120520 and 25120718) from the Ministry of Education, Culture, Sports, Science and Technology (Japan), JSPS KAKENHI (No. 15H05956) to Y.T. In vitro protein synthesis was adapted from Nomoto and Tada, 2018a and 2018b and Mabuchi et al., 2018. AlphaScreen for protein-DNA interaction was adapted from Mabuchi et al., 2018.
Competing interests
The authors declare no conflicts of interest or competing interests.
References
- Chalfie. M., Tu, Y., Euskirchen, G., Ward, W. W. and Prasher, D. C. (1994). Green fluorescent protein as a marker for gene expression. Science 263(5148): 802-805.
- Mabuchi, K., Maki, H., Itaya, T., Suzuki, T., Nomoto, M., Sakaoka, S., Morikami, A., Higashiyama, T., Tada, Y., Busch, W. and Tsukagoshi, H. (2018). MYB30 links ROS signaling, root cell elongation, and plant immune responses. Proc Natl Acad Sci U S A 115(20): E4710-E4719.
- Nomoto, M. and Tada, Y. (2018a). Cloning-free template DNA preparation for cell-free protein synthesis via two-step PCR using versatile primer designs with short 3'-UTR. Genes Cells 23(1): 46-53.
- Nomoto, M. and Tada, Y. (2018b). Cell-free protein synthesis of plant transcription factors. Methods MolBiol 1830: 337-349.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Nomoto, M., Tada, Y. and Tsukagoshi, H. (2019). In vitro Protein-DNA Binding Assay (AlphaScreen® Technology). Bio-protocol 9(3): e3155. DOI: 10.21769/BioProtoc.3155.
Category
Molecular Biology > Protein > Detection
Plant Science > Plant molecular biology > DNA > Gene expression
Molecular Biology > DNA > DNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.