- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

flySAM Transgenic CRISPRa System Manual

Published: Vol 9, Iss 2, Jan 20, 2019 DOI: 10.21769/BioProtoc.3147 Views: 9619
Reviewed by: Gal HaimovichAbhijit KaleAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Powerful and general methods that can enhance gene expression are useful to systematically study gene function. To date, compared with the methods in generating loss-of-function mutants, methods to achieve gain-of-function are limited. The entire field in Drosophila has relied heavily on the Gal4/UAS:cDNA overexpression system developed over two decades ago. It is laborious and expensive to clone the coding DNA sequence (CDS) of a gene, especially those of large size. In addition, side effects of this method are often observed because of the ectopic expression. Also, simultaneous activation of two genes with the traditional method is often time-consuming, and few are achievable for three or more genes. In this protocol, we describe how to build an effective and convenient targeting activator system, flySAM, to activate endogenous genes in Drosophila melanogaster based on the structure-guided engineering of CRISPR-Cas9 complex.
Keywords: CRISPR-Cas nucleaseBackground
Currently, most of the genes in Drosophila can be knocked down or knocked out through powerful genetic tools and several loss-of-function (LOF) resources have been established based on these tools (Ren et al., 2013; Qiao et al., 2018). A similar gain-of-function (GOF) resource is valuable to study gene function as it can be a complement to LOF. However, we do not have a genome-wide GOF resource until now mainly because of technical difficulties. The traditional gene overexpression system in Drosophila is the Gal4/UAS:cDNA system. It needs to clone the coding DNA sequence (CDS) of a gene downstream of the upstream activation sequence (UAS) which can be controlled by Gal4 transcription factor. It is laborious, expensive and sometimes unachievable. Therefore, an easier way to realize GOF is urgently needed. The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system is an effective genome-editing tool. The transcriptional activation system in Drosophila based on the CRISPR/Cas9 technique, termed “CRISPR transcriptional activation (CRISPRa) system”, has been developed in recent years (Konermann et al., 2015; Lin et al., 2015; Ewen-Campen et al., 2017). Cas9 has HNH and RuvC nuclease domains which are responsible for the cutting of complementary and non-complementary DNA strand, respectively. Through introducing single amino acid mutation within HNH and RuvC domains of Cas9 (dCas9), nuclease activity of Cas9 is abolished without affecting its DNA binding ability (Dominguez et al., 2016). The CRISPR transcriptional activation CRISPRa system is generated by fusing several activation domains to dCas9 and can be recruited to the upstream of the Transcription Start Site (TSS) through single-guide RNAs (sgRNAs). The specificity of CRISPRa system is controlled by 20 bp guide sequence in sgRNA, thus one can simply use the 20 bp guide sequence against different targets. Previously, VPR-based CRISPRa system has been built in Drosophila in vivo and in vitro (Lin et al., 2015; Ewen-Campen et al., 2017). In this system, three activation domains, VP64, P65, Rta (VPR), tandemly following the dCas9. However, due to low efficiency, this system relies on two sgRNAs to boost gene transcription, which not only increased the complicacy and cost of the cloning procedure, but also increased toxicity and the chances of off-target effects. In addition, although it works both in vitro and in vivo, it often produces weaker phenotypes than the traditional Gal4/UAS:cDNA system.
To improve the efficiency, reduce the toxicity, and streamline the cloning procedure, this protocol introduces how to construct transcriptional activation lines based on flySAM system. In this system, the dCas9-VP64 and MCP-p65-HSF1 is controlled by 10x UAS and sgRNA2.0 which is driven by U6:2 promoter (Jia et al., 2018). Unlike the previous methods, single sgRNA can successfully activate gene transcription. In addition, to study the intrinsic and extrinsic roles of genes with redundant functions or involved in protein complex, we can easily clone multiple sgRNAs to target different genes.
Materials and Reagents
- 1.5 ml MaxyClear Microtubes (Axygen, catalog number: MCT-150-C)
- 0.2 ml Polypropylene PCR Tube (Axygen, catalog number: PCR-0208-C)
- Pipette tips (Corning, Axygen®)
- 0.22 μm Millipore filters (Millipore, catalog number: R7MA69670)
- Fly stocks (All Drosophila strains are stored in Tsinghua Fly Center)
- y sc v nanos-integrase; attP40 (TB00016)
- y sc v nanos-integrase;; attP2 (TB00018)
- y sc v (TB00077)
- y sc v; Gla Bc/CyO (TB00023)
- y sc v;; Dr,e/TM3,Sb (TB00139)
- Trans5α Chemically Competent Cell (Transgen Biotech, catalog number: CD201-01)
- flySAM (Ampicillin resistant, plasmid map is shown in Figure 1)
- BbsI (New England Biolabs, catalog number: R0539L)
- NheI (New England Biolabs, catalog number: R3131L)
- SpeI (New England Biolabs, catalog number: R3133L)
- T4 DNA ligase (New England Biolabs, catalog number: M0202L)
- Alkaline phosphatase, calf intestinal (CIP) (New England Biolabs, catalog number: M0290S)
- AxyPrep Plasmid Miniprep Kit (AXYGEN, catalog number: AP-MN-P-250)
- AxyPrep DNA Gel Extraction Kit (AXYGEN, catalog number: AP-GX-250)
- PurePlasmid Mini Kit (CWBiotech, catalog number: CW0500M)
- Forward Primer 1: 5’-TCAACAAACGaacaataggacac-3’
- Reverse Primer 1: 5’-aAAAAAGCACCGACTCGGTG-3’
- Forward Primer 2: 5’-ACATCAGGAAAGAGCAGTTGAG-3’
- Reverse Primer 2: 5’-TTGCTCACCTGTGATTGCTCC-3’
- 50x TAE (Double Helix, catalog number: P0309A)
- Agarose (Invitrogen, catalog number: 0000602315)
- LB powder (BD, catalog number: 2171199)
- KCl (AMRESCO, catalog number: 7447-40-7)
- Tris-HCl (AMRESCO, catalog number: 1185-53-1)
- EDTA (AMRESCO, catalog number: 60-00-4)
- NaCl (AMRESCO, catalog number: 7647-14-5)
- Ampicillin (Solarbio, catalog number: A8180)
- Ethidium bromide (Sigma, catalog number: E8751)
- GoTag Green Master Mix, 2x (Promega, catalog number: 000179370)
- Sodium phosphate buffer (see Recipes)
- 10x Injection buffer (see Recipes)
- 10x Annealing buffer (see Recipes)
- LB (Luria Bertani) medium (see Recipes)
- 1x TAE (see Recipes)
- 1.5% agarose gel (see Recipes)
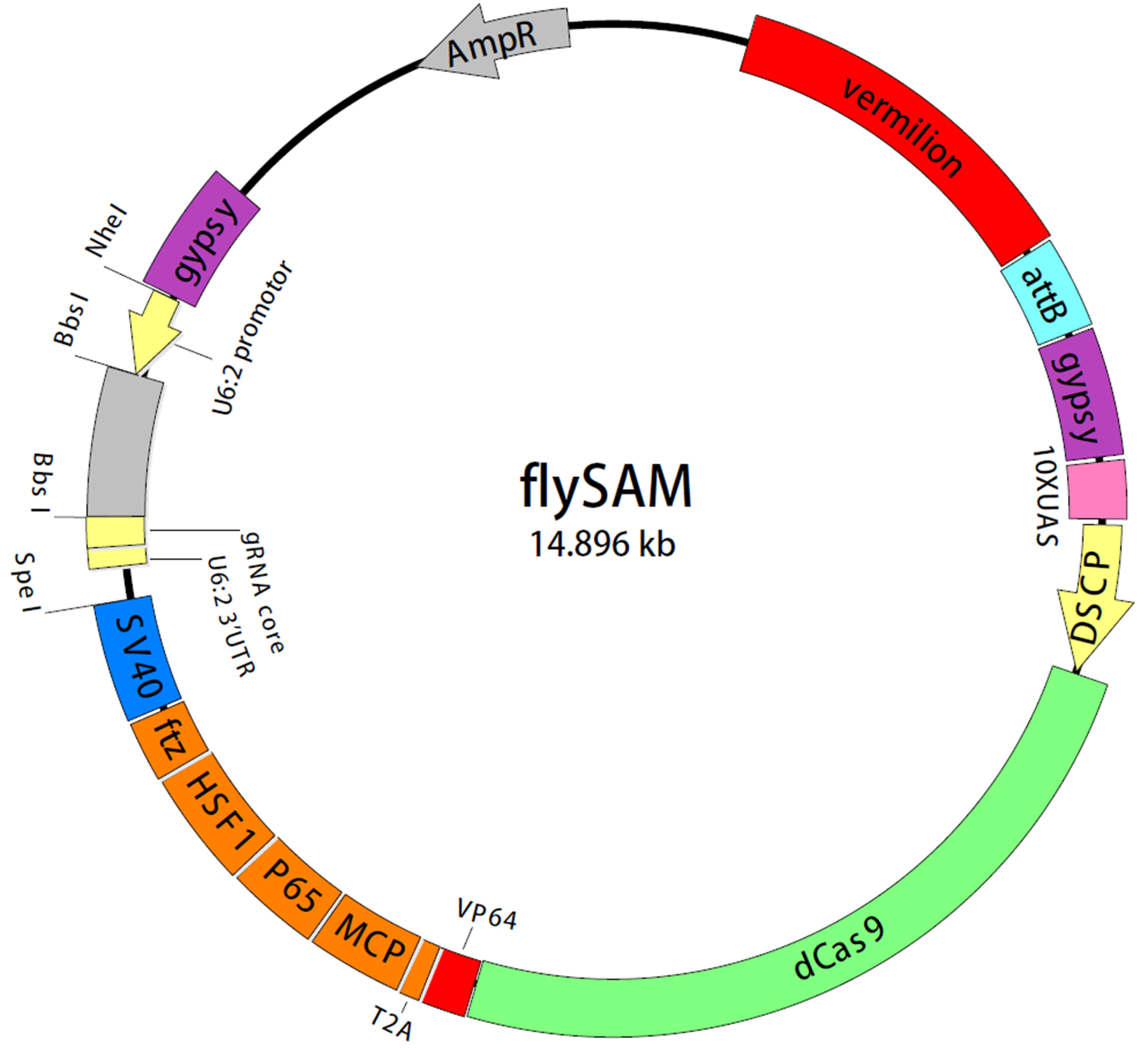
Figure 1. The map of flySAM construct. The dCas9 is under the control of 10 x UAS and Drosophila synthetic core promoter (DSCP) and followed by VP64. dCas9-VP64 and MCP-P65-HSF1 are mediated and separated by T2A self-cleaving peptide. ftz intron is located between CRISPRa components and SV40 polyA tail. The grey block between two BbsI sites is a marker to detect whether the sgRNA is under the control of U6:2 regulatory sequence. Two gypsy elements which can boost gene expression are used to flank the CRISPRa components. The attB sequence and a vermillion+ marker are also included in this plasmid.
Equipment
- Pipette (Eppendorf)
- Microwave (SANYO, model: EM-2509EB1)
- Autoclave (SANYO, model: MLS-3780)
- PCR Thermal Cycler (Eppendorf, Mastercycler nexus GSX1)
- Microcentrifuge (Eppendorf, model: 5417R)
- DNA electrophoresis apparatus (Bio-Rad)
- NanoDrop 2000 Spectrophotometer (Thermo Scientific)
Procedure
- Oligo designation
- Select 20 nt length sgRNA upstream of NGG within -100 to -400 of the target gene’s TSS
The following parameters should be considered to ensure the quality of sgRNAs:- TTTT sequence should not be in 20 nt sgRNA sequence because it is the transcriptional termination signal.
- To avoid off-target, ≥ 18 nt of the targeting sequence of sgRNA should not match to other genomic loci.
- The optimal sgRNA normally carries high GC content, usually over 50%.
- To synthesize the 20 nt DNA sequence, the overhang sequence 5’-TTCG-3’ from BbsI should be added into forward primer and 5’-aaac-3’ to the reverse complement primer.
e.g., 5’-NNNAGCAATCGACATGCAAGCGGCTCGGAGCCAGGGCACCTGCNNN-3’
(Underlined AGG is the PAM sequence)
Forward: 5’-ttcgCATGCAAGCGGCTCGGAGCC-3’
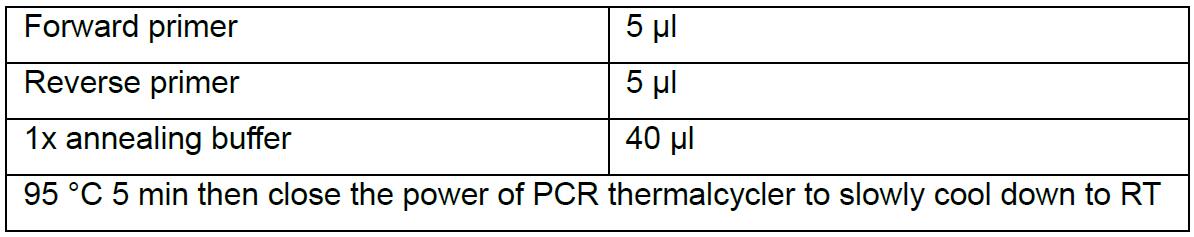
Reverse: 5’-aaacGGCTCCGAGCCGCTTGCATG-3’ - Resuspend oligos with ddH2O to final 20 µM and set up the annealing reaction:
- Select 20 nt length sgRNA upstream of NGG within -100 to -400 of the target gene’s TSS
- Backbone preparation
- BbsI is used to linearize flySAM vector.
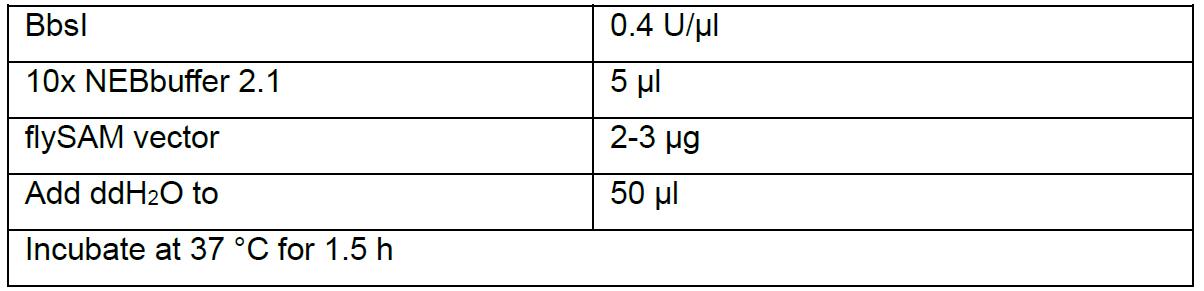
- Run 1.5% agarose gel. Two bands should be visible (Figure 2), one is about 700 bp (this spacer can help to evaluate the cutting efficiency and later on to select correct clones by PCR, as the size of PCR product from correct clone is different from incorrect clone), another is about 14 kb. Cut and collect the 14 kb band and extract the backbone by AxyPrep DNA Gel Extraction kit following the standard protocol. Elute DNA with 50 µl ddH2O, measure the DNA concentration with NanoDrop 2000, should be around 50 ng/µl.
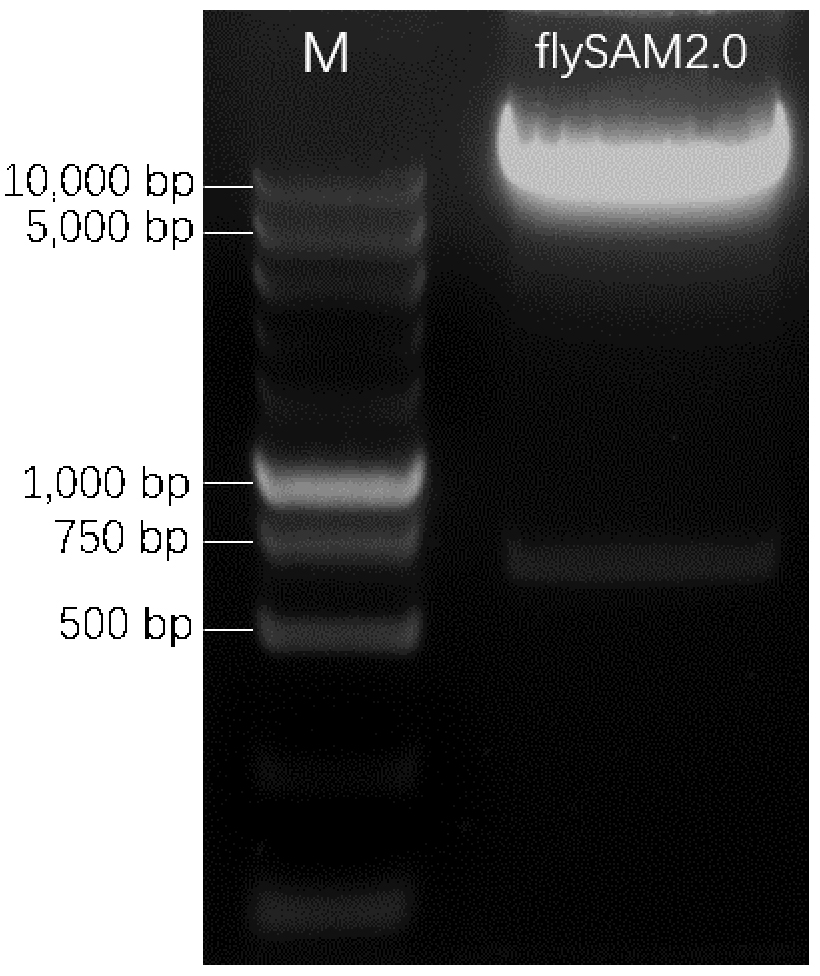
Figure 2. An example gel of BbsI-digested flySAM2.0. M is DNA marker. flySAM2.0 is the BbsI-digested product.
- BbsI is used to linearize flySAM vector.
- Ligation
- Ligate the annealed sgRNA product with linearized flySAM vector
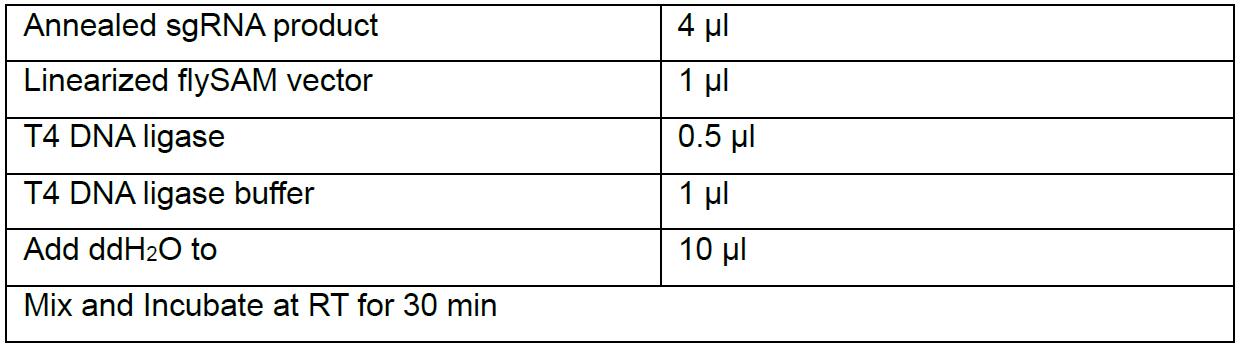
Transform 2-5 µl of the ligation product into 10 µl Trans5α Chemically Competent Cells following the standard protocol, and spread on an LB-agar plate containing 100 µg/ml ampicillin. Incubate these plates at 37 °C overnight in an inverted state. - Use the following primers to perform colony PCR:
Forward Primer 1: 5’-TCAACAAACGaacaataggacac-3’
Reverse Primer 1: 5’-aAAAAAGCACCGACTCGGTG-3’
Thermal cycling condition is: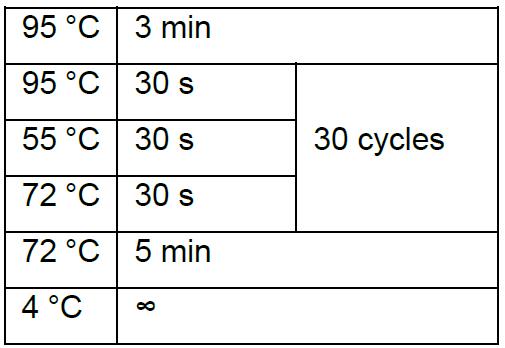
The size of PCR product from correct clone is about 300 bp, whereas either no band or 1,000 bp band is incorrect. - Inoculate correct clone in LB medium with 100 µg/ml ampicillin.
- Culture bacteria and miniprep plasmid by using AxyPrep Plasmid Miniprep Kit.
- Ligate the annealed sgRNA product with linearized flySAM vector
- Sanger sequencing to further confirm the insertion
Confirm flySAM-sgRNA plasmid by Sanger sequencing using below primer:
Forward Primer 1: 5’-TCAACAAACGaacaataggacac-3’
Notes:- If only one sgRNA needs to be cloned, the next step is to go directly to Procedure J.
- (Optional) If two or more genes need to be activated, in other words, two or more sgRNAs need to be cloned, the next procedures can be performed after Procedure D:
- Fragment preparation
- Double digest flySAM (one sgRNA has been cloned into it) by NheI and SpeI.
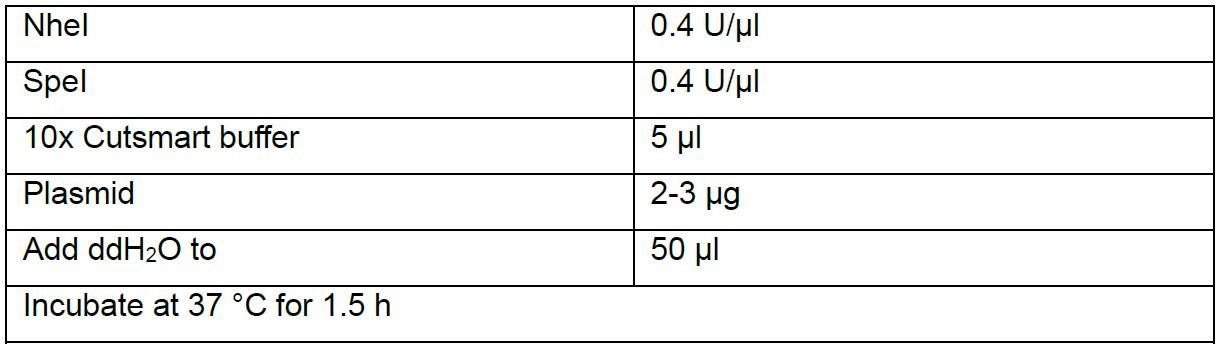
- Run agarose gel. Two bands should be visible, one is about 1 kb, another is about 14 kb. Cut and collect the 1 kb band and extract the fragment by AxyPrep DNA Gel Extraction kit following the standard protocol. Elute DNA with 50 µl ddH2O, measure the DNA concentration by NanoDrop 2000, should be around 40 ng/µl.
- Double digest flySAM (one sgRNA has been cloned into it) by NheI and SpeI.
- Backbone preparation
- Digest flySAM (another sgRNA which is different with the one in Procedure E has been cloned into it) by NheI or SpeI.
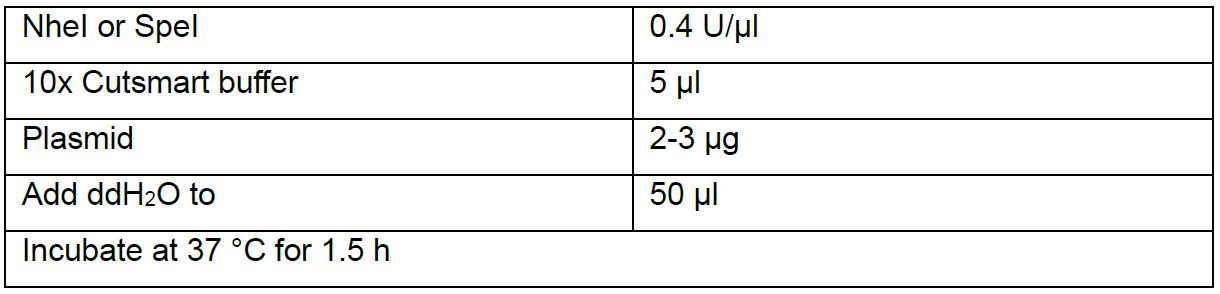
- Add 0.2 µl CIP and incubate at 37 °C for 8 min.
- Purify the digested product by AxyPrep DNA Gel Extraction kit.
- Digest flySAM (another sgRNA which is different with the one in Procedure E has been cloned into it) by NheI or SpeI.
- Ligation and transformation
Ligate the fragment from Procedure E to backbone from Procedure F. The procedure is similar to Procedure C. - Colony identification
- The following primers are used for colony PCR:
Forward Primer 2: 5’-ACATCAGGAAAGAGCAGTTGAG-3’
Reverse Primer 2: 5’-TTGCTCACCTGTGATTGCTCC-3’
Thermal cycling condition is: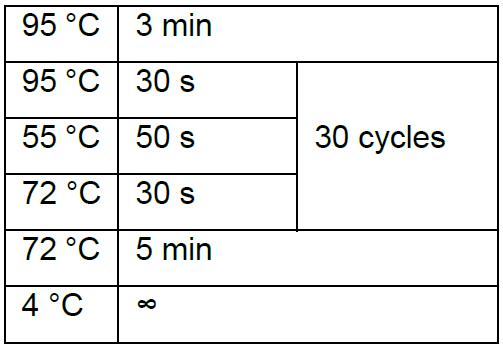
The correct clone is about 800 bp. - Inoculate the correct clone in LB medium with 100 µg/ml ampicillin.
- Culture bacteria and miniprep plasmid by using AxyPrep Plasmid Miniprep Kit.
- The following primers are used for colony PCR:
- Further confirm the correct clone by restriction enzymes digestion
- Double digest the plasmid by NheI and SpeI.
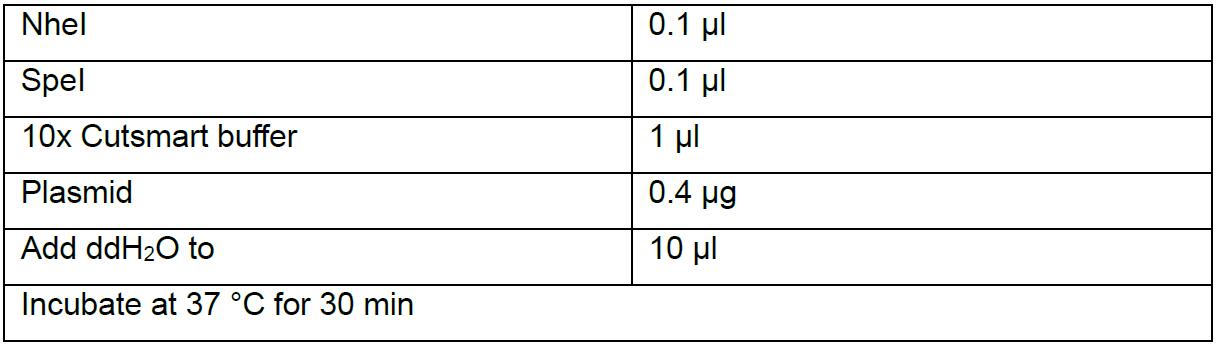
- Run agarose gel, one band should be about 2 kb (if two sgRNAs together), another is about 13 kb if the plasmid is right.
- Double digest the plasmid by NheI and SpeI.
- Purification and microinjection
- For microinjection, the plasmid (~10 mg) should be purified. PurePlasmid Mini Kit is used according to the producer’s specifications. The plasmid DNA is finally eluted in 70 µl of 1x Injection Buffer. The appropriate concentration of each sample is 100 ng/µl-200 ng/µl, which can be determined by NanoDrop 2000.
- Prepare the injected embryos and inject the transgene vector into the embryos of tool flies as standard procedure (Chromosome II y sc v nanos-integrase; attP40. Chromosome III y sc v nanos-integrase;; attP2) (Ni et al., 2011).
- The injected embryos should be kept at 25 °C and 60% humidity to adulthood (G0).
- Selection for transgenic fly
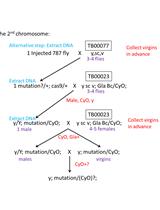
Both ♂ G0 and ♀ G0 should be crossed to opposite sex of y sc v;;. Since the flySAM transgenes carry a copy of vermillion (v+), which rescues the mutant vermillion (v) allele inherited from the injection stock. Thus transformants will have a dark red wild type (v+) eye color and the v+ G1 should be picked (Figure 3).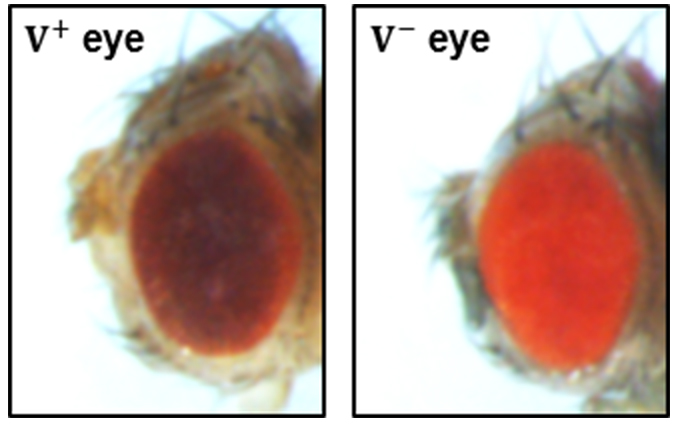
Figure 3. Eye color screening marker. The transformants have a dark red wild type (v+) eye color (left) while flies that haven’t been genetically modified have a light red (v-) eye color (right).- For G1♂ transformants from ♂ G0 follow these sequential steps
- Cross the siblings to the virgin ♀ of y sc v;; Dr, e/TM3, sb (for 3rd chromosome insertions) or y sc v; Gla Bc/CyO (for 2nd chromosome insertions).
- Collect ♂ and virgin ♀ siblings that have v+ (wild type) eye color and CyO or TM3 and cross them to each other.
- Keep the homozygous flies (keep the stock as balanced heterozygotes in case the homozygous flies are unhealthy).
- For G1♂ transformants from ♀ G0 follow these sequential steps
- Cross to virgin ♀ of y sc v;;
- Cross the v+ ♂ to the virgin ♀ of y sc v;; Dr, e/TM3, sb (for 3rd chromosome insertions) or y sc v; Gla Bc/CyO (for 2nd chromosome insertions).
- Collect ♂ and virgin ♀ siblings that have v+ (wild type) eye color and CyO or TM3 and cross them to each other.
- Keep the homozygous flies (keep the stock as balanced heterozygotes in case the homozygous flies are unhealthy).
- For ♀ transformants from ♂ or ♀ G0 follow these sequential steps
- Cross to ♂ of y sc v;;
- Cross the v+ ♂ to the virgin ♀ of y sc v;;
- Cross the v+ ♂ to the virgin ♀ of y sc v;; Dr, e/TM3, sb (for 3rd chromosome insertions) or y sc v; Gla Bc/CyO (for 2nd chromosome insertions).
- Collect ♂ and virgin ♀ siblings that have v+ (wild type) eye color and CyO or TM3 and cross them to each other.
- Keep the homozygous flies (keep the stock as balanced heterozygotes in case the homozygous flies are unhealthy).
- For G1♂ transformants from ♂ G0 follow these sequential steps
Data analysis
If we want to activate two or more genes, the primer used for DNA sequencing in Procedure D cannot be used to sequence because two U6:2 promoters are in one vector. Colony identification and enzymes digestion are more efficient to identify the right clone.
Recipes
- 0.2 M Sodium phosphate buffer (pH 6.8) (stock solution)
51 ml 0.2 M NaH2PO4
49 ml 0.2 M Na2HPO4
Store at room temperature- 0.2 M Na2HPO4
Dissolve 71.6 g Na2HPO4•12H2O in 1 L of ddH2O, mix thoroughly
Store at room temperature - 0.2 M NaH2PO4
Dissolve 31.2 g Na2HPO2•2H2O in 1 L of ddH2O, mix thoroughly
Store at room temperature
- 0.2 M Na2HPO4
- 10x Injection buffer
1 mM sodium phosphate buffer (pH 6.8)
50 mM KCl
Prepare in sterile distilled water and filter through 0.22 μm Millipore filters
It can be stored at -20 °C for one year - 10x Annealing buffer
100 mM Tris-HCl (pH 7.5)
10 mM EDTA
1 M NaCl
It can be stored at room temperature for one year - LB (Luria Bertani) medium
Dissolve 25 g of the powder in 1 L of ddH2O, mix thoroughly
Autoclave at 121 °C for 15 min - 1x TAE
20 ml 50x TAE
980 ml ddH2O
Store at room temperature - 1.5% agarose gel
1.5 g agarose
100 ml 1x TAE
Heat solution to dissolve agarose in microwave
Add ethidium bromide to a final concentration of 0.2 μg/ml
Acknowledgments
This work was supported by NIH Grants R01GM084947 and R24OD021997 and the NIH’s Ruth L. Kirschstein National Research Service Award F32GM113395 from the NIH General Medical Sciences Division. This work was supported by the National Key Technology Research and Development Program of the Ministry of Science and Technology of the People's Republic of China (2015BAI09B03, 2016YFE0113700), and the National Natural Science Foundation of China (31571320).
Competing interests
The authors do not have any possible conflicts of interest.
References
- Qiao, H. H., Wang, F., Xu, R. G., Sun, J., Zhu, R., Mao, D., Ren, X., Wang, X., Jia, Y., Peng, P., Shen, D., Liu, L. P., Chang, Z., Wang, G., Li, S., Ji, J. Y., Liu, Q. and Ni, J. Q. (2018). An efficient and multiple target transgenic RNAi technique with low toxicity in Drosophila. Nat Commun 9(1): 4160.
- Ren, X., Sun, J., Housden, B. E., Hu, Y., Roesel, C., Lin, S., Liu, L. P., Yang, Z., Mao, D., Sun, L., Wu, Q., Ji, J. Y., Xi, J., Mohr, S. E., Xu, J., Perrimon, N. and Ni, J. Q. (2013). Optimized gene editing technology for Drosophila melanogaster using germ line-specific Cas9. Proc Natl Acad Sci U S A 110(47): 19012-19017.
- Dominguez, A. A., Lim, W. A. and Qi, L. S. (2016). Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol 17(1): 5-15.
- Ewen-Campen, B., Yang-Zhou, D., Fernandes, V. R., Gonzalez, D. P., Liu, L. P., Tao, R., Ren, X., Sun, J., Hu, Y., Zirin, J., Mohr, S. E., Ni, J. Q. and Perrimon, N. (2017). Optimized strategy for in vivo Cas9-activation in Drosophila. Proc Natl Acad Sci U S A 114(35): 9409-9414.
- Jia, Y., Xu, R. G., Ren, X., Ewen-Campen, B., Rajakumar, R., Zirin, J., Yang-Zhou, D., Zhu, R., Wang, F., Mao, D., Peng, P., Qiao, H. H., Wang, X., Liu, L. P., Xu, B., Ji, J. Y., Liu, Q., Sun, J., Perrimon, N. and Ni, J. Q. (2018). Next-generation CRISPR/Cas9 transcriptional activation in Drosophila using flySAM. Proc Natl Acad Sci U S A 115(18): 4719-4724.
- Konermann, S., Brigham, M. D., Trevino, A. E., Joung, J., Abudayyeh, O. O., Barcena, C., Hsu, P. D., Habib, N., Gootenberg, J. S., Nishimasu, H., Nureki, O. and Zhang, F. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517(7536): 583-588.
- Lin, S., Ewen-Campen, B., Ni, X., Housden, B. E. and Perrimon, N. (2015). In vivo transcriptional activation using CRISPR/Cas9 in Drosophila. Genetics 201(2): 433-442.
- Ni, J. Q., Zhou, R., Czech, B., Liu, L. P., Holderbaum, L., Yang-Zhou, D., Shim, H. S., Tao, R., Handler, D., Karpowicz, P., Binari, R., Booker, M., Brennecke, J., Perkins, L. A., Hannon, G. J. and Perrimon, N. (2011). A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat Methods 8(5): 405-407.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Jia, Y., Shen, D., Wang, X., Sun, J., Peng, P., Xu, R., Xu, B. and Ni, J. (2019). flySAM Transgenic CRISPRa System Manual. Bio-protocol 9(2): e3147. DOI: 10.21769/BioProtoc.3147.
Category
Molecular Biology > DNA > Gene expression
Molecular Biology > DNA > DNA modification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.