- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Generation of CRISPR-Cas9 Complexes with Covalently Bound Repair Templates for Genome Editing in Mammalian Cells
(*contributed equally to this work) Published: Vol 9, Iss 1, Jan 5, 2019 DOI: 10.21769/BioProtoc.3136 Views: 11925
Reviewed by: Gal HaimovichAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The CRISPR-Cas9 system is a powerful genome-editing tool that promises application for gene editing therapies. The Cas9 nuclease is directed to the DNA by a programmable single guide (sg)RNA, and introduces a site-specific double-stranded break (DSB). In mammalian cells, DSBs are either repaired by non-homologous end joining (NHEJ), generating small insertion/deletion (indel) mutations, or by homology-directed repair (HDR). If ectopic donor templates are provided, the latter mechanism allows editing with single-nucleotide precision. The preference of mammalian cells to repair DSBs by NHEJ rather than HDR, however, limits the potential of CRISPR-Cas9 for applications where precise editing is needed. To enhance the efficiency of DSB repair by HDR from donor templates, we recently engineered a CRISPR-Cas9 system where the template DNA is bound to the Cas9 enzyme. In short, single-stranded oligonucleotides were labeled with O6-benzylguanine (BG), and covalently linked to a Cas9-SNAP-tag fusion protein to form a ribonucleoprotein-DNA (RNPD) complex consisting of the Cas9 nuclease, the sgRNA, and the repair template. Here, we provide a detailed protocol how to generate O6-benzylguanine (BG)-linked DNA repair templates, produce recombinant Cas9-SNAP-tag fusion proteins, in vitro transcribe single guide RNAs, and transfect RNPDs into various mammalian cells.
Keywords: CRISPR-Cas9Background
The CRISPR-Cas9 system efficiently induces site-directed DSBs, which are repaired by cell-autonomous mechanisms. Since mammalian cells predominantly repair DSBs by NHEJ, the CRISPR-Cas9 system mainly causes indel mutations at targeted loci. For many applications, precise repair via HDR from template DNA is however desired. Hence, several attempts have been made to increase the efficiency of accurate DSB repair, including biochemical alteration the repair pathways (Chu et al., 2015; Maruyama et al., 2015; Yu et al., 2015; Song et al., 2016), limiting DSB-induction to the S/G2 phase of the cell cycle where HDR is active (Lin et al., 2014; Gutschner et al., 2016; Howden et al., 2016), and optimizing length and symmetry of the repair template (Hendel et al., 2015; Rahdar et al., 2015; Yin et al., 2017; Richardson et al., 2016; Liang et al., 2017). In addition, several groups have recently demonstrated that temporal and spatial co-localization of the repair template to the Cas9 complex enhances HDR rates (Carlson-Stevermer et al., 2017; Aird et al., 2018; Gu et al. 2018; Savić et al., 2018). Importantly, these approaches do not involve any potentially harmful chemical treatment to alter endogenous cellular processes, and due to the shorter half-life of ribonucleoprotein complexes compared to DNA, they exhibit reduced risks of generating off-target mutations (Kim et al., 2014). In addition, procedures for clinical-grade recombinant protein production are well established, and progress has been made to deliver Cas9 RNP complexes in vivo in animal models (Zuris et al., 2015; Wang et al., 2016; Lee et al., 2017; Staahl et al., 2017). In summary, using Cas9 complexes with linked DNA repair templates is a promising approach for gene editing therapies, where high rates of precise repair is desired.
Here, we provide a detailed protocol for linking the DNA repair template to Cas9 as demonstrated in Savić et al., 2018. The described approach is based on the SNAP-tag technology, which enables the generation of a covalently linked Cas9-DNA complexes in vitro. We explain how to label repair-oligonucleotides with Benzylguanine (BG), how to produce Cas9-SNAP fusion proteins, how to produce single guide (sg)RNAs, how to generate DNA-ribonucleoprotein complexes in vitro, how to transfect them into mammalian cells, and how to analyze correction efficiencies (Figure 1).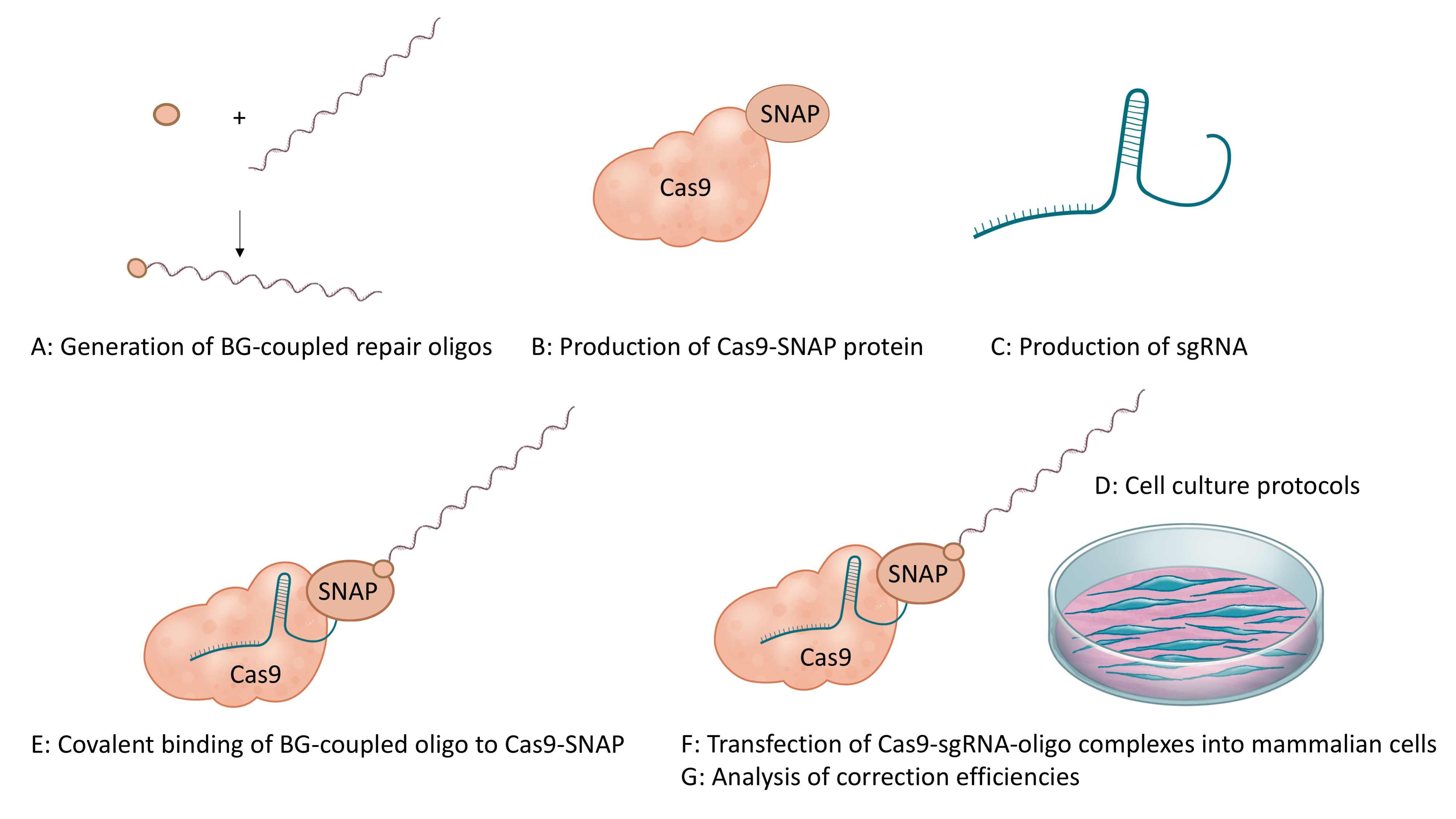
Figure 1. Schematic overview of the workflow of the protocol
Materials and Reagents
- Materials
- Syringe Luer 1 ml (Codan, catalog number: 3.7420.01)
- Syringe Luer 10 ml (Codan, catalog number: 3.7410.06)
- Hypodermic needle Luer, 22 G x 1¼" (BD MicrolaneTM, catalog number: 613-3928)
- 24-well cell culture plate Cellstar® (Greiner Bio-One, catalog number: 7662160)
- 6-well cell culture plate Cellstar® (Greiner Bio-One, catalog number: 7657160)
- 10 cm tissue culture dish Cellstar® (Greiner Bio-One, catalog number: 7664160)
- µ-Slide 8-well glass bottom (Vitaris, Ibidi®, catalog number: 80827-IBI)
- Centrifugation tube ED Safe-Lock 1.5 ml (Eppendorf, catalog number: 0030120.086)
- 10 μl, 20 μl, 200 μl, 1,000 μl pipette filter tips (Axon Lab AG, catalog numbers: AL60010, AL60020, AL60200, AL61000)
- Centrifugation tubes PP 15 ml (Greiner Bio-One, catalog number: 7188271)
- 5 ml sterile polystyrene round bottom test tubes with cell strainer cap (12 x 75 mm) (Corning Life Sciences, Falcon®, catalog number: 352235)
- Safe-Lock microcentrifuge tubes (Eppendorf, catalog number: 0030 120.086)
- 5 ml, 10 ml, 25 ml, 50 ml polystyrene serological pipettes (SARSTEDT, catalog numbers: 86.1253.001, 86.1254.001, 86.1685.001, 86.1256.001)
- Tubes and flat caps, strips of 8 (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: AB1182)
- Tubes, 0.2 ml, flat cap, individual tubes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: AB0620)
- 1.5 ml tube (SARSTEDT, catalog number: 72.690.001)
- 1,000 ml NalgeneTM PPCO centrifuge bottles (Thermo ScientificTM, catalog number: 3120-1000)
- NalgeneTM Oak Ridge high-speed PPCO centrifuge tubes (Thermo ScientificTM, catalog number: 3119-0050)
- Deep well plate, 96-well (VWR, catalog number: 732-0585)
- Ni-NTA Superflow (QIAGEN, catalog number: 30430)
- XK16/20 empty column (GE Life Sciences, catalog number: 28988937)
- HiTrap Heparin HP (GE Healthcare, catalog number: 17-0407-01)
- HiLoad 16/600 Superdex 200 pg (GE Healthcare, catalog number: 28989335)
- Amicon Ultra-15, PLHK Ultracel-PL Membran, 100 kDa (Merck, catalog number: UFC910096)
- 1 ml polypropylene tubes, CRYO.S (e.g., Greiner Bio-one, catalog number: 123279)
- 1.5 ml screw neck vials and 200 µl glass inserts (e.g., BGB, catalog numbers: 080400 and 110501)
- Waters XBridge Oligonucleotide BEH C18 column, 10 x 50 mm, 2.5 μm (Waters, catalog number: 186008212)
- 0.2 µm vacuum filter (TPP, catalog number: 99255)
- Glen Gel-Pak 1.0 desalting column (Glen research, catalog number: 61-5010)
- Cells
- Human embryonic kidney (HEK293T) (ATCC, catalog number: CRL-3216)
- Human blood (chronic myelogenous leukemia) (K562) (Sigma-Aldrich, catalog number: 89121407)
- Mouse Embryonic Stem Cells (mESC, WT E14 line) (ATCC, catalog number: CRL-1821)
- RosettaTM 2 (DE3) SinglesTM Competent Cells (Novagen, catalog number: 71400-3)
- Reagents
- pNS20-SpCas9-SNAP plasmid (Addgene, catalog number: 113717)
- Synthetic oligonucleotides with a 5’-Amino Modifier C6 functional group (Integrated DNA Technologies)
- Benzylguanine-GLA-NHS (New England Biolabs, catalog number: S9151S)
- HEPES (Sigma-Aldrich, catalog number: H4034-500G)
- N,N-Dimethylformamide (DMF) (Sigma-Aldrich, catalog number: 227056-250ML)
- TrypLETM Express Enzyme (1x), phenol red (Thermo Fisher Scientific, GibcoTM, catalog number: 12605010)
- SytoxTM Red Dead Cell Stain (Thermo Fisher Scientific, InvitrogenTM, catalog number: S34859)
- Recombinant protein Streptococcus pyogenes (SpCas9-SNAP) (Savić et al., 2018)
- Recombinant protein Staphylococcus aureus (SadCas9-SNAP) (Savić et al., 2018)
- Phosphate buffered saline (PBS), pH 7.4 (Thermo Fisher Scientific, GibcoTM, catalog number: 10010023)
- Penicillin-streptomycin solution (Pen-Strep) (10,000 U/ml) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122)
- Fetal bovine serum (FBS) (Sigma-Aldrich, catalog number: F7524)
- Fetal bovine serum (FBS) (Thermo Fisher Scientific, Life Technologies, catalog number: 10270106) for mESC culture
- Dulbecco’s modified Eagle’s medium (high glucose) (DMEM) (Thermo Fisher Scientific, GibcoTM, catalog number: 31966021) for HEK293T culture
- Dulbecco’s modified Eagle’s media (DMEM) (Sigma-Aldrich, catalog number: D6429-500ML) for mESC culture
- RPMI 1640 medium, GlutaMAXTM supplement (Thermo Fisher Scientific, GibcoTM, catalog number: 61870010) for K562 culture
- LIF (Millipore, catalog number: ESG1107)
- 2-β-mercaptoethanol (Thermo Fisher Scientific, Life Technologies, catalog number: 31350-010)
- Gelatin from porcine skin powder gel (Sigma-Aldrich, catalog number: G1890-500G)
- Poly-L-lysine (Sigma, catalog number: P1524)
- Trypsin-EDTA (0.05%), phenol red (Thermo Fisher Scientific, GibcoTM, catalog number: 25300054)
- LipofectamineTM 2000 transfection reagent (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11668019)
- Opti-MEMTM I reduced serum medium, no phenol red (Thermo Fisher Scientific, GibcoTM, catalog number: 11058021)
- Hoechst 33342 solution (20 mM) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 62249)
- Glycogen (Roche, catalog number: 10901393001)
- 3 M sodium acetate buffer solution (NaAc) (Sigma-Aldrich, catalog number: S7899-100ML)
- Ethanol (Merck, catalog number: 100983)
- Isopropanol (Merck, catalog number: 109634)
- Water, 0.1 μm filtered, double distilled, deionized, nuclease/protease-free (Sigma-Aldrich, catalog number: W4502)
- EDTA disodium salt 2-hydrate for molecular biology (AppliChem, catalog number: A2937)
- GelRed nucleic acid gel stain 10,000x in DMSO (Biotium, catalog number: 41002)
- IVT primers (Integrated DNA Technologies, 100 nmole DNA oligos)
- Phusion polymerase (New England Biolabs, catalog number: M0530)
- Deoxynucleotide (dNTP) Solution Mix (New England Biolabs, catalog number: M0447)
- QIAquick PCR Purification Kit (QIAGEN, catalog number: 28106)
- Gel 6x Loading Dye, Purple (New England Biolabs, catalog number: B7024S)
- LE agarose, multi-purpose agarose (Clever Scientific, catalog number: CSL-AG)
- DEPC-treated water (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9916)
- RNA Polymerase, T7 (Roche, catalog number: 10881767001)
- NTP Set, 100 mM solution (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0481)
- RNase-free DNase set (QIAGEN, catalog number: 79254)
- Ammonium acetate solution, BioUltra 5M (Sigma-Aldrich, catalog number: 09691)
- RNA Clean & Concentrator (Zymo Research, catalog number: R1017)
- RNase AWAY (Sigma-Aldrich, catalog number: 83931)
- Ambion® NorthernMax® formaldehyde load dye (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM8552)
- Formaldehyde 37% in H2O (Sigma-Aldrich, catalog number: 252549)
- Low Range ssRNA ladder (New England Biolabs, catalog number: N0264)
- SYBRTM Gold nucleic acid gel stain, 10,000x Concentrate in DMSO (Thermo Fisher Scientific, InvitrogenTM, catalog number: S11494)
- 1,1,1,3,3,3-Hexafluoro-2-propanol (Fluorochem, catalog number: 003409)
- RNase-free ultrapure H2O (purified on our EMD Millipore Synergy UV system)
- Methanol (VWR, catalog number: 20864.320)
- Triethylamine puriss, p.a. > 99.5% (Sigma-Aldrich, catalog number: 90340)
- Acetic acid puriss, p.a. > 99.8% (Sigma-Aldrich, catalog number: 33209)
- DNeasy Blood and Tissue kit (QIAGEN, catalog number: 69506)
- NEBNext® high-fidelity 2x PCR master mix (New England Biolabs, catalog number: M0541L)
- Agencourt AMPure XP reagent (Beckman Coulter, catalog number: A63881)
- Index adaptor sequences, HPLC quality (Integrated DNA Technologies)
- LB-agar (Luria/Miller) (Carl Roth, catalog number: X969.2)
- LB medium (Miller) pulver (Faust Laborbedarf AG, catalog number: 6.271 000)
- Kanamycinsulfat (Carl Roth, catalog number: T832.4)
- Isopropyl-β-D-thiogalactopyranosid (IPTG) (Chemie Brunschwig AG, catalog number: BP1755-100)
- HEPES 200 mM pH 8.5 (see Recipes)
- HEK293T medium (see Recipes)
- K562 medium (see Recipes)
- mESC proliferative growth medium (see Recipes)
- Gelatin solution 0.2% (see Recipes)
- Poly-L-lysine solution 0.2% (see Recipes)
- FACS buffer (see Recipes)
- EDTA buffer 0.5 M pH 8.0 (see Recipes)
- Protein buffer (see Recipes)
- KCl buffer 500 mM (see Recipes)
- 3-(N-morpholino) propanesulfonic acid (MOPS) 10x running buffer (see Recipes)
- Tris-borate-EDTA (TBE) 10x buffer (see Recipes)
- Tris buffer 1 M (see Recipes)
- TE buffer pH 8.0 (see Recipes)
- HPLC eluent A (see Recipes)
- LC/MS eluent A (see Recipes)
- Chloramphenicol 1,000x (34 mg/ml stock) (see Recipes)
- Kanamycin 1,000x (50 mg/ml stock) (see Recipes)
- IPTG 1 M (see Recipes)
- NaCl 5 M (see Recipes)
- DTT 1 M (see Recipes)
- Imidazole 2.5 M pH 8.0 (see Recipes)
- HEPES 1 M pH 7.5 (see Recipes)
- Protease inhibitor (1:1,000) (see Recipes)
- KCl 4 M (see Recipes)
- Tris buffer 1 M pH 6.8 (see Recipes)
- Lysis buffer (see Recipes)
- Wash buffer (see Recipes)
- Elution buffer (see Recipes)
- Heparin buffer A (see Recipes)
- Heparin buffer B (see Recipes)
- SEC buffer (see Recipes)
- 1x Laemmli buffer (see Recipes)
- 5x Laemmli buffer (see Recipes)
Equipment
- 250 ml Erlenmeyer flask (Huberlab AG, catalog number: 9.0460.63)
- 2 L Erlenmeyer flask (Carl Roth, catalog number: C141.2)
- Pipetman G P2G, P10G, P20G, P200G, P1000G (Gilson, catalog numbers: F144054P, F144055M, F144056P, F144058P, F144059P)
- Pipette controller, Pipet-Aid® Hood Mate® (Drummond, catalog number: 4-000-302)
- Laminar flow hood SafeFAST Premium (FASTER S.r.l, catalog number: F00024900000)
- T100TM thermal cycler (Bio-Rad, catalog number: 1861096)
- NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM 2000, catalog number: ND-2000)
- Tissue culture incubator set at 37 °C, 5% CO2 (Thermo Fisher Scientific, Thermo ScientificTM, model: HeracellTM 150i, catalog number: 51026283)
- Vortex-Genie 2 (Scientific Industries, model: Vortex-Genie 2, catalog number: SI-0276)
- AREX digital PRO magnetic stirrer with hot plate (Velp Scientifica, catalog number: SA20500411)
- Centrifuge Micro Star 17 (VWR, catalog number: 521-1646)
- Microcentrifuge, MiniStar silverline (VWR, catalog number: 521-2845)
- Microlitre centrifuge (Hermle Labortechnik, catalog number: Z216MK)
- Countess automated cell counter (Thermo Fisher Scientific, InvitrogenTM, catalog number: C10227)
- Countess® cell counting chamber slides (Thermo Fisher Scientific, InvitrogenTM, catalog number: C10228)
- BD LSR Fortessa cell analyzer (Becton-Dickinson)
- Thermoshaker, cooling thermal shake touch (VWR, catalog number: 460-0198)
- Microwave oven (Primotecq, catalog number: MW 5920)
- TW12 water bath (Julabo, catalog number: 9550112)
- Wide Mini-Sub® Cell GT horizontal electrophoresis system, 15 x 10 cm tray (Bio-Rad, catalog number: 1704468)
- ChemiDoc XRS gel imaging system (Bio-Rad, catalog number: 1708265)
- Liquid chromatography-mass spectrometry system and software for peak analysis (e.g., Agilent 1200 Series HPLC [Agilent Technologies, model: 1200 Infinity Series] connected to an Agilent 6130 Series Quadrupole [Agilent Technologies, model: 6130 Series])
- Waters Acquity UPLC C18 column, 2.1 x 50 mm, 1.7 μm (Waters, catalog number 186003949)
- Centrifugal concentrator (e.g., Genevac, miVac Duo, catalog number: DUC-23050-B00)
- Water purification system (e.g., EMD Millipore Synergy UV equipped with a Biopak polisher [Merck, catalog number: CDUFBI001])
- Semi-preparative high-performance liquid chromatography (HPLC) system equipped with an autosampler, multiple wavelength detector and a fraction collector (e.g., Agilent 1200 Series) and software for peak analysis
- New BrunswickTM Innova® 42 incubator (Eppendorf, New BrunswickTM, model: Innova® 42, catalog number: M1335-0002)
- Sorvall LYNX 4000 Superspeed Centrifuge (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 75006580) or an equivalent centrifuge that can be cooled down to 4 °C and can perform up to 6,000 x g
- New BrunswickTM Innova® 44/44R (Eppendorf, New BrunswickTM, model: Innova® 44/44R, catalog number: M1282-0002) or any equivalent shaker incubator where the temperature can be set at 37 °C and 18 °C
- Cell density meter UltrospecTM 10 (GE Healthcare, catalog number: 80-2116-30), or equivalent spectrophotometer that can measure the density of cells in suspension at 600 nm
- SONOPULS HD 3200 Sonifier (BANDELIN electronic, model: HD 3200) with VS 70 T Sonotrode (BANDELIN electronic, model: VS 70 T) or alternatively a French Pressure Cell (French Press) for cell lysis
- Peristaltic pump P-1 with connectors for 5 ml HisTrap HP (GE Healthcare, catalog number: 18111091 )
- Tubing Connectors for Use with Peristaltic Pump P-1 (GE Healthcare, catalog number: 11300082 )
- ÄKTApurifier 10 FPLC system (GE Healthcare, catalog number: 28406264 )
- Sorvall LYNX 4000 Superspeed Centrifuge (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 75006580) or an equivalent centrifuge that can be cooled down to 4 °C and can perform up to 30,000 x g
- pH meter ProLine B210 (QiS)
- Filter holder assembly for filtration (Merck, catalog number: XX1014700 or Nalgene, Thermo Fisher Scientific, Thermo ScientificTM, catalog number: DS0320-2545)
- Diaphragm Vacuum Pumps LABOPORT® N 820 (ABM van Zijl B.V, catalog number: ABMK N8203FT18)
- Mini-PROTEAN Tetra cell vertical electrophoresis system (Bio-Rad Laboratories, model: Mini-PROTEAN Tetra Cell, catalog number: 1658004EDU)
- Perfection V850 Pro scanner (Epson) for SDS-PAGE gel imaging
Software
- ChemStation for LC, Rev. B. 03.02 (Agilent Technologies)
- ChemStation for LC/MS, Rev. B. 04.03-SP2 (Agilent Technologies)
- Leica LAS X SP8 (version 1.0)
- Leica LAS AF (Lite) software (version 3.3) (available for free)
- ImageJ software (version 1.51 n) (available for free)
- FlowJo software (version 10.2) for FACS analysis
- GraphPad’s Prism7 software for statistical analyses
To purchase software, please visit the respective company webpage.
Procedure
- Generation of Benzylguanine (BG)-coupled repair template oligos
- BG oligo coupling reaction
Notes:- Starting with a 5 nmol reaction results in approximately 3 nmol BG-coupled oligo after HPLC purification. For a transfection of HEK293T cells in a single 24-well 2.2 pmols are needed.
- The ratio 100:1 BG-GLA-NHS: amino-modified oligo has been optimized to achieve maximal coupling for 81 base long oligos. For shorter oligos, smaller ratios of BG-GLA-NHS: amino-modified oligo could be applied.
- Dissolve 2 mg BG-GLA-NHS in 207 μl DMF. This gives a stock concentration of 20 mM. Syringe and needle are needed for the DMF, when this is packaged with a Sure/Seal. Before starting the experiment, prepare a BG-GLA-NHS working stock solution of 3.3 mM in DMF.
- Mix the following components in 1.5 ml Eppendorf tubes in a flow hood (see the following Table for amounts, this example is for 5 nmols reaction in the ratio 100:1 BG-GLA-NHS: amino-modified oligo):
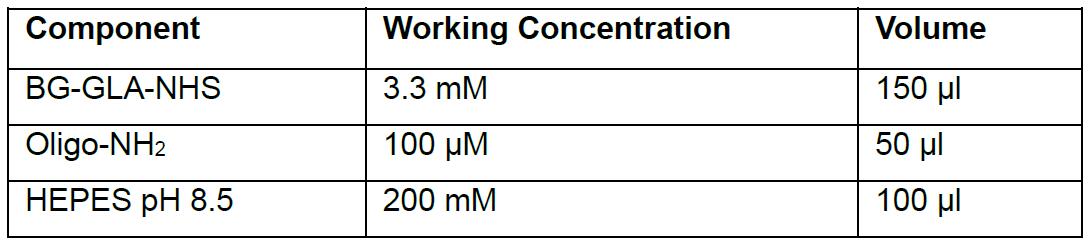
- Mix the components properly by pipetting the up and down 5 times.
- Incubate the BG coupling reaction at 30 °C for 60 min.
- Ethanol precipitation on BG coupling reactions
- Add the following components to the BG coupling reaction:
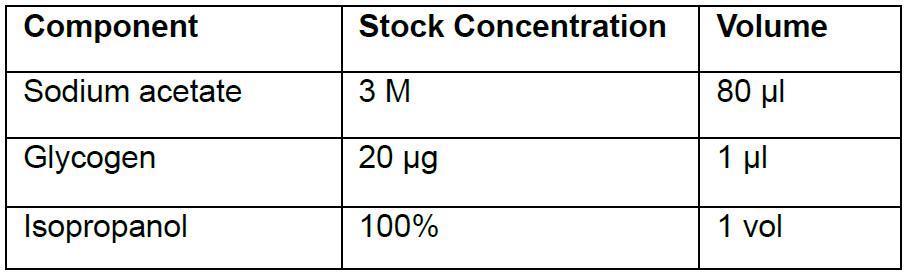
- Vortex the BG coupling reaction briefly and store the Eppendorf tube(s) at -20 °C for 4-6 h or overnight (PAUSE POINT).
- Spin the Eppendorf tube(s) for 15 min at 13,500 x g at 4 °C.
- Remove the supernatant and add 500 μl 100% ethanol (-20 °C) to wash the pellet.
- Spin for 10 min at 13,500 x g at 4 °C.
- Remove the supernatant and add 500 μl 80% ethanol (-20 °C) to wash the pellet.
- Spin for 10 min at 13,500 x g at 4 °C.
- Remove the supernatant and air dry the pellet for 10-15 min.
- Dissolve the pellet in 200 µl RNase-free sterile water when proceeding with purification of the BG-coupled DNA on reversed phase HPLC. When using the Glen Gel-Pak cartridge purification method, dissolve the pellet in 1,500 µl RNase-free sterile water.
- Add the following components to the BG coupling reaction:
- Purification of BG-coupled DNA on reversed phase HPLC
- Dilute reaction mixture to 200 µl with RNase-free ultrapure H2O.
- Determine the threshold for UV absorbance based fraction collection:
Inject 10 µl of the crude reaction mixture and record chromatogram at 260 nm. Perform purification on an Agilent 1200 Series HPLC system (see item 26 in the equipment list) fitted with a Waters XBridge Oligonucleotide BEH C18 column (10 x 50 mm, 2.5 μm) at 65 °C using a gradient of 5%-25% methanol (Table 1) over 8 min with a flow rate of 5 ml min-1. HPLC eluent A is 0.1 M triethylammonium acetate, pH 8.0 (see Recipes). - Estimate a suitable threshold for bulk product purification according to the following empirical formula:
- Bulk product purification:
Inject residual 190 µl of the reaction mixture and collect the product containing fraction using the threshold estimated above. - Remove solvent under vacuum from product containing fractions using a centrifugal concentrator, such as Genevac miVac Duo, at 35 °C and re-dissolve the purified product in 100 µl RNase-free ultrapure H2O. A representative HPLC chromatogram of the separation of BG-coupled DNA from unreacted 5’-amino DNA is shown in Figure 2.
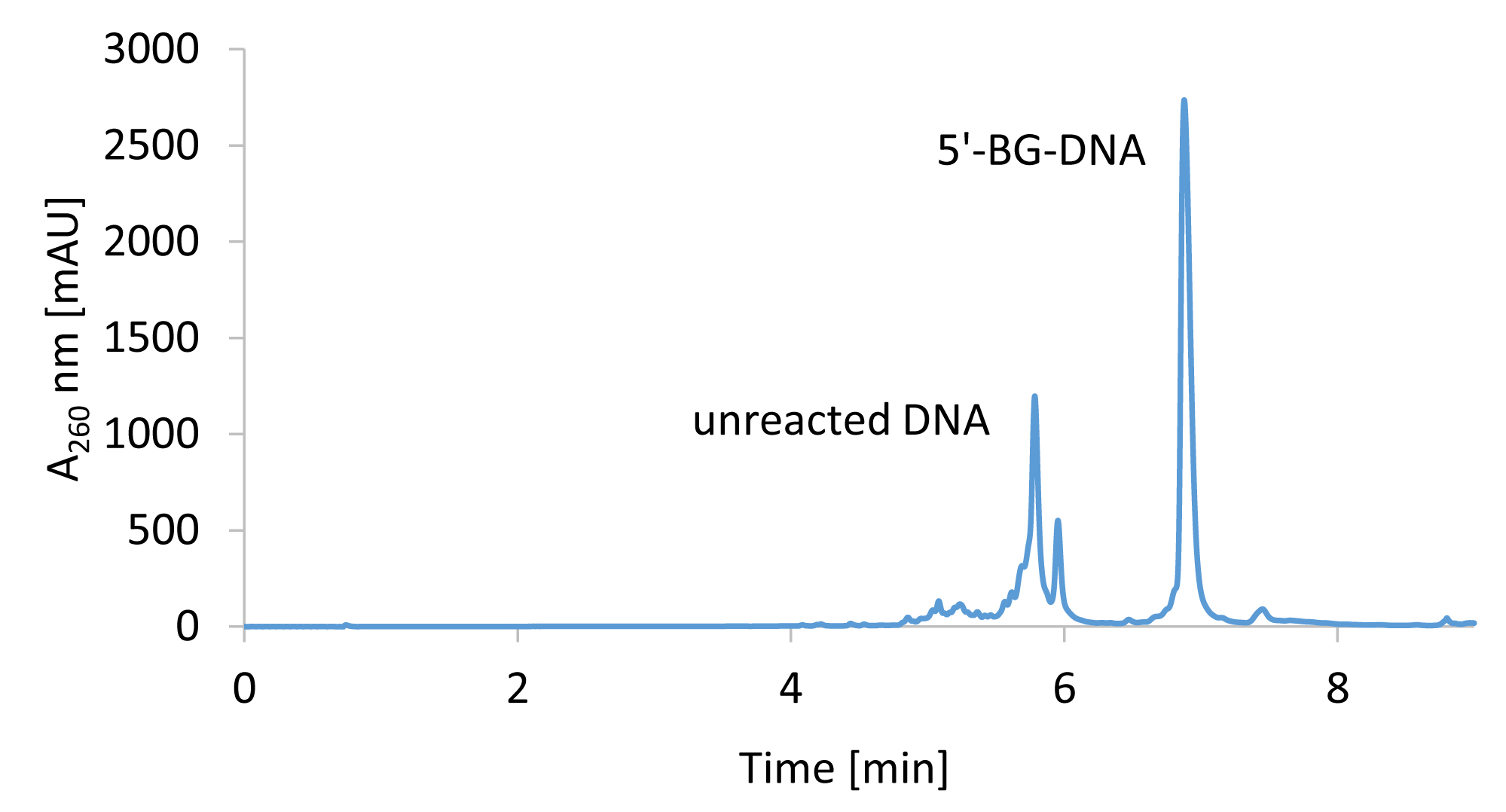
Figure 2. Representative chromatogram of the separation of unreacted DNA from 5’-BG-linked DNA. 5’-BG-DNA was purified using a gradient of 5%-25% methanol over 8 min. BG conjugation was performed under non-optimized conditions.
Table 1. Gradient for HPLC purification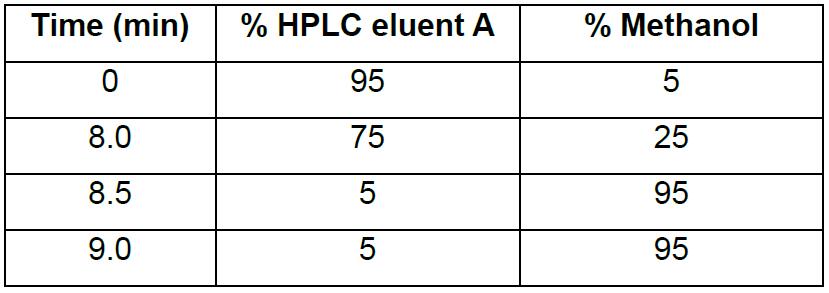
where,
V1 = volume of test injection
V2 = volume of main injection
A1 = absorbance threshold suitable for test injection
A2 = absorbance threshold suitable for main injection
Note: If the lab has no access to a suitable HPLC system, the ethanol precipitated BG-coupled oligos can also be further separated from unreacted BG by using a size-exclusion based desalting column, such as Glen Gel-Pak 1.0. For Glen Gel-Pak based purification follow the manufacturer’s protocol. In our hands, the combination of ethanol precipitation and Glen Gel-Pak based purification resulted in only a slightly lower coupling efficiency compared to the purification on reversed phase HPLC (Figure 3). In addition, when we functionally tested RNPD complexes purified with different methods in fluorescent HEK reporter cells, we found that the correction efficiencies were comparable (Figure 4).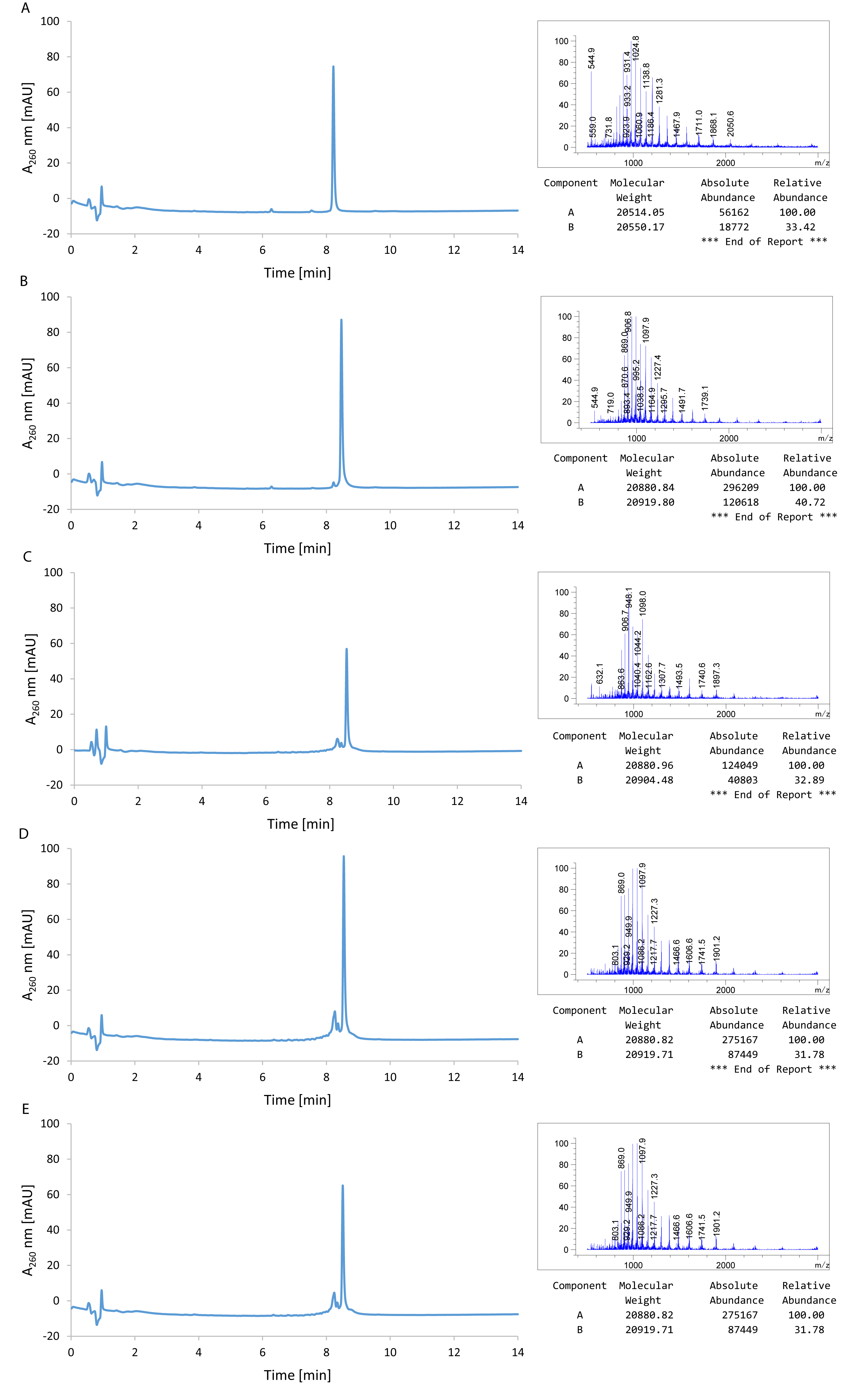
Figure 3. LC/MS analysis of BG-coupled and uncoupled DNA after different methods of purification. A. Uncoupled RFP DNA repair templates after HPLC purification (M = 20516.4). B. BG-coupled RFP repair templates after HPLC purification (M = 20882.8). C. BG-coupled repair templates after desalting on Glen Gel-Pak cartridge only. D. BG-coupled repair templates after ethanol precipitation only. E. BG-coupled repair templates after ethanol precipitation followed by Gel-Pak cartridge purification. BG conjugation was performed under optimized conditions for quantitative coupling.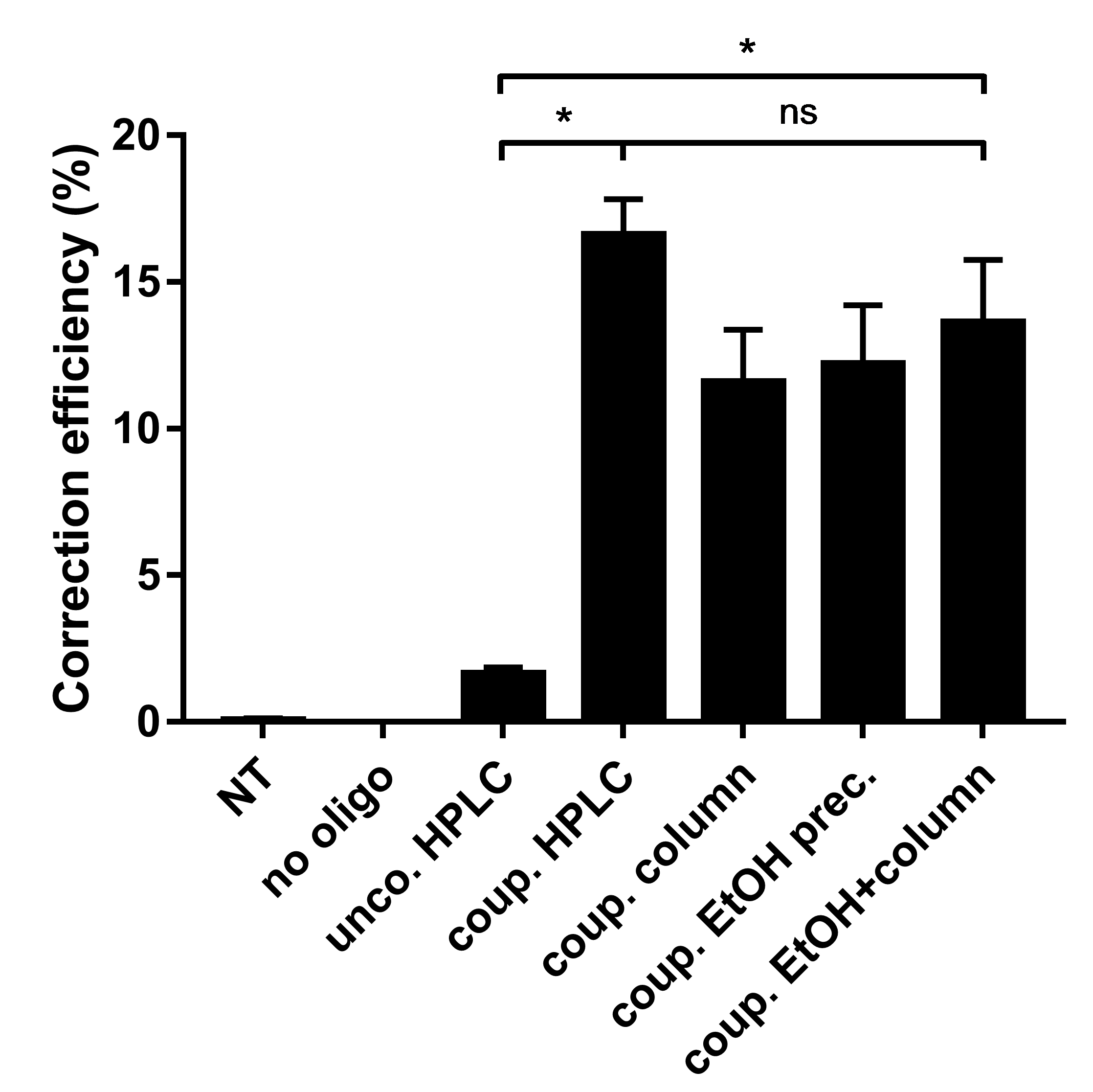
Figure 4. Correction efficiency of RNPD systems with differently purified BG-coupled DNA. Results show that repair oligos can also be purified from excess BG using Gel-Pak cartridge purification in combination with EtOH purification. FACS analysis of the fluorescent reporter cells transfected with different RNPD systems: Non-treated, no oligo, uncoupled RFP oligo after HPLC purification, coupled RFP oligo after HPLC purification, coupled RFP oligo after desalting on Glen Gel-Pak cartridge only, coupled RFP oligo after ethanol precipitation only, coupled RFP oligo after ethanol precipitation followed by Gel-Pak cartridge purification. - Dilute reaction mixture to 200 µl with RNase-free ultrapure H2O.
- Quality control of purified BG-coupled DNA by LC/MS
- Mix 1 µl purified BG-DNA with 30 µl RNase-free ultrapure H2O in a 1.5 ml screw neck vial with a 200 µl insert.
- Inject 30 µl and record absorbance at 260 nM and total ion chromatogram (TIC). Perform LC/MS analysis on an Agilent 1200 Series HPLC fitted with a Waters Acquity UPLC OST C18 column (2.1 x 50 mm, 1.7 μm) connected to an Agilent 6130 Series Quadrupole (or equivalent) using a gradient of 5%-45% methanol (see Table 2) over 14 min with a flow rate of 0.3 ml min-1 at 65 °C. LC/MS eluent A is aqueous 0.4 M 1,1,1,3,3,3-hexafluoro-2-propanol containing 15 mM triethylamine.
MS parameters (negative mode) are as follows:- Mass range: m/z 500-3000
- Capillary voltage: 3500V
- Drying gas flow: 13.0 L/min
- Drying gas temperature: 350 °C
- Nebulizer pressure: 45 psi
Table 2. Gradient for LC/MS analysis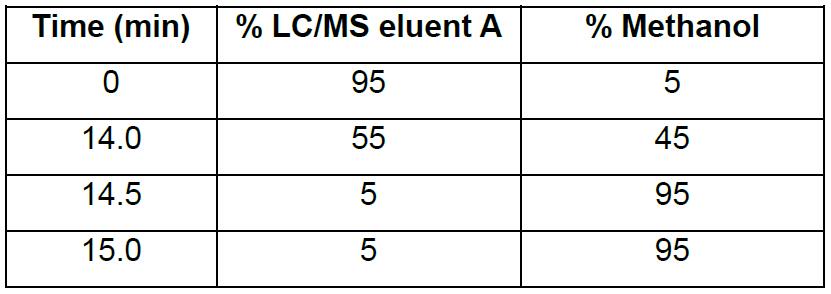
- BG oligo coupling reaction
- Production of Cas9-SNAP protein
- Expression of SpCas9-SNAP in E. coli
- Transformation (Day 1)
- Add 1 ng of pNS20-SpCas9-SNAP plasmid (Addgene #113717) to 50 μl of chemically competent E. coli RosettaTM 2 (DE3) cells.
- Tap gently to mix and incubate tube on ice for 30 min.
- Incubate cells at 42 °C for 45 s in a heat block.
- Immediately place tube back on ice for 3 min.
- Add 350 µl of room temperature sterile LB medium.
- Incubate at 37 °C while shaking for 60 min.
- Plate 100 µl of the mixture on an LB agar plate containing 50 µg/ml of kanamycin and 33 µg/ml of chloramphenicol. Use sterile glass beads or a Drigalski spatula for spreading the cells.
- Incubate LB agar plate overnight at 37 °C.
- Preparation of media and stock solutions (Day 1)
- Prepare 8 x 750 ml LB media in 2 L Erlenmeyer flasks.
- Prepare 1 x 100 ml LB media in a 250 ml Erlenmeyer flask for pre-culture.
- Prepare antibiotic stock solutions.
- Start pre-culture (Day 2)
- Add 100 µl of 50 mg/ml kanamycin stock solution and 33 µl of 50 mg/ml chloramphenicol stock solution to 100 ml of sterile LB medium prepared on Day 1.
- Pick a single colony from the LB agar plate of E. coli Rosetta 2 (DE3) cells transformed with P120 on Day 1. For picking, you can use a sterile pipette tip or an autoclaved tooth pick.
- Incubate pre-culture at 37 °C overnight while shaking at 220 rpm. Start pre-culture in the late afternoon.
- Prepare 10 ml of IPTG stock solution.
- Expression of SpCas9-SNAP in E. coli RosettaTM 2 (DE3) (Day 3)
- Add 750 µl of 50 mg/ml kanamycin stock solution and 750 µl of 33 mg/ml chloramphenicol stock solution to each of the 750 ml LB media from Day 1.
- Inoculate each flask with 10 ml of the overnight preculture prepared on Day 2.
- Incubate the cultures at 37 °C while shaking at 110 rpm.
- Check the optical density at a wavelength of 600 nm (OD600) of the culture every hour until it reaches 0.6-0.8.
- Reduce the temperature in the incubator to 18 °C.
- Remove a 1 ml sample of the culture and centrifuge for 10 min at 17,000 x g. Discard the supernatant and resuspend the pellet in 1x Laemmli SDS loading buffer according to the measured OD600, by adding 10 µl of 1x Laemmli buffer per 0.1 absorbance (i.e., if OD600 is 0.7 add 70 µl of 1x Laemmli buffer). Store the sample at -20 °C for later analysis by SDS-PAGE (“uninduced culture” sample).
- To induce the expression of SpCas9-SNAP, add 150 µl of 1 M ITPG to each flask (0.2 mM final concentration).
- Incubate culture overnight at 18 °C (~16 h), shaking at 110 rpm.
- Prepare 100 ml of Lysis buffer, 1 L of Wash buffer and 1 L of Elution buffer.
- Harvesting of bacterial culture (Day 4)
- Before harvesting the cells, remove a 200 µl sample from overnight culture for SDS-PAGE analysis (“induced culture” sample), add 800 µl LB medium and determine the OD600. Centrifuge for 10 min at 17,000 x g. Discard the supernatant and resuspend the pellet according to the measured OD600 (same procedure like with the uninduced sample as described in Step B1d vi). Store at -20 °C.
- Transfer overnight cultures from Day 3 in 1 L centrifuge bottles.
- Pellet the cells by centrifuging at 5,000 x g at 4 °C for 20 min.
- Discard supernatant.
- Cell pellets can be stored at -20 °C (pellets can be also frozen and stored after resuspension in Lysis buffer).
- Prepare buffers for Ion exchange and Size exclusion chromatography (500 ml of Heparin buffer A, 500 ml of Heparin buffer B, 1 L of SEC buffer).
- Transformation (Day 1)
- SpCas9-SNAP purification
Purification of SpCas9-SNAP: Part I (Day 4)- If cell pellets were frozen after harvesting, thaw the cell pellets on ice.
- Resuspend pellets in a total volume of 40 ml of Lysis buffer (~2.5-5 ml Lysis buffer per gram of cell pellet).
- To lyse the cells use a sonicator with the following settings: 6 min total time, 1 s on, 2 s off, 20% amplitude. The cell suspension noticeably changes color and viscosity during lysis.
- Transfer the lysed cells into Oak Ridge centrifuge tubes and centrifuge for 45 min at 30,000 x g at 4 °C.
- Remove a 5 µl sample of the cleared lysate for SDS-PAGE analysis (“clarified lysate” sample, Figure 8). Mix with 20 µl 1x Laemmli buffer. Store at -20 °C.
- Use a peristaltic pump to wash a 10-ml self-packed Ni-NTA Superflow column (or two 5 ml QIAGEN Ni-NTA Superflow prepacked cartridges connected in series) with 5 column volumes of water at a flow rate of 3 ml/min.
- Wash the column with another 5 column volumes of Wash buffer.
- Load the cleared lysate (~50 ml) onto the equilibrated column with a flow rate of 3 ml/min.
Note: There might be still unbound SpCas9-SNAP protein left in the flow-through. It can be loaded again on the Ni-NTA column for a second round of purification. Equilibrate the Ni-NTA column in Wash buffer before reloading the flow-through. - Remove a 5 µl sample of the flow-through for SDS-PAGE analysis (“flow-through” sample, Figure 8) and mix it with 20 µl 1x Laemmli buffer. Store at -20 °C.
- Wash the pumps of the Äkta FPLC system with Wash buffer (pump A) and Elution buffer (pump B) and flush the flow-path of the system with Wash buffer until the absorbance signal (280 nm wavelength, A280) in the UV detector reaches a stable baseline.
- Connect the loaded Ni-NTA column to the Äkta FPLC and wash the column with Wash buffer with a flow rate of 3 ml/min until A280 reaches baseline level (Figure 5).
- Elute the protein with 50% Elution buffer and collect in 1.8 ml fractions in a 96-well 2 ml deep-well block, or in tubes with appropriate capacity (Figure 5).
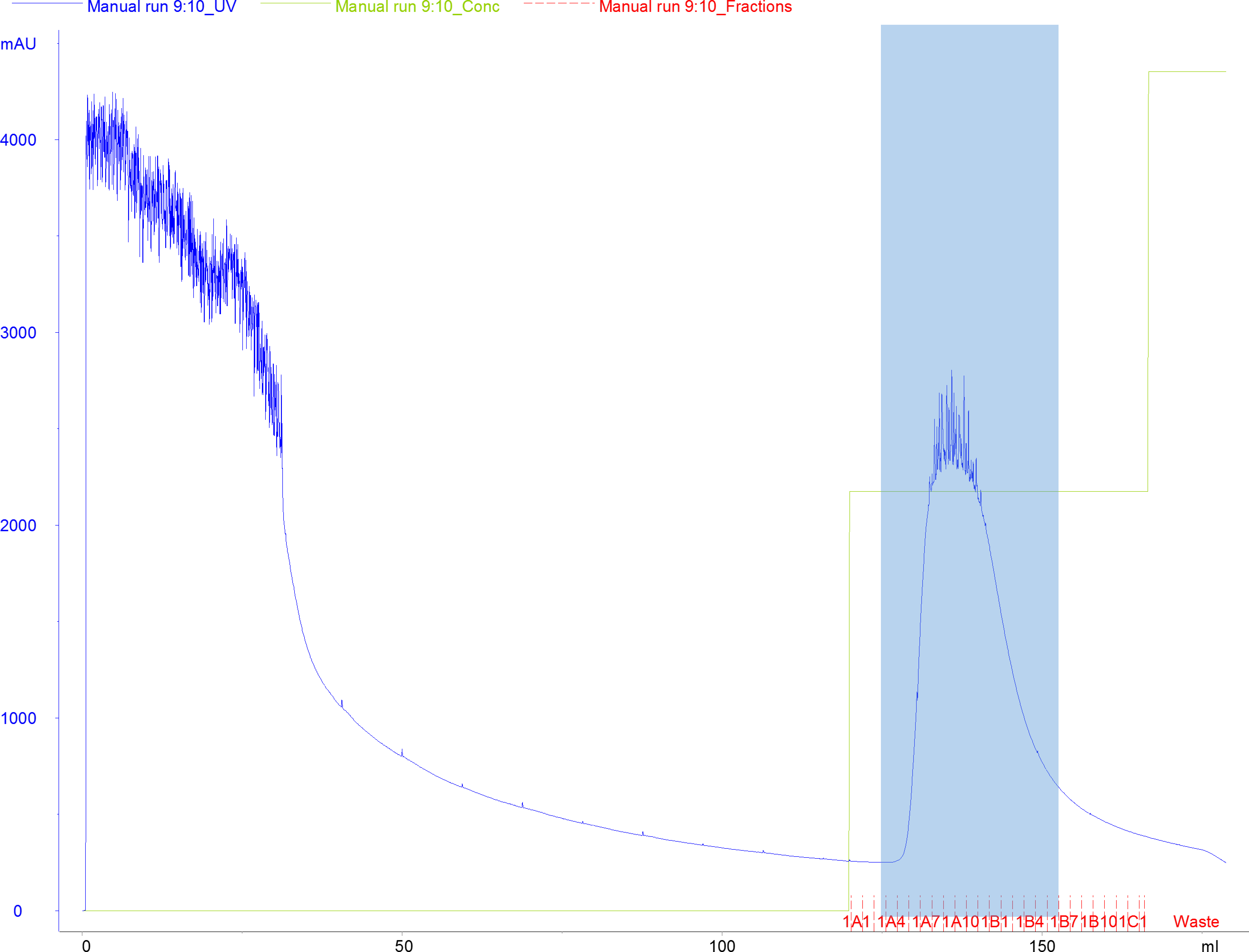
Figure 5. Representative chromatogram of Cas9 SNAP purified by HisTrap HP (10 ml) affinity purification. Blue line denotes absorbance signal at 280 nm (A280), green line denotes the concentration of the Elution buffer, and the fractions are indicated with red tick marks. The blue shaded area indicates fractions collected for dialysis and subsequent purification steps. - Pool the peak fractions in a clean 50 ml tube and determine the concentration by measuring the absorbance at 280 nm using a NanoDrop UV-VIS spectrophotometer. The extinction coefficient of His6-MBP-TEV-SpCas9-SNAP is 1416140 M-1 cm-1.
- Remove a 5 µl sample of the pooled fractions for SDS-PAGE analysis (“Ni-NTA elution” sample, Figure 8) and mix it with 20 µl 1x Laemmli buffer. Store at -20 °C.
- Add 1 mg of recombinant, His-tagged TEV per 50 mg of Cas9 SNAP (as determined from absorbance measurement).
- Incubate overnight at 4 °C.
Purification of Cas9 SNAP: Part II (Day 5)- Remove a 5 µl sample of the overnight TEV cleavage reaction for SDS-PAGE analysis (“TEV cleavage” sample, Figure 8) and mix it with 20 µl 1x Laemmli buffer. Store at -20 °C.
- Heat the collected samples for SDS-PAGE analysis for 5 min at 95 °C.
- Load 3 µl on a 12% SDS PAGE gel with 15 wells to check if cleavage is complete.
- Connect a 5 ml Heparin FF column to a peristaltic pump and wash with at least 5 column volumes of water at a flow rate of 3 ml/min.
- Equilibrate the column with at least 5 column volumes of Heparin buffer A.
- Load the overnight cleavage reaction onto the column and collect the flow-through in a clean 50 ml tube.
- Remove a 5 µl sample of the flow-through for SDS-PAGE analysis (“Heparin flow-through” sample, Figure 8) and mix it with 20 µl 1x Laemmli buffer. Store at -20 °C.
- Wash the pumps of the Äkta FPLC with Heparin buffer A and Heparin buffer B and flush the flow path of the system with Heparin buffer A until it reaches a stable baseline at the absorbance of 280 nm.
- Connect the loaded Heparin FF column to the Äkta FPLC system and wash the column with Heparin buffer A at a flow rate of 3 ml/min until the A280 signal reaches a steady baseline (Figure 6).
- Elute with a linear gradient to 50% Heparin buffer B over 20 column volumes (100 ml).
- Collect eluted protein in 1.8 ml fractions in a 96-well 2 ml deep-well block, or in tubes with appropriate capacity (Figure 6).
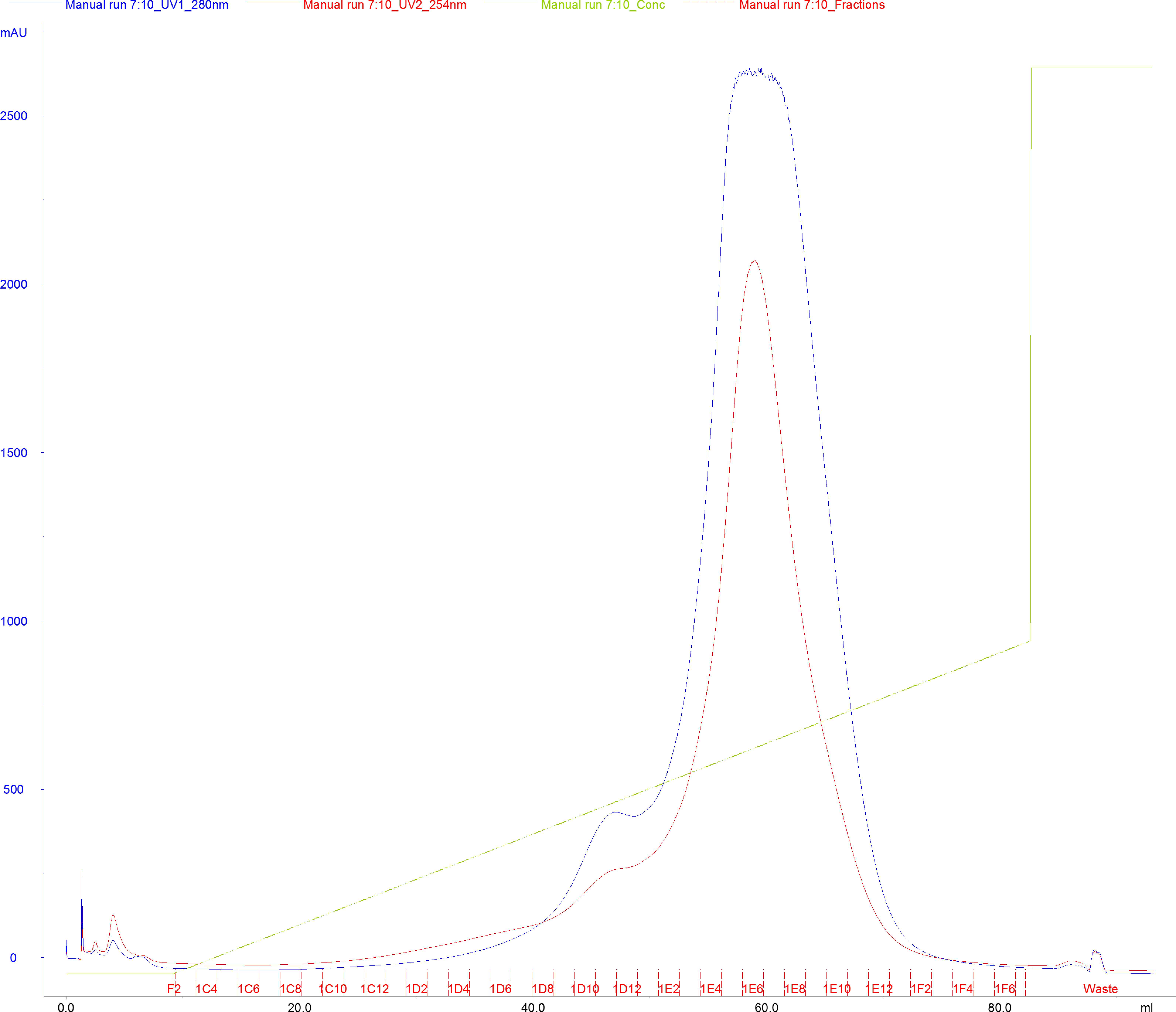
Figure 6. Representative chromatogram of Cas9 SNAP purified by Heparin FF (5 ml) affinity purification. Blue line denotes absorbance signal at 280 nm (A280), red line denotes absorbance signal at 260 nm (A260), green line denotes concentration of the Elution Buffer. Fractions are indicated with red tick marks. The blue shaded area indicates fractions collected for subsequent purification steps. - Pool the peak fractions containing SpCas9-SNAP (Figure 6).
- Remove a 20 µl sample of the pooled fractions for SDS-PAGE analysis (“Heparin peak” sample, Figure 8) and mix it with 5 µl 5x Laemmli buffer. Store at -20 °C.
- Transfer the pooled fractions to an Amicon Ultra-15 Centrifugal Filter Unit (MWCO of 100 kDa). Centrifuge the sample at 4,500 x g at 4 °C and concentrate to a final volume of < 5 ml. Mix the concentrate periodically throughout the concentration procedure by gently pipetting the solution up and down.
- Transfer concentrated sample into a 15 ml tube and centrifuge for 5 min at 4,500 x g at 4 °C to remove any precipitated material.
- In the meantime, equilibrate a HiLoad 16/600 Superdex 200 pg with 1 column volume of SEC buffer on the Äkta FPLC at a flow rate of 1 ml/min.
- Prepare a 5 ml injection loop by washing with 15 ml of SEC buffer on the Äkta FPLC. Flush the injection port with 10 ml of SEC buffer using a 10 ml syringe.
- Load the concentrated sample on the HiLoad 16/600 Superdex 200 pg via the 5 ml injection loop at a flow rate of 1 ml/min. Elute with SEC buffer at a flow rate of 1 ml/min (Figure 7).
- Collect elution in 1.8 ml fractions in a 96-well 2 ml deep-well block, or in tubes with appropriate capacity (Figure 7).
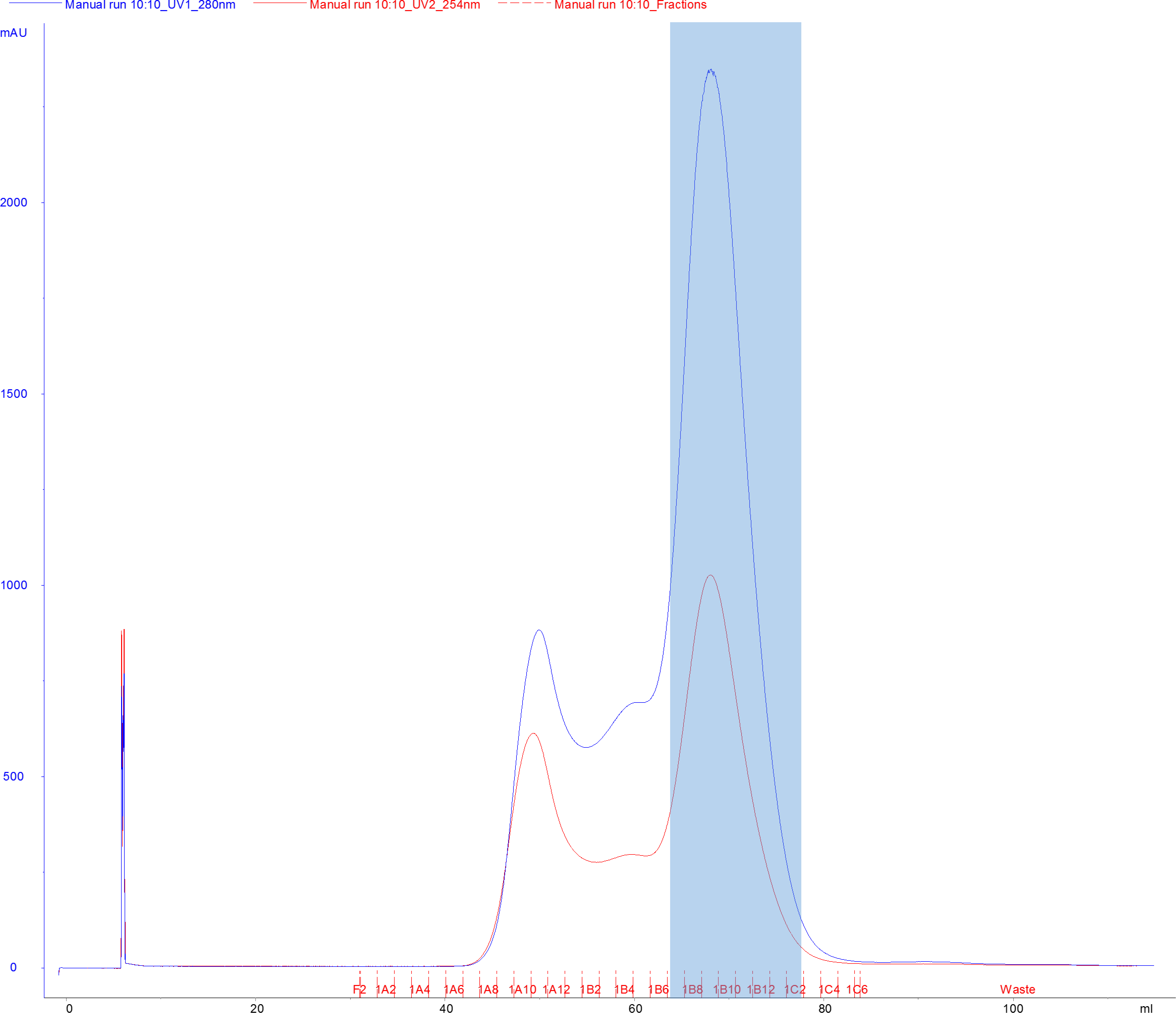
Figure 7. Representative chromatogram of Cas9 SNAP resolved on a HiLoad 16/600 Superdex 200 pg size-exclusion column. Blue line denotes absorbance signal at 280 nm (A280), red line denotes absorbance signal at 260 nm (A260), and fractions are indicated with red tick marks. The blue shaded area indicates fractions pooled for final concentration. - Pool the peak fractions and determine the concentration by measuring the absorbance at 280 nm using the NanoDrop UV-VIS spectrophotometer. Remove a 20 µl sample of the pooled fractions for SDS-PAGE analysis (“SEC peak” sample, Figure 8) and mix it with 5 µl 5x Laemmli Buffer. Store at -20 °C.
- Concentrate the SpCas9-SNAP protein to the required concentration (~1 mg/ml) by using an Amicon Ultra-15 Centrifugal Filter Unit (MWCO of 100 kDa). SpCas9-SNAP can be concentrated up to 10-20 mg/ml as long as the SEC buffer contains at least 500 mM KCl.
- Prepare 50 µl aliquots of purified SpCas9-SNAP protein, freeze in liquid nitrogen and store at -80 °C.

Figure 8. Representative analysis of SpCas9-SNAP fractions on a 10% SDS-PAGE gel. M: PageRuler Plus Prestained Protein Ladder. Marker band sizes are indicated in kDa. The gel is stained with Coomassie Brilliant Blue.
- Expression of SpCas9-SNAP in E. coli
- Production of sgRNA
Guide RNAs can be produced via in vitro transcription from a template DNA (see protocol below), or purchased from various sources (e.g., Integrated DNA Technologies).- sgRNA production
- Order IVT primers that generate the DNA template when annealed:
- sgRNA specific forward primer with T7 promoter:
GAAATTAATACGACTCACTATA(GGN18-20)GTTTTAGAGCTAGAAATAGC
Note: An example from the Savić et al. publication–sgRNASpCas9 (mutRFP) for: GAAATTAATACGACTCACTATAGGTGGCTACCAGCTTCATGCTGTTTTAGAGCTAGAAATAGC (Savić et al., 2018). - SpCas9 common reverse primer containing the sequence of the sgRNA scaffold: AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC
- sgRNA specific forward primer with T7 promoter:
- Perform the PCR with Phusion polymerase using a Thermal cycler
Reaction setup: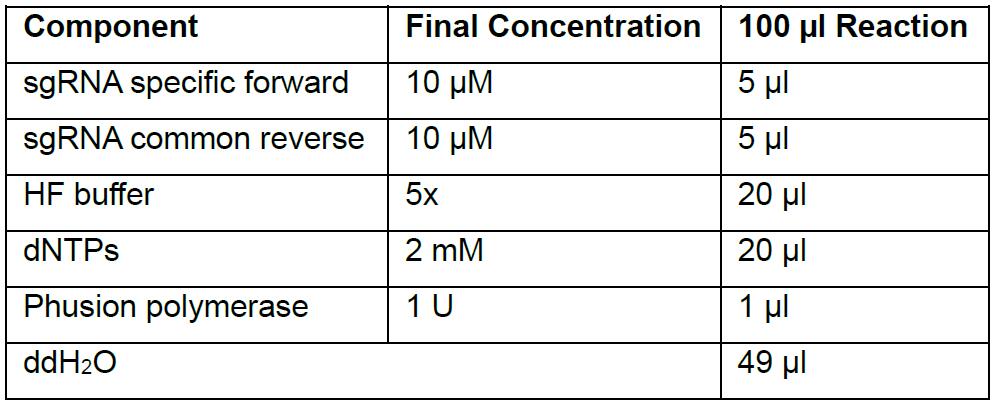
Thermocycling conditions: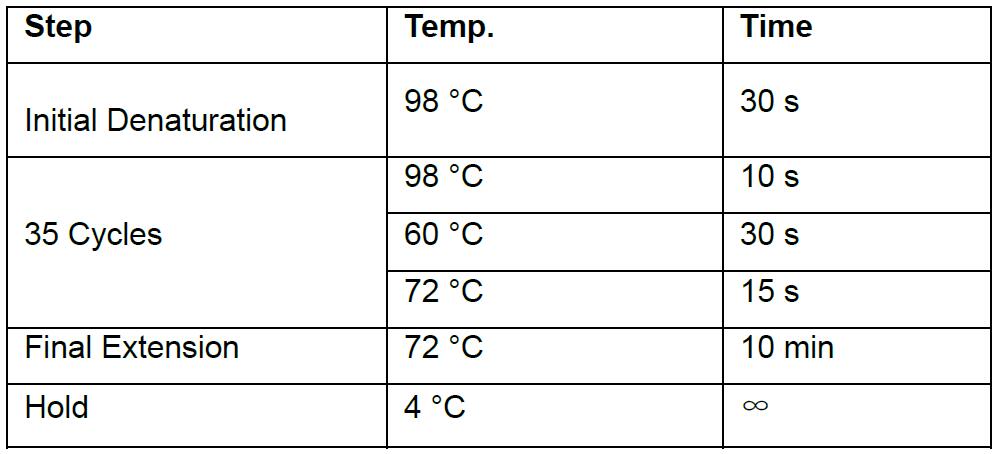
- Purify the DNA template using QIAquick purification kit, according to the manufacturer’s protocol and elute in 50 µl DEPC-treated water. Measure the eluted DNA concentration on a NanoDrop spectrophotometer.
- Run 500 ng of the purified PCR products mixed with 5 volumes of 6x loading dye on a 2% agarose gel containing 1x GelRed Nucleic Acid Stain. Separate bands by electrophoresis in 1x TBE buffer at 100 V for 1 h using Horizontal Electrophoresis System. Image using a Gel imaging System to confirm the concentration and the correct amplicon size.
Important: From here on, work RNase free! - In vitro transcribe the template DNA using the T7 RNA polymerase to obtain the guide RNA.
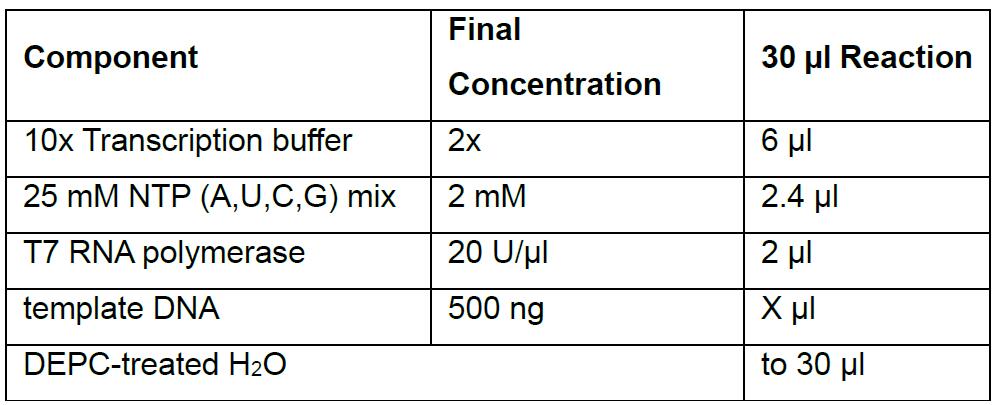
Incubate reaction overnight at 37 °C in the Thermal Cycler.
- Order IVT primers that generate the DNA template when annealed:
- sgRNA purification
- Purify the sgRNA by DNase I treatment followed by ethanol precipitation:
- Add 1 µl DNase I and incubate for 15 min at 37 °C in the Thermal Cycler.
- Add DEPC-treated water up to 100 µl.
- Add 10 µl 5 M Ammonium acetate and 330 µl 100% ethanol and incubate overnight at -20 °C.
- Add 1 µl Glycogen (20 µg), vortex and centrifuge at 17,000 x g for 15 min at 4 °C using Microlitre Centrifuge.
- Wash the pellet with 500 µl 75% ethanol and centrifuge at 17,000 x g for 15 min at 4 °C using Microlitre Centrifuge.
- Dissolve the pellet in 20-30 µl DEPC-treated water.
- Purify the sgRNAs further using RNA Clean & Concentrators, according to the manufacturer’s protocol. Measure the eluted RNA concentration on NanoDrop spectrophotometer. Store the RNA at -80 °C.
- Purify the sgRNA by DNase I treatment followed by ethanol precipitation:
- sgRNA control on a denaturing MOPS gel
- Control the sgRNA on a denaturing 2% MOPS gel:
- Prepare the gel
Heat 2 g agarose in 80 ml double distilled and deionized water using a microwave oven until dissolved, and then cool to 60 °C. Add 10 ml 10x MOPS running buffer, and 10 ml 37% formaldehyde (12.3 M), pre-warmed to 60 °C using a water bath. Mix and pour the gel using a comb that will form wells large enough to accommodate at least 25 µl. Let the gel settle for 30 min. Assemble the gel in the tank, and add enough 1x MOPS running buffer to cover the gel by a few millimeters. Remove the comb.
Note: Prewash the chamber, tray, and comb with soapy water/tween/SDS, wipe with RNase AWAY, and remove the leftover RNase AWAY with a paper tissue. Use autoclaved or UV-treated Erlenmeyer flask.
Warning: Formaldehyde is toxic through skin contact and inhalation of vapors. Manipulations involving formaldehyde should be done in a chemical fume hood. - Prepare RNA samples and ladder1)Add 3 volumes of Formaldehyde Load Dye to 1 µg (500 ng/U) sgRNA. Adjust total volume to 10 µl with DEPC-treated water. Use 2 µg of Low Range ssRNA Ladder.2)Heat denature samples at 65 °C for 15 min using a Thermoshaker.3)Briefly spin down samples in a microcentrifuge and place on ice.
- RNA electrophoresis
Load the gel and run at 5-6 V/cm (approx. 80 V) until the bromophenol blue (the faster-migrating dye) has migrated at least 2-3 cm into the gel, then raise to 120 V and let run as far as 2/3 the length of the gel. - Stain the RNA gel using Sybr Gold1)Dissolve 15 µl of Sybr Gold (10,000x) in 150 ml TE buffer pH 8.0.Note: Protect the gel from the light during incubation.2)Incubate for 45 min, shaking at room temperature. Wash 1 x 5 min in TE buffer followed by 1 x 5-min wash in double distilled and deionized water.
- Image on ChemiDoc imaging System to confirm the concentration and the RNA quality.
Note: An example of denaturing RNA gel with in vitro transcribed sgRNAs can be found in Figure 3–figure supplement 1 g from Savić et al., 2018.
- Prepare the gel
- Control the sgRNA on a denaturing 2% MOPS gel:
- sgRNA production
- Cell culturing
- Culture HEK293T line
Notes:- Change cell culture medium (for HEK293T medium, see Recipes) every other day.
- Passage cells before they become fully confluent, approximately every 4 days.
- Cells grow at 37 °C in a humidified 5% CO2 environment.
- Aspirate the medium from dish.
- Add 1 ml of TrypLE Express per 10 cm (for a 10 cm dish).
- Place the plate in the 37 °C incubator for 5 min.
- Once the cells detach, add 9 ml of DMEM medium and transfer the cells to a 15 ml conical tube.
- Centrifuge at 250 x g for 5 min at 4 °C.
- Aspirate the medium from the conical tube.
- Add 10 ml of fresh DMEM medium and split cells at a 1:10-1:15 dilution factor on to a new 10 cm dish and add enough additional medium to each plate for 10 ml total.
- Culture K562 line (suspension)
Notes:- Change cell culture medium (for K562 medium, see Recipes) every other day.
- Passage cells before they become fully confluent, approximately every 4 days.
- Cells grow at 37 °C in a humidified 5% CO2 environment.
- Collect the K562 cells from the dish in a 15 ml conical tube.
- Centrifuge at 250 x g for 5 min at 4 °C.
- Aspirate the medium from the tube.
- Add 10 ml of fresh RPMI medium and split cells at a 1:5-1:10 dilution factor on to a new 10 cm dish and add enough additional medium to each plate for 10 ml total.
- Culture mouse embryonic stem cell line
Notes:- Change cell culture medium (for mESC proliferative growth medium, see Recipes) daily.
- Passage cells every 2-3 days depending upon the growth rate of cells. The optimal condition is to maintain cells at approximately 80% confluency on Day 2 or 3.
- Cells grow at 37 °C in a humidified 8% CO2 environment.
- mECS grow on 0.2% gelatin-coated flasks/plates in absence of feeder cells.
- Aspirate the medium from the T75 flask.
- Rinse the T75 flask or dish with PBS pH 7.4 (1x) without CaCl2 and MgCl2.
- Add 1 ml 0.05 % Trypsin containing EDTA to the flask.
- Place the flask in the 37 °C incubator for 5 min.
- Once cells begin to dissociate from the flask, transfer the detached cell aggregates to a 15 ml conical tube containing 5 ml mESC DMEM growth medium.
- Rinse the flask with an additional 3 ml of DMEM growth medium to collect any remaining aggregates. Add the rinse to the conical tube that contains the cells.
- Centrifuge the conical tube at 250 x g for 2 min at room temperature.
- Aspirate the medium from the conical tube and add the desired amount of growth medium. Split the cells at a 1:4 to 1:10 dilution factor into a new flask or dish and add enough additional growth medium to each flask or plate.
- Culture HEK293T line
- Covalent binding of Cas9-SNAP protein and BG-coupled oligonucleotide
Complex the BG-coupled repair oligo templates with Cas9-SNAP proteins directly before performing the transfection.- Thaw the SpCas9-SNAP protein on wet ice.
Note: Very small samples such as 10 μl aliquots can be thawed by holding the tube in your hand. - Once thawed, gently mix the sample using a micropipettor with a polypropylene tip to make sure the solution is homogeneous. Be careful not to introduce bubbles into the solution. When necessary, dilute the protein in Protein buffer.
- Mix 2.2 pmols BG-coupled oligos together with 2.2 pmols SpCas9-SNAP protein in 1.5 ml Safe-Lock microcentrifuge tubes.
- Mix by flicking the microcentrifuge tubes.
- Briefly spin down samples in a microcentrifuge.
- Incubate for 60 min at 30 °C using Thermoshaker.
- Briefly spin down samples in a microcentrifuge.
- Place the sample on ice until used for cell transfection.
- Thaw the SpCas9-SNAP protein on wet ice.
- Transfecting cell lines with RNPD complexes
- Seeding HEK293T cells
One day prior to transfection (24 h): Seed HEK293T cells at a density of 120,000-140,000 cells per well in 24-well plates.- Aspirate the medium from dish.
- Add 1 ml of TrypLE Express (for a 10 cm dish).
- Place in a 37 °C incubator with 5% CO2 environment for 5 min.
- Once the cells detach, add 9 ml of DMEM medium and transfer the cells to a 15 ml conical tube.
- Pipette up and down several times with 5 ml Serological pipette to ensure single cell suspension.
- Perform a cell count using a hemocytometer or automated cell counter.
- Dilute the cells with DMEM medium at the desired final concentration of 120,000-140,000 cells/well.
- Seeding mES cells
One day prior to transfection (24 h): Seed mES cells at a density of 180,000-190,000 cells per well in 6-well plates.- Aspirate the medium from a flask or dish.
- Rinse the T75 flask or dish with PBS pH 7.4 (1x) without CaCl2 and MgCl2.
- Add 1 ml 0.05% Trypsin containing EDTA to the flask or dish.
- Place the flask in the 37 °C incubator with 5% CO2 environment for 5 min.
- Once cells begin to dissociate from the flask, transfer the detached cell aggregates to a 15 ml conical tube containing 5 ml mESC growth medium.
- Rinse the flask with an additional 3 ml of growth medium to collect any remaining aggregates. Add the rinse to the conical tube containing the cells.
- Perform cell count using a hemocytometer or automated cell counter.
- Dilute the cells with DMEM growth medium without PenStrep at the desired final concentration of 180,000-190,000 cells/well.
- Seeding K562 cells
On the day of transfection: K562 cells (suspension culture) can be distributed 6 h prior to transfection in 24-well plates at a density of 220,000-240,000 cells per well.- Collect the K562 cells from the dish in a 15 ml conical tube.
- Centrifuge at 250 x g for 5 min at 4 °C.
- Aspirate the medium from the tube.
- Add fresh RPMI medium and pipette up and down several times with 5 ml Serological pipette to ensure a homogeneous cell suspension.
- Perform cell count using a hemocytometer or automated cell counter. Dilute the cells with RPMI medium at the desired final concentration of 120,000-140,000 cells/well.
- Preparation of transfection reactions
Prepare transfection reactions by mixing following components in a laminar flow hood (see Table 3 for amounts, this example is for 24-well plate format):
Table 3. Transfection format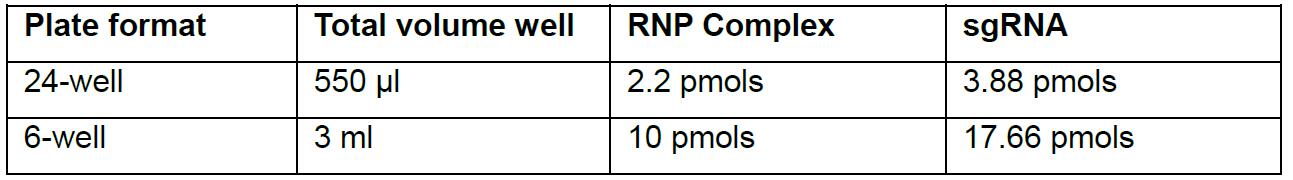
- 1.5 ml Eppendorf Tube A: Add selected RNP complex (2.2 pmols), and sgRNA (3.88 pmols) to Opti-MEM. Total volume in reaction Tube A should be 50 µl after adding components.
- 1.5 ml Eppendorf Tube B: Add 3 µl Lipofectamine 2000 to the 47 µl Opti-MEM. Total volume in the reaction Tube B should be 50 µl.
- Briefly vortex Tube A and B, and spin down samples in a microcentrifuge.
- Incubate for 5 min at room temperature.
- Add the content of Eppendorf Tube B to Eppendorf Tube A, briefly vortex and spin the content down in a microcentrifuge.
- Incubate the complexes at room temperature for 15 min, to allow lipid particle formation.
- Add the transfection mixture to the desired wells, by slowly dropping the mixture on the medium.
- Gently mix the media via back and forth motion.
Note: The medium of the mES transfected cells should be replaced to mES culture medium with Pen-Strep 8 h post-transfection. - Incubate at 37 °C with 5% CO2 environment for 24 h.
- Next day (post-transfection), HEK293T and K562 cells can be transferred to 10 cm dish for further expansion of the transfected cells.
- The mES cells can be collected after 48 h post transfection for extracting genomic DNA.
- At Day 5 post-transfection, HEK293T and K562 cells can be analyzed by different methods. When a reporter-line is used, the correction and indel formation events can be visualized by fluorescence imaging and quantified by FACS. In case no reporter line used, cells can be collected for DNA extraction and analysis via next-generation sequencing (NGS).
- Correction efficiency analysis
The protocols described here allow to assess the correction rates of the RNPD system in the cell line of interest. The protocols are designed for HEK293T cells, and would have to be adapted for the cell line of interest.- Microscopy imaging
As mentioned above, when a fluorescent reporter cell line is used, correction and indel formation events can be visualized by either fluorescence imaging or flow cytometry. One day post-transfection, the HEK reporter cell line can be transferred to Poly-L-lysine coated glass chamber slides and can be analyzed 5-7 days post-transfection.
Conformation and visualization by microscope imaging can be achieved as followed:- A few hours before transferring the transfected cells, coat the glass chamber slides with Poly-L-lysine. Pour a few hundred microliters of 0.2% Poly-L-lysine solution on the bottom of the chamber slides, distribute homogeneously (the entire surface must be covered, e.g., 150 µl for 8-well chamber slide), incubate for minimum 1 h at 37 °C and finally remove the leftover Poly-L-lysine solution that is on the chamber slides.
Note: Store the chamber slides until use in a 37 °C incubator when using them on the same day or else store the chamber slides at 4 °C. - Aspirate the medium of the HEK293T cells that got transfected the day before.
- Add 100 µl of TrypLE Express to each well (in case of 24-well plate).
- Pipette up and down several times with a P200 pipet to ensure single cell suspension.
- Add 240 µl pre-warmed DMEM medium (HEK293T cells) to the coated chamber slides.
- Take 10 µl of the trypsinized HEK293T suspension and add this to the coated chamber slide. Let the HEK293T cells grow until they reach a confluency of 80%-90%. This will be probably around Days 5-7 post-transfection.
- Prior to the image session, add Hoechst 33342 to the cell culture medium to a final concentration of 0.1 µg/ml.
- Incubate the cells for 10 min at 37 °C with 5% CO2 environment.
- Start imaging session.
- A few hours before transferring the transfected cells, coat the glass chamber slides with Poly-L-lysine. Pour a few hundred microliters of 0.2% Poly-L-lysine solution on the bottom of the chamber slides, distribute homogeneously (the entire surface must be covered, e.g., 150 µl for 8-well chamber slide), incubate for minimum 1 h at 37 °C and finally remove the leftover Poly-L-lysine solution that is on the chamber slides.
- Flow cytometry analysis
Depending on the type of experiment, the best time point for flow cytometry analysis should be determined by the experimenter. In this protocol, we analyze HEK293T reporter cells at 5 days post-transfection. Important note when performing flow cytometry: Make use of proper controls for setting up your gating.
Quantification of correction and indel formation events by flow cytometry analysis can be achieved as followed:- Aspirate the medium of dishes.
- Add 1 ml of TrypLE Express per 10 cm dish.
- Incubate for 3 min at 37 °C.
- Once the cells detach, add 4 ml of DMEM medium and transfer the cells to a 15 ml conical tube.
- Centrifuge at 250 x g for 5 min at 4 °C.
- Aspirate the supernatant.
- Add 1 ml FACS buffer with or without Sytox Red to the cell pellet and pipette up and down several times with P1000 pipet to ensure single cell suspension. Sytox Red can be added for excluding dead cells from the populations.
- Transfer the cell suspension to 5 ml test tubes with cell strainer cap.
- Place samples on ice until starting with the flow cytometry analysis on the BD LSR Fortessa cell analyzer.
- Next Generation Sequencing (NGS)
For the library preparation, collected cell pellets should be extracted by using for example a DNeasy Blood and Tissue kit, according to the manufacturer’s instructions. Preparation of the library:- PCR to amplify the target region
- For each 50 µl reaction mix, combine the following in 0.2 ml tubes:
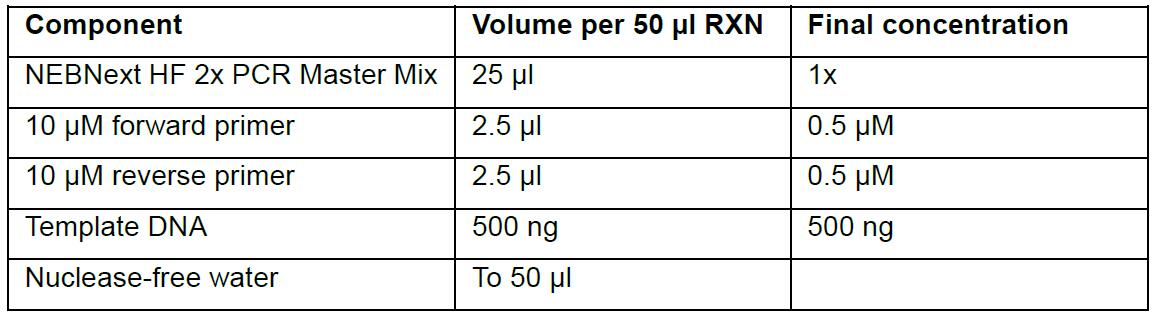
- Gently mix the reaction and briefly spin down content in a microcentrifuge.
- PCR cycling conditions used in this protocol:
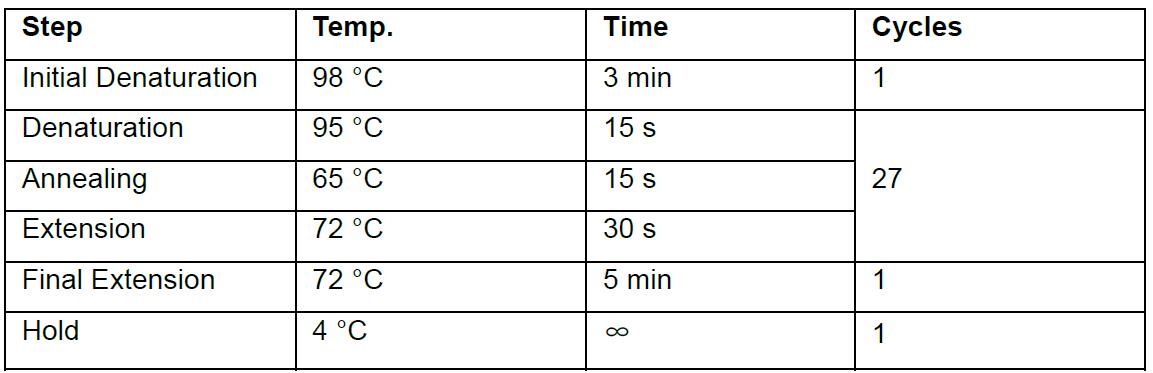
Note: Annealing temperatures should be optimized for each primer set to ensure that a single amplicon will be produced. - Run 10 µl of the PCR products on agarose gel (2%) to confirm correct amplicon size.
- Purify PCR amplicons by solid phase reversible immobilization (SPRI) bead cleanup using Agencourt AMPure XP reagent, according to the manufacturer’s protocol.
- Run 5 µl of the purified samples on agarose gel (2%) to confirm the purification step succeeded.
- For each 50 µl reaction mix, combine the following in 0.2 ml tubes:
- Generation of pooled sequencing libraries
For generating the libraries, order Index Adaptor Sequences for the second amplification step. In this protocol, TruSeq Index Adaptor Sequences were used from Illumina (see Publication Savić et al., 2018).- For each 50 µl reaction mix, combine the following in 0.2 ml tubes:
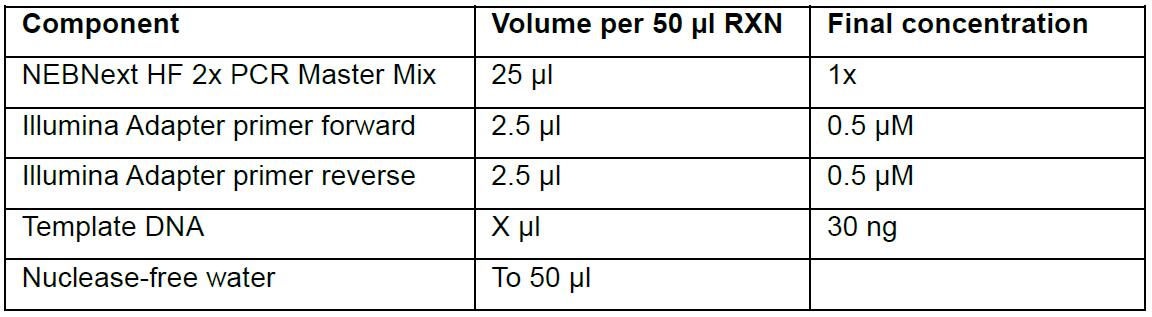
- Gently mix the reaction and briefly spin down content in a microcentrifuge.
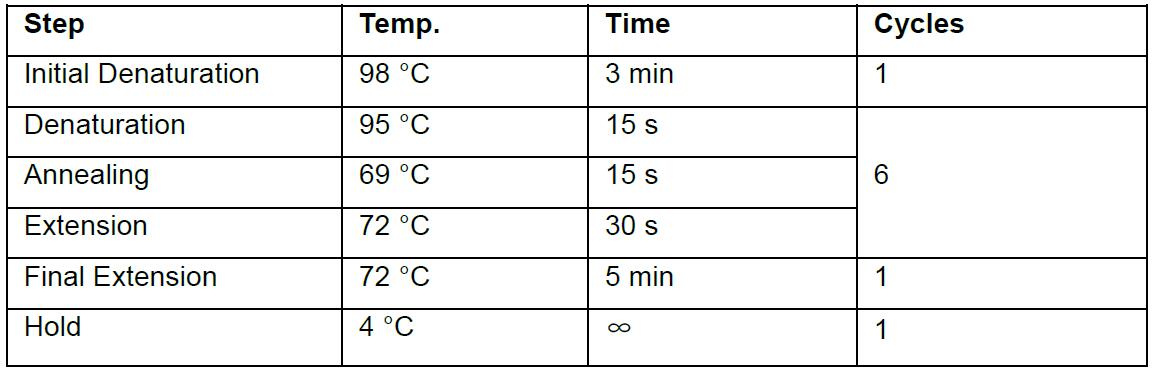
- PCR cycling conditions used were as follows in this protocol:
- Purify the PCR products (PCR amplicons with adaptors) by solid phase reversible immobilization (SPRI) bead cleanup using Agencourt AMPure XP reagent, according to the manufacturer’s protocol.
- Run 5 µl of the purified samples on an agarose gel (2%) to confirm the purification step succeeded and that the adaptors are added. Additional tip: run one PCR product from the first PCR along the PCR samples of the second PCR on the agarose gel. This will show a clear difference when the Index Adapters are successfully added to the PCR amplicons.
Note: Before sequencing the libraries, it is recommended to perform a quality check on, e.g., Agilent 2200 TapeStation system.
- For each 50 µl reaction mix, combine the following in 0.2 ml tubes:
- PCR to amplify the target region
- Microscopy imaging
- Seeding HEK293T cells
Data analysis
Correction efficiencies in HEK293T fluorescent reporter cells are available in Savić et al., 2018. For obtaining correction rates in other cell types the here described protocols would have to be adapted.
- Flow cytometry
- Example gating strategy
- Forward versus side scatter (FSC-A vs. SSC-A) gating should be used to identify cells of interest.
- Doublets should be excluded using the forward scatter height versus forward scatter area density plot (FSC-H vs. FSC-A).
- Live cells should be gated based on Sytox-Red-negative staining.
- Live-gated cells should be further used to quantify the percentage of eGFP negative and turboRFP positive populations, in this example.
- Correction efficiency calculation
Correction efficiency (percentage of corrections in edited cells):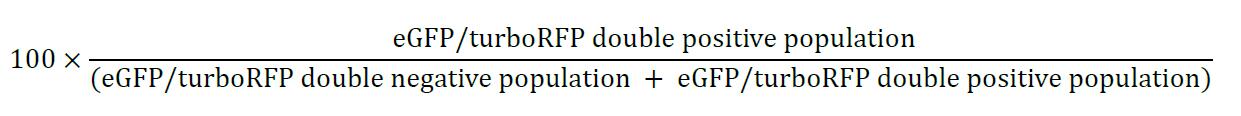
- Example gating strategy
- NGS
The Scripts for mapping sequencing data, counting mutations and generating plots are available at https://github.com/HLindsay/Savic_CRISPR_HDR.
Correction efficiency calculation:
Percentage of edited alleles is calculated as: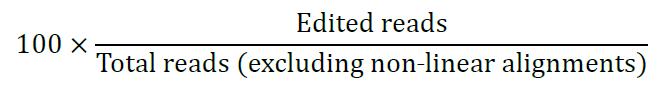
Correction efficiency is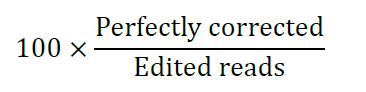
Recipes
- HEPES 200 mM pH 8.5
Dissolve 4.77 g of HEPES in 75 ml of dH2O
Adjust to desired pH using 10 M NaOH
Fill to final volume of 100 ml with dH2O
Autoclave and store at 4 °C - HEK293T medium
Dulbecco modified Eagle’s medium (high glucose) with GlutaMAXTM (DMEM), supplemented with 1% Pen-Strep and 10% FBS
Store at 4 °C - K562 medium
RPMI 1640 Medium with GlutaMAXTM, supplemented with 1% Pen-Strep and 10% FBS
Store at 4 °C - mESC proliferative growth medium
Dulbecco’s modified Eagle media (DMEM) (Sigma) supplemented with 15% FBS, 100 U/ml LIF, 0.1 mM 2-β-mercaptoethanol and 1% Pen-Strep - Gelatin solution 0.2% (w/v) (500 ml)
Dilute 1 g of gelatin from porcine skin with 500 ml of autoclaved water
Mix well and then autoclave the solution
Store at 4 °C for 1 week - Poly-L-lysine solution 0.2%
Prepare a stock solution by dissolving 100 mg poly-L-lysine in 100 ml water and filter sterilize through a 0.22-μm filter - FACS buffer
PBS supplemented with 1% FBS and 5 mM EDTA - EDTA buffer 0.5 M pH 8.0
Dissolve 93.06 g of EDTA in 400 ml of dH2O
Adjust to desired pH using 10 M NaOH or solid NaOH pellets
Fill to final volume of 500 ml with dH2O
Filter the solution through a 0.5 μm filter
Autoclave and store at room temperature - Protein buffer
20 mM HEPES and 500 mM KCl, pH 7.5 - KCl buffer 500 mM
Dissolve 1864 g of KCl in 500 ml dH2O - 3-(N-morpholino) propanesulfonic acid (MOPS) 10x running buffer
Dissolve 42 g of MOPS, 8.2 g of sodium acetate, 3.72 g EDTA in 1 L dH2O, pH 7.0
Filter sterilize or autoclave
Store at room temperature protected from light - Tris-borate-EDTA (TBE) 10x buffer
Dissolve 108 g Tris base (121 g/mol), and 55 g Boric acid (61.8 g/mol)
Add 40 ml of 0.5 M EDTA (pH 8.0)
Adjust final volume to 1 L with double distilled and deionized water
Store at room temperature - Tris buffer 1 M
Dissolve 60.57 g Tris (hydroxymethyl) aminomethane (121.4 g/mol) in 0.5 L dH2O
Adjust pH to 8.0 using HCl - TE buffer pH 8.0
10 mM Tris, bring to pH 8.0 with HCl, 1 mM EDTA
To sterilize, autoclave the solution on a liquid cycle
Store at room temperature - HPLC eluent A
0.1 M triethylammonium acetate in RNase-free ultrapure H2O, pH 8.0
50.5 g triethylamine, 30 g acetic acid, add RNase-free ultrapure H2O to 5 L
Adjust pH to 8.0 using triethylamine and acetic acid
Filter sterilize - LC/MS eluent A
0.4 M 1,1,1,3,3,3-hexafluoro-2-propanol, 15 mM triethylamine
Dissolve 67.2 g 1,1,1,3,3,3-hexafluoro-2-propanol, 1.5 g triethylamine, in 1 L RNase-free ultrapure H2O
Filter sterilize - Chloramphenicol 1,000x (34 mg/ml stock)
Dissolve 0.34 g Chloramphenicol in 10 ml of 100% ethanol
Filter sterilize through a 0.22-μm filter
Store at -20 °C - Kanamycin 1,000x (50 mg/ml stock)
Dissolve 0.5 g kanamycin sulfate in 10 ml of sterile water
Filter sterilize through a 0.22-μm filter
Store at -20 °C - IPTG 1 M (IsoPropyl-1-Thio-β-D-Galactopyranoside)
Dissolve 2.38 g IPTG in 10 ml of sterile water
Filter sterilize through a 0.22-μm filter
Store at -20 °C - NaCl 5 M
Dissolve 292.2 g NaCl in 1 L of double distilled and deionized water
Filter sterilize through a 0.22-μm filter - DTT 1 M (Dithiothreitol)
Dissolve 1.54 g DTT in 10 ml of double distilled and deionized water
Filter sterilize through a 0.22-μm filter
Store at -20 °C - Imidazole 2.5 M pH 8.0
Dissolve 170.19 g Imidazole in 750 ml of dH2O
Adjust to desired pH using 10 M NaOH
Fill to final volume of 1 L with dH2O
Filter sterilize through a 0.22-μm filter - HEPES 1 M pH 7.5
Dissolve 238.3 g of HEPES in 750 ml of dH2O
Adjust to desired pH using 10 M NaOH
Fill to final volume of 1 L with dH2O
Filter sterilize through a 0.22-μm filter - Protease inhibitor (1:1,000)
Dissolve 5 mg Pepstatin A, 500 mg AEBSF Hydrochloride in 50 ml 100% EtOH - KCl 4 M
Dissolve 298.2 g KCl in 1 L of dH2O
Filter sterilize through a 0.22-μm filter - Tris buffer 1 M pH 6.8
Dissolve 6.06 g Tris (hydroxymethyl) aminomethane (121.4 g/mol) in 40 ml dH2O
Adjust pH to 6.8 using HCl
Fill to final volume of 1 L with dH2O
Filter sterilize through a 0.22-μm filter - Lysis buffer
2 ml Tris pH 8.0 (20 mM)
10 ml NaCl (500 mM)
0.2 ml Imidazole pH 8.0 (5 mM)
Protease inhibitor (1:1,000)
Fill to final volume of 100 ml with dH2O
Filter sterilize through a 0.22-μm filter - Wash buffer
20 ml Tris pH 8.0 (20 mM)
100 ml NaCl (500 mM)
2 ml Imidazole pH 8.0 (5 mM)
Fill to final volume of 1 L with dH2O
Filter sterilize through a 0.22-μm filter - Elution buffer
20 ml Tris pH 8.0 (20 mM)
50 ml NaCl (250 mM)
80 ml Imidazole pH 8.0 (200mM)
Fill to final volume of 1 L with dH2O
Filter sterilize through a 0.22-μm filter - Heparin buffer A
10 ml Hepes pH 7.5 (20 mM)
12.5 ml KCl (100 mM)
Fill to final volume of 500 ml with dH2O
Filter sterilize through a 0.22-μm filter - Heparin buffer B
10 ml HEPES pH 7.5 (20 mM)
250 ml KCl (2 M)
Fill to final volume of 500 ml with dH2O
Filter sterilize through a 0.22-μm filter - SEC buffer
20 ml HEPES pH 7.5 (20 mM)
125 ml KCl (250 mM)
Fill to final volume of 1 L with dH2O
Filter sterilize through a 0.22-μm filter - 1x Laemmli buffer
Dilute 100 μl of 5x Laemmli Buffer with 400 µl of dH2O - 5x Laemmli buffer
2.25 ml Tris pH 6.8
5 ml glycerol (100%)
0.5 g SDS
5 mg Bromophenol blue
2.5ml DTT
Final volume will be 10 ml
Filter sterilize through a 0.22-μm filter
Store at -20 °C
Acknowledgments
This work has been funded by the Swiss National Science Foundation PMPDP3_171388 (to NS), 31003A_160230 (to GS), 31003A_149393 (to MJ) and by the Vallee Scholar Award from Vallee Foundation (to MJ).
Competing interests
The authors declare no competing interests.
References
- Aird, E. J., Lovendahl, K. N., St. Martin, A., Harris, R. S. and Gordon, W. R. (2018). Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Communications Biology 1(1): 54.
- Carlson-Stevermer, J., Abdeen, A. A., Kohlenberg, L., Goedland, M., Molugu, K., Lou, M. and Saha, K. (2017). Assembly of CRISPR ribonucleoproteins with biotinylated oligonucleotides via an RNA aptamer for precise gene editing. Nat Commun 8(1): 1711.
- Chu, V. T., Weber, T., Wefers, B., Wurst, W., Sander, S., Rajewsky, K. and Kuhn, R. (2015). Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol 33(5): 543-548.
- Gu, B., Posfai, E. and Rossant, J. (2018). Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat Biotechnol 36(7): 632-637.
- Gutschner, T., Haemmerle, M., Genovese, G., Draetta, G. F. and Chin, L. (2016). Post-translational regulation of Cas9 during G1 enhances homology-directed repair. Cell Rep 14(6): 1555-1566.
- Hendel, A., Bak, R. O., Clark, J. T., Kennedy, A. B., Ryan, D. E., Roy, S., Steinfeld, I., Lunstad, B. D., Kaiser, R. J., Wilkens, A. B., Bacchetta, R., Tsalenko, A., Dellinger, D., Bruhn, L. and Porteus, M. H. (2015). Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol 33(9): 985-989.
- Howden, S. E., McColl, B., Glaser, A., Vadolas, J., Petrou, S., Little, M. H., Elefanty, A. G. and Stanley, E. G. (2016). A Cas9 variant for efficient generation of indel-free knockin or gene-corrected human pluripotent stem cells. Stem Cell Reports 7(3): 508-517.
- Kim, S., Kim, D., Cho, S. W., Kim, J. and Kim, J. S. (2014). Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24(6): 1012-1019.
- Lee, K., Conboy, M., Park, H. M., Jiang, F., Kim, H. J., Dewitt, M. A., Mackley, V. A., Chang, K., Rao, A., Skinner, C., Shobha, T., Mehdipour, M., Liu, H., Huang, W. C., Lan, F., Bray, N. L., Li, S., Corn, J. E., Kataoka, K., Doudna, J. A., Conboy, I. and Murthy, N. (2017). Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat Biomed Eng 1: 889-901.
- Liang, X., Potter, J., Kumar, S., Ravinder, N. and Chesnut, J. D. (2017). Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J Biotechnol 241: 136-146.
- Lin, S., Staahl, B. T., Alla, R. K. and Doudna, J. A. (2014). Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 3: e04766.
- Maruyama, T., Dougan, S. K., Truttmann, M. C., Bilate, A. M., Ingram, J. R. and Ploegh, H. L. (2015). Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol 33(5): 538-542.
- Rahdar, M., McMahon, M. A., Prakash, T. P., Swayze, E. E., Bennett, C. F. and Cleveland, D. W. (2015). Synthetic CRISPR RNA-Cas9-guided genome editing in human cells. Proc Natl Acad Sci U S A 112(51): E7110-7117.
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L. and Corn, J. E. (2016). Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol 34(3): 339-344.
- Savić, N., Ringnalda, F. C., Lindsay, H., Berk, C., Bargsten, K., Li, Y., Neri, D., Robinson, M. D., Ciaudo, C., Hall, J., Jinek, M. and Schwank, G. (2018). Covalent linkage of the DNA repair template to the CRISPR-Cas9 nuclease enhances homology-directed repair. Elife 7: e33761.
- Song, J., Yang, D., Xu, J., Zhu, T., Chen, Y. E. and Zhang, J. (2016). RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat Commun 7: 10548.
- Staahl, B. T., Benekareddy, M., Coulon-Bainier, C., Banfal, A. A., Floor, S. N., Sabo, J. K., Urnes, C., Munares, G. A., Ghosh, A. and Doudna, J. A. (2017). Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat Biotechnol 35(5): 431-434.
- Wang, M., Zuris, J. A., Meng, F., Rees, H., Sun, S., Deng, P., Han, Y., Gao, X., Pouli, D., Wu, Q., Georgakoudi, I., Liu, D. R. and Xu, Q. (2016). Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc Natl Acad Sci U S A 113(11): 2868-2873.
- Yin, H., Song, C. Q., Suresh, S., Wu, Q., Walsh, S., Rhym, L. H., Mintzer, E., Bolukbasi, M. F., Zhu, L. J., Kauffman, K., Mou, H., Oberholzer, A., Ding, J., Kwan, S. Y., Bogorad, R. L., Zatsepin, T., Koteliansky, V., Wolfe, S. A., Xue, W., Langer, R. and Anderson, D. G. (2017). Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat Biotechnol 35(12): 1179-1187.
- Yu, C., Liu, Y., Ma, T., Liu, K., Xu, S., Zhang, Y., Liu, H., La Russa, M., Xie, M., Ding, S. and Qi, L. S. (2015). Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 16(2): 142-147.
- Zuris, J. A., Thompson, D. B., Shu, Y., Guilinger, J. P., Bessen, J. L., Hu, J. H., Maeder, M. L., Joung, J. K., Chen, Z. Y. and Liu, D. R. (2015). Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol 33(1): 73-80.
Article Information
Copyright
![]() Savić et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Savić et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Savić, N., Ringnalda, F. C., Berk, C., Bargsten, K., Hall, J., Jinek, M. and Schwank, G. (2019). In vitro Generation of CRISPR-Cas9 Complexes with Covalently Bound Repair Templates for Genome Editing in Mammalian Cells. Bio-protocol 9(1): e3136. DOI: 10.21769/BioProtoc.3136.
- Savić, N., Ringnalda, F. C., Lindsay, H., Berk, C., Bargsten, K., Li, Y., Neri, D., Robinson, M. D., Ciaudo, C., Hall, J., Jinek, M. and Schwank, G. (2018). Covalent linkage of the DNA repair template to the CRISPR-Cas9 nuclease enhances homology-directed repair. Elife 7: e33761.
Category
Molecular Biology > DNA > Mutagenesis
Cell Biology > Cell engineering > CRISPR-cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.