- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Microirradiation for Precise, Double-strand Break Induction in vivo in Caenorhabditis elegans
Published: Vol 8, Iss 24, Dec 20, 2018 DOI: 10.21769/BioProtoc.3130 Views: 5412
Reviewed by: Neelanjan BoseLivia UlicnaJuan Facundo Rodriguez Ayala
Original research article
The authors used this protocol in:

Jan 2018
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
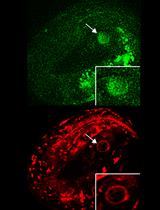

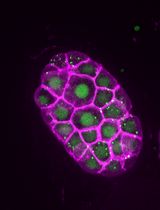
Abstract
DNA double-strand breaks (DSBs) are toxic lesions that every cell must accurately repair in order to survive. The repair of DSBs is an integral part of a cell life cycle and can lead to lethality if repaired incorrectly. Laser microirradiation is an established technique which has been used in yeast, mammalian cell culture, and Drosophila cell culture to study the regulation of DSB repair. Up to our studies, this method has not been adapted for use in a whole, live, multicellular organism to study this repair in vivo. We have recently shown that this system can be used for study of the recruitment of vital repair proteins to microirradiation-induced breaks in the transparent nematode Caenorhabditis elegans. With the integration of microirradiation and imaging technology, we can precisely induce DSBs in target nuclei and study the recruitment of fluorescently tagged repair proteins from the time of damage induction. Whole, live worms are plated and immobilized for targeting of nuclei, and immediately following induction the targeted region can be imaged for up to an hour and a half post-microirradiation. This method is the first that allows for study of DNA repair protein kinetics in vivo in an intact organism, which can be adapted in numerous ways to allow for study of repair kinetics in various aspects of the repair process.
Keywords: MicroirradiationBackground
DNA double-strand breaks (DSBs) are one of the most toxic forms of DNA damage and can be induced exogenously (e.g., UV damage) or endogenously (e.g., SPO-11-induced meiotic DSBs). Studying the recruitment of proteins to the sites of DSBs provides valuable information regarding how the process, and specific proteins involved, are regulated. Microirradiation has been extensively used in cell culture as a means of studying DNA repair (Aten et al., 2004; Kong et al., 2009). This method, coupled with fluorescently-labeled proteins and time-lapse imaging, has provided critical information regarding the regulation of proteins in specific cellular contexts. However, this incredibly tractable method had not been adapted for use in a whole, live, multicellular organism.
In our paper, we describe a laser microirradiation method which applies this technology to intact, live worms (Koury et al., 2018). With the use of a UVA 365 nm pulsed laser, and fluorescently tagged repair proteins RPA-1 and RAD-51, we were able to precisely induce DSBs in specific regions of the C. elegans germline to study DSB repair kinetics in meiotic tissue in vivo. RPA-1 and RAD-51 are ssDNA binding proteins that are essential for DSB repair using homologous recombination. The transparent nature of worms allows for live imaging of fluorescent proteins without any form of dissection or gonad extrusion, and no pre-sensitization of the worms is required for efficient damage induction. Not only is this method straight-forward, but it can be adapted for, and applied to, numerous questions in the field of DSB repair and regulation in both meiotic and somatic tissues.
Materials and Reagents
Note: We indicated our vendors and cat numbers for lab reagents such as tips, tubes, and chemicals, however, any vendor will likely be suitable and sufficient.
- 0.2-20 μl Pipette tips (VWR, catalog number: 89079-438)
- Microscope slides, 1” x 3” x 0.4” (Surgipath-Leica, catalog number: 3800240)
- Petri dishes, any 60 x 15 mm plate with ventilation ribs (Kord-Valmark, catalog number: 2901)
- Glass bottom dishes: 35 mm dish with 14 mm glass microwell (No. 1.5 coverglass, MatTek, catalog number: P35G-0.170-14-C)
- Parafilm, 4” x 125’ (Parafilm M, catalog number: PM996)
- Pasteur pipet, 5 ¾” Flint Glass (Fisher Scientific, catalog number: 50-930-565)
- General-Purpose Laboratory Labeling Tape, ¾” (VWR, catalog number: 89098-004)
- 12 mm tube (any type of tube with a 12 mm diameter would work; VWR, catalog number: 20170-579)
- Worm pick, hand-made by attaching platinum wire to a Pasteur pipette using fire
- 0.1-μm polystyrene beads (Polybead, Polysciences, catalog number: 00876, store at 4 °C)
- Dissection blade (Stainless Steel Surgical Blades, 4-311)
- Escherichia coli OP50 (from the Caenorhabditis Genetics Center)
- Agarose (any brand of Agarose that is typically used for gel electrophoresis, e.g., VWR, catalog number: 9012-36-6)
- Immersion oil, 100x/1.4 NA (Leica Microsystems, catalog number: 11513859)
- Sodium chloride (NaCl) (RPI, catalog number: 7647-14-5)
- Peptone (RPI, catalog number: 73049-73-7)
- Cholesterol (Sigma-Aldrich, catalog number: 57-88-5)
- Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: 10035-04-8)
- Magnesium sulfate (MgSO4) (Sigma-Aldrich, catalog number: 7487-88-9)
- Agar (RPI, catalog number: 9002-18-0)
- Potassium dihydrogen phosphate (KH2PO4) (RPI, catalog number: 7778-77-0)
- Sodium phosphate dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: 7558-80-7)
- Ethanol
- Nematode growth medium (NGM) agar plate (see Recipes)
- M9 buffer (see Recipes)
- 10% agarose pads (see Recipes)
Equipment
- Microscope suitable for live imaging (such as Leica DMi8) with Andor MicroPoint 365 nm pulsed laser (Leica, model: Leica DMi8)
- Humidity chamber (box with wet paper towels and elevated surface)
- Microwave (any brand should work, we use Panasonic Inverter, model: NN-S543BFR)
- Stereo microscope (Leica Microsystems, model: KL200 LED)
- 20 °C incubator (Nor-Lake Scientific Refrigerated Incubator, LRI201WWW/0)
- 37 °C incubator (Precision Scientific Thelco Incubator, model: 31483)
- Autoclave
- 4 °C refrigerator
Software
- MetaMorph Software (Molecular Devices, LLC, version 7.8.12.0)
- Fiji (Fiji is just ImageJ) (Free to download at imagej.net/Fiji) (Schindelin et al., 2012)
- GraphPad Prism v6 (GraphPad Software, La Jolla California USA, www.graphpad.com/)
Procedure
- Worm preparation
Worm husbandry instruction can be found in WormBook (Stiernagle, 2006).- Pour 10 ml of NGM agar media (see Recipes) into 60 x 15 mm Petri dishes and allow to set for 2 days before seeding with E. coli OP50.
- Seed with OP50 from a fresh starter and incubate at 37 °C overnight. Culture C. elegans strains to be used for live imaging experiments on these plates and maintain at 20 °C.
- Pick L4 stage worms of the desired strain the day before the planned live imaging experiment so that worms are one-day old adults for the experiment. We move worms using a pick and identify the L4 stage by the shape of the vulva. However other methods such as synchronization by bleaching can be used (Porta-de-la-Riva et al., 2012).
- Live Imaging Sample Preparation
See Figure 1 for a visual representation of the following 3 steps.- Place 6 μl of M9 buffer in the center of the glass bottom dish to prepare the live imaging sample. Pick the desired number of worms and place into the drop of M9, taking care to transfer as little bacteria as possible.
Note: We typically place a maximum of 5 worms in the M9 buffer due to the limited number of stages we can save on the Leica DMi8. - Gently remove as much of the M9 buffer as possible without drying out the worm and taking care not to suck up the worm into the pipette tip.
- Place 0.75 μl of polystyrene beads from stock onto the worm. Gently place a small, circular cut-out of the 10% agarose pad onto the worm(s), taking care not to drag the worms in the process, that will cover most of the exposed coverslip. We use a 12 mm tube to cut these circles. 10% agarose pad preparation is detailed under Recipe 3.
Note: This step is best done quickly to avoid desiccation of the worm(s). We use a dissection blade to lift and place the agarose pad. - Observe the worms under a dissection microscope. Once worms have stopped moving underneath the pad (typically ~3 min), place the lid on top of the glass bottom dish and parafilm the lid onto the Petri dish (this prevents drying out of the agarose pad and the worm). Draw a vertical mark on the parafilm to use as a reference for setting of coordinates on the microscope (Figure 2).
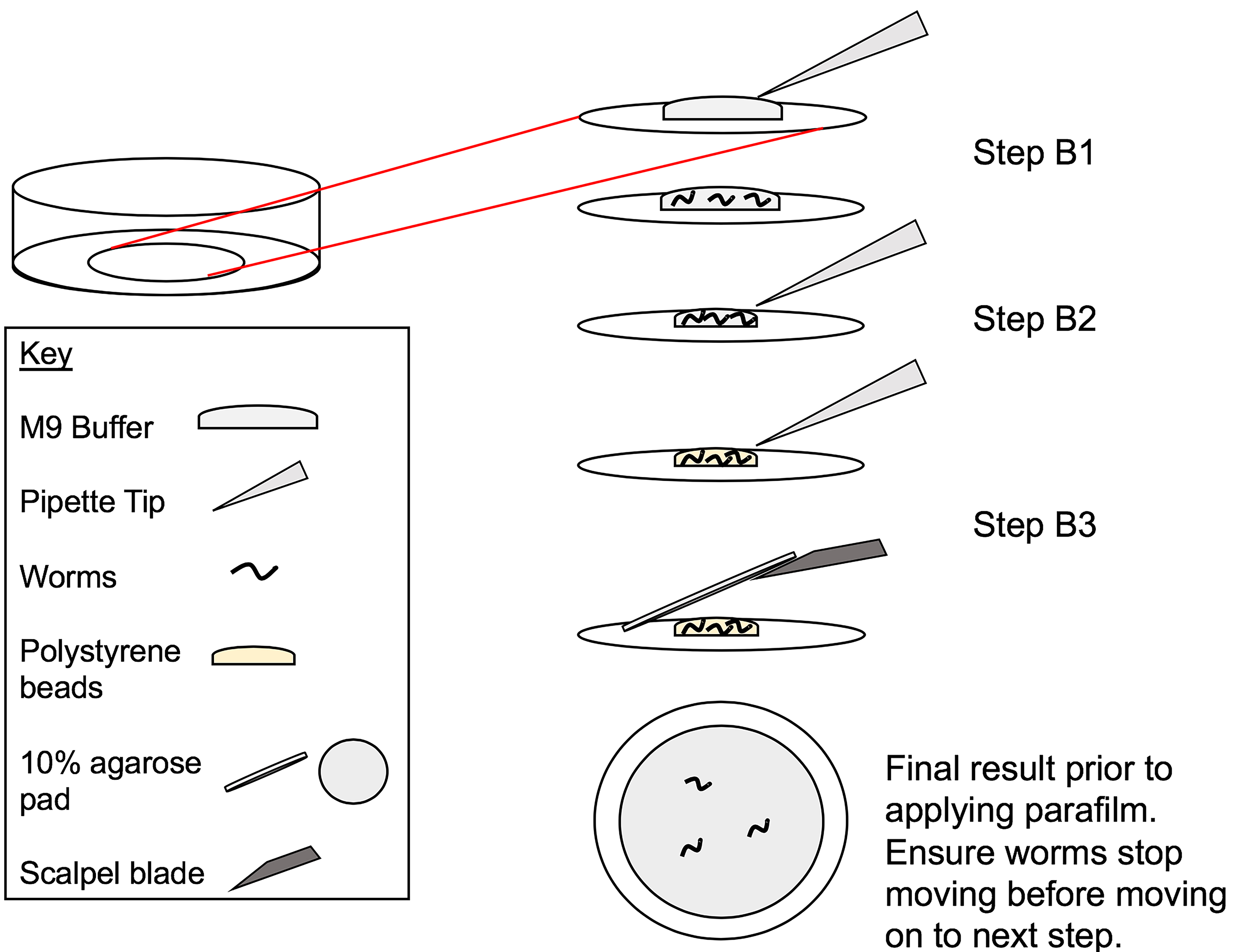
Figure 1. Live imaging sample preparation. Steps B1-B3 from Section B of Procedures illustrating the steps for proper live imaging preparation prior to applying parafilm to the glass-bottom dish. Steps B1-B3 refer to the specific step in the procedures that the image is illustrating.
Figure 2. Glass bottom dish set-up. A. Glass bottom dish ready for imaging. B. Glass bottom dish on the microscope. - Place 6 μl of M9 buffer in the center of the glass bottom dish to prepare the live imaging sample. Pick the desired number of worms and place into the drop of M9, taking care to transfer as little bacteria as possible.
- MetaMorph imaging setup and parameters
- Imaging should be done in a room that maintains constant temperature at 20-23 °C and in the dark.
- Turn on the Leica Inverted Microscope, as well as the Andor MicroPoint software. Ensure that the scope is fully on before opening the MetaMorph software.
- Place the glass bottom dish on the scope and align your vertical reference mark on the parafilm to a location on the scope platform so that any XY-coordinates saved will be the same after the objective change (Figure 2B). Locate the worms using the 20x objective and save their XY-coordinates on the Leica Inverted Microscope. Remove the glass bottom dish before changing objectives.
- Set the objective to 100x and place a small drop of 100x/1.4 NA immersion oil directly onto the objective lens. Ensure that magnification in the MetaMorph toolbar is set to 100x. Otherwise the microirradiation technology will not work. This will automatically raise the objective. Lower it before replacing the glass bottom dish on the platform.
- Place glass bottom dish back onto the scope platform, using your vertical reference mark on the parafilm to orient the glass bottom dish exactly where it was before, ensuring that your saved XY-coordinates will be the same for all of your worms.
- In MetaMorph, open up Apps > Multi Dimensional Acquisition (MDA). This will open a window that allows you to set the parameters for your time-lapse imaging (Figure 3).
- Under the “Wavelengths” tab, specify the number of wavelengths for the experiment (e.g., 2 wavelengths, one for brightfield (BF) and one for GFP). A corresponding tab for each of these wavelengths will open and under each specify the exposure times. We use 20 ms for BF and 500 ms for GFP. Check the box for “Z series with the wavelength” to ensure that the Z series you set for the experiment will be acquired all the way through the set range in that specific wavelength (Figure 3C).
- Set intensity of the light source to 5%.
- To visualize the worm on the computer within the MetaMorph program, hit the button with the small camera and play icon (Figure 3E). This will pull up a live feed from the microscope which you can then use to set the appropriate stages.
- Under the “Stages” tab of the MDA window set the stage, or location, of the area of the worm to target in X, Y, and Z planes. These stages can be saved to be revisited for adjustments, and these will be the stages that are automatically moved to and imaged during the acquisition period (Figure 3B).
- Under the “Z Series” tab, specify the number of steps to be acquired around the central Z plane specified when setting the stage (the middle of the nucleus, in our case). The number of steps and the size of each step through the Z plane can be set under this tab (Figure 3D).
- Under the “Timelapse” tab set the time interval and duration of experiment/number of time points (e.g., every 2 min for 60 min; Figure 3A). Be sure to take into account the number of stages you have and how many Z planes are acquired at each stage and allow for enough time between each interval acquisition for each stage to be acquired. It takes time to go through the planes at each step, and even more time if multiple wavelengths are selected.
- Specify where the output from the program will be saved in the “Saving” tab in the MDA window.
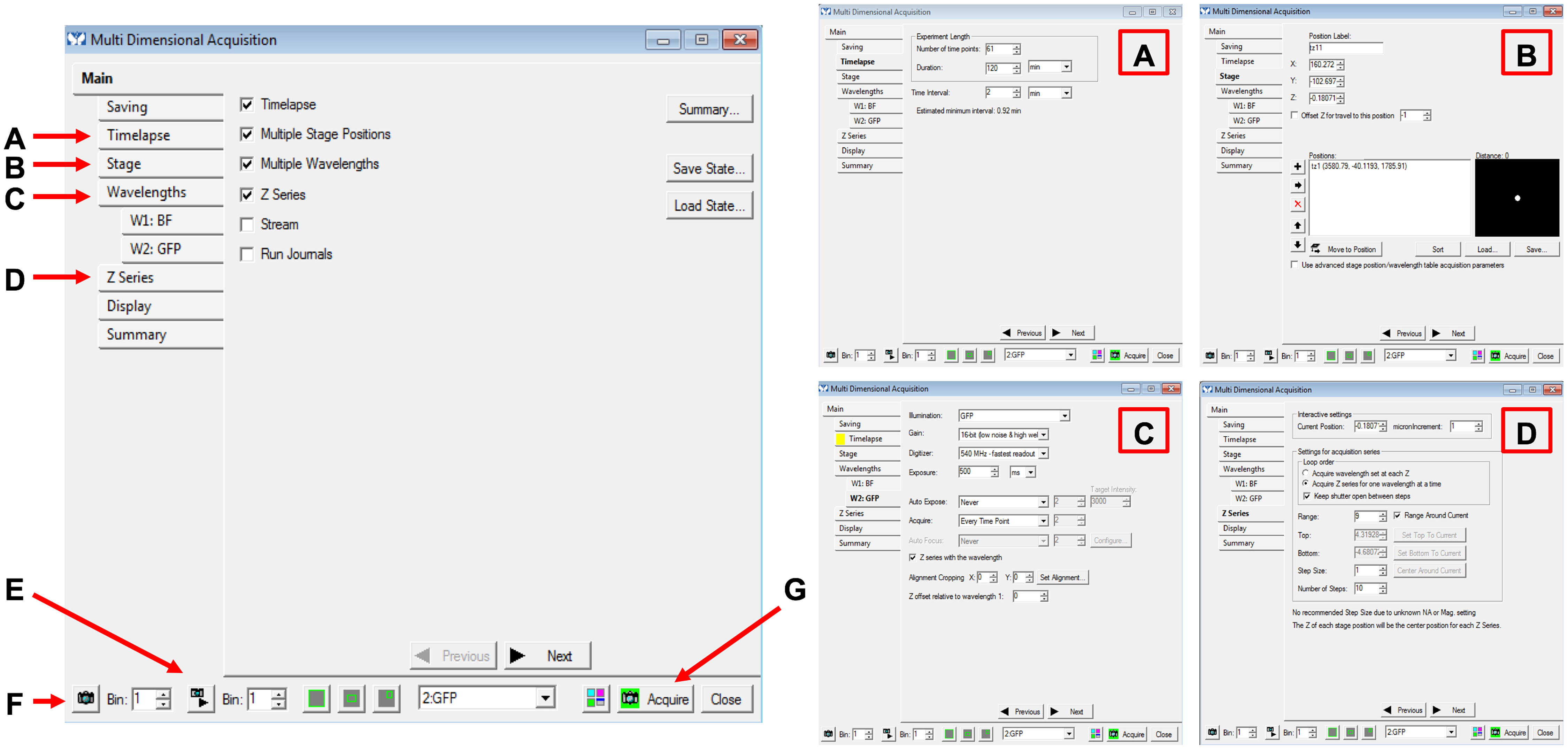
Figure 3. Multi Dimensional Acquisition. Screenshot of the Main configuration for movie acquisition (left) with corresponding tabs expanded (right).
- Microirradiation
- In MetaMorph, open up Devices > MicroPoint Targeted Illumination. This is used for microirradiation of the nuclei and can be set at various attenuations according to the type of DNA damage desired (e.g., Koury et al. [2018] uses 15% attenuation, however, microirradiation-induced foci can be observed with as low as 5%, although in reduced numbers).
- For Koury et al. (2018) the following parameters were set:
- “Configuration” tab (Figure 4A)
- Illumination setting (during pulse): Micropoint
- Coordinate system setting (required): 100x
- Display images during calibration (box checked)
- Range for calibration point (% of maximum range): 40%
- Number of pulses: 1
- Attenuation plate: 15% of transmission
- “Location” tab (Figure 4B)
- Coordinate system setting: 100x
- Target location: All regions
- Transfer target location to acquired image (box checked)
- “Timelapse” tab
- # of points: 6
- Interval: 1
- Units: microseconds
- Acquire to stack with current settings
- “Configuration” tab (Figure 4A)
- In the MDA window, once all desired parameters have been set, hit “Acquire” (Figure 3G).
- After the first time point is recorded, immediately pause the program. Visit each saved stage, take a snapshot (camera icon in the toolbar), and then use the circle selection tool from the toolbar to target selected nuclei. Hit pulse (“Location” tab, Figure 4B) after nuclei selection for each stage. Select nuclei and pulse for each stage before moving on to the next. Resume time lapse imaging and allow program to run through desired time frame.
Note: Perform this step as quickly as possible to ensure that all stages were microirradiated at around the same time.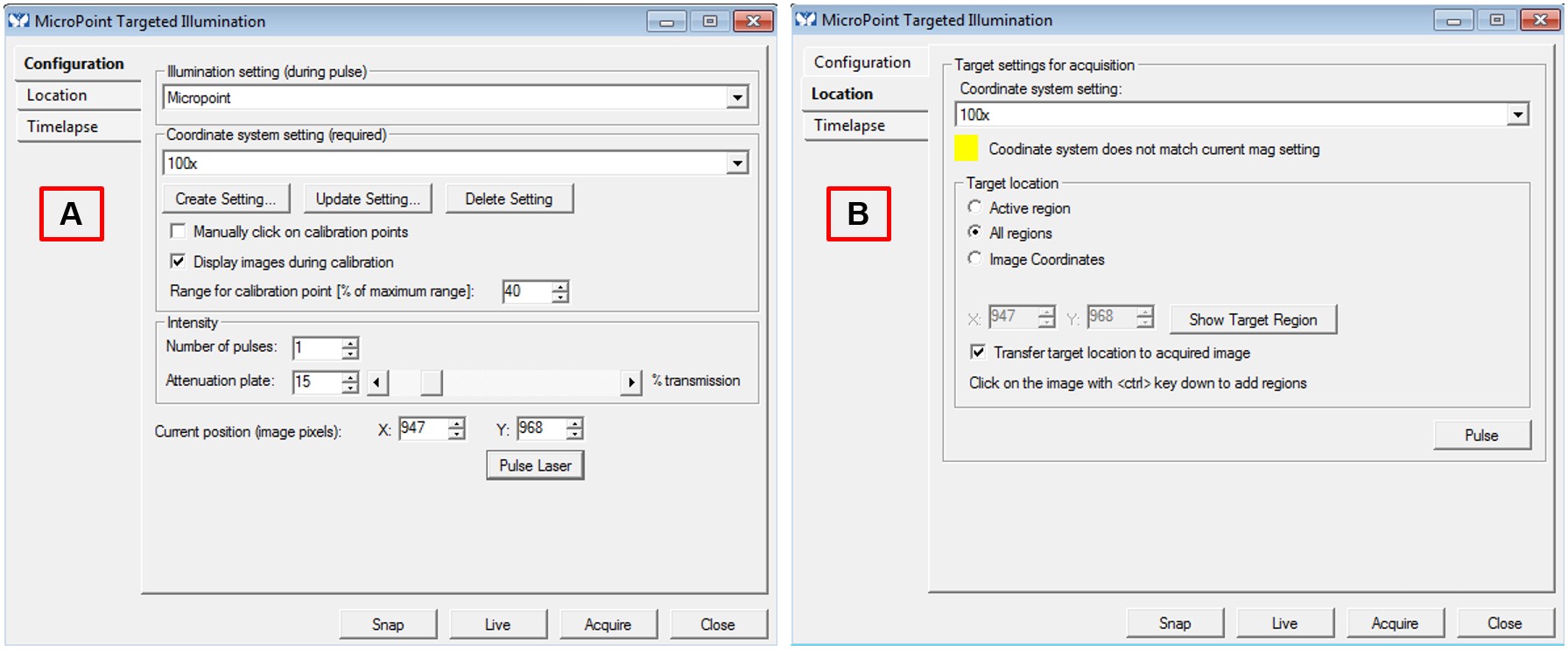
Figure 4. MicroPoint Targeted Illumination. Screenshot of the “Configuration” (A) and “Location” (B) tabs for the MicroPoint Targeted Illumination parameters. Settings for microirradiation power and location of damage induction are configured in these two windows.
- Recovery (optional)
Worms can only be viably maintained on the glass bottom dish for up to 2 h. Many experiments require analysis in later time points or analysis by other methods (e.g., dissection for immunofluorescence staining-based experiments). In this case, you will need to recover the worms.- Remove the parafilm from the glass bottom dishes to recover worms after microirradiation. Place 350 μl of M9 over the agarose pad.
- Gently lift the edge of the pad to allow M9 to wash underneath and lift the pad off of the worm(s).
Note: We use the dissection blade mentioned in the glass bottom dish Preparation procedure to lift the agarose pad at this step. - Use a glass Pasteur pipet to transfer the worm(s) to an NGM plate, to be used for any follow-up experiments.
Data analysis
- Image export
- In MetaMorph, open Apps > Review Multi Dimensional Acquisition. Under this window highlight the fluorescent channel you wish to review. Number of time points and which Z planes to include in the exported file can be specified here.
- Once desired time points and Z planes are chosen, hit the button “Create Rotation” under the “Z Projection” tab. Hit “Create” in the second window prompt. Save the selection as a TIFF for further analysis in Fiji (File > Save). See Video 1 for the resulting file.
 Video 1. GFP::RAD-51 in spo-11 background. Example output of the gfp::rad-51; spo-11 strain before (first frame) and after microirradiation (all following frames). Green = GFP::RAD-51. A sample analysis of this same movie is presented in Figure 5.
Video 1. GFP::RAD-51 in spo-11 background. Example output of the gfp::rad-51; spo-11 strain before (first frame) and after microirradiation (all following frames). Green = GFP::RAD-51. A sample analysis of this same movie is presented in Figure 5.
- Statistical analyses
Following manual quantification of foci in Fiji-ImageJ (see Figure 5), perform an unpaired nonparametric test (we used the Mann-Whitney test). If multiple comparisons are made in one experiment, the Kruskal-Wallis test should be performed to determine if any of the groups are significantly different than the others before performing pair-wise comparisons. Statistics can be performed with the GraphPad Prism software.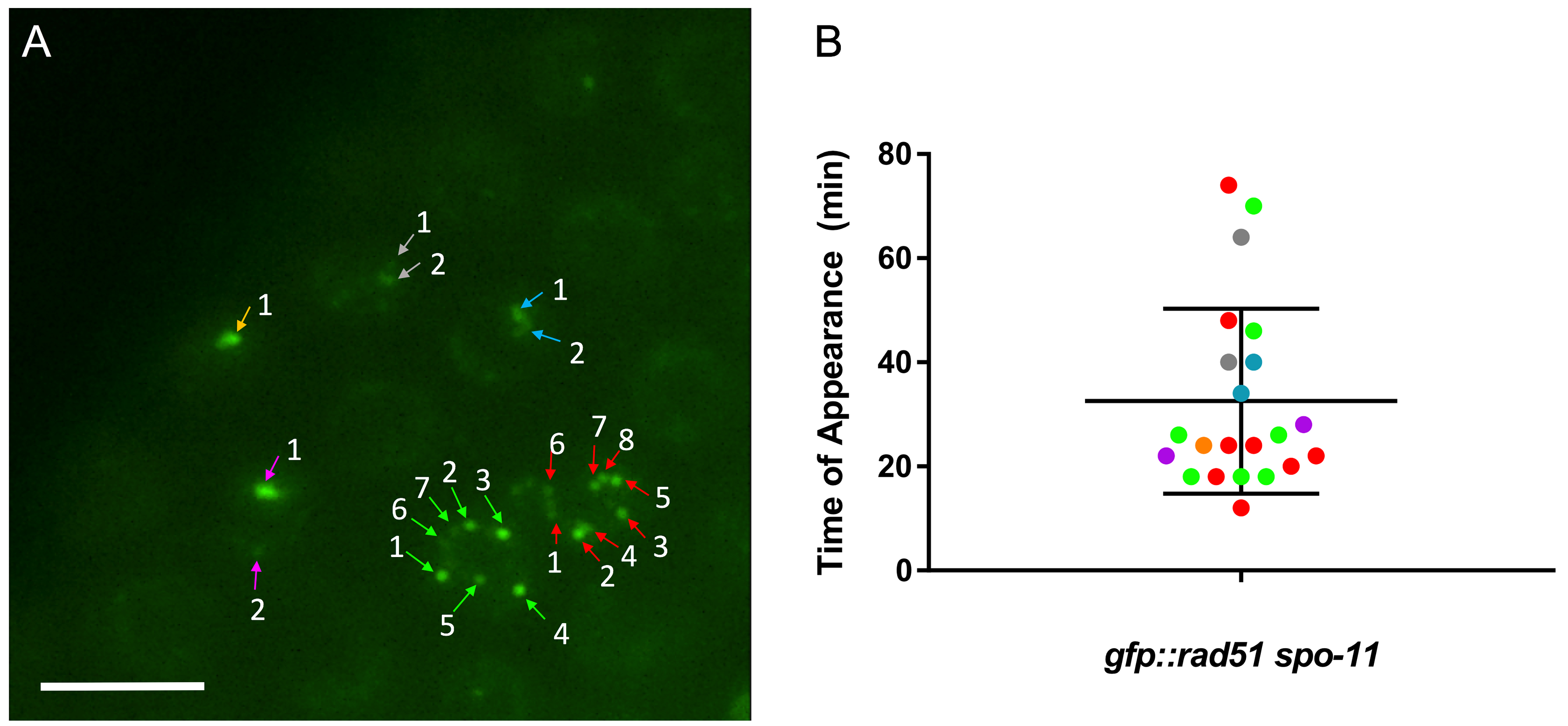
Figure 5. Example data set. A. Following data export from MetaMorph, images such as the one above can be analyzed in Fiji-ImageJ. Numbers indicate order of appearance of foci in each nucleus. See Video 1 for full video. Scale bar = 6 μm. B. Quantification of time of appearance of each focus (in minutes) in all 6 microirradiated nuclei for the strain gfp::rad-51 spo-11. Colors correspond to the nucleus that the measurement was taken from in A. Error bars are average ± SD (SEM can also be used). For more examples of quantified data output, see Koury et al. (2018).
Notes
- Limit exposure of the worms to the GFP channel when setting stages. Over-exposure can bleach the worms and your desired fluorescently-tagged protein if exposed for too long. Try to stay in bright field as much as possible. We typically see bleaching around an hour and a half into imaging with exposure to GFP occurring every 2 min.
- Up to five worms can be imaged at the same time on the same glass bottom dishes at 2-min intervals, however any more than 5 worms, or at intervals less than 2 min, will likely result in bleaching of the worms before the end of the desired time interval. For shorter intervals, you may need to use less worms.
Recipes
Note: For more detail see Stiernagle, (2006).
- Nematode growth medium (NGM) agar plates
3 g NaCl
2.5 g Peptone
1 ml of 5 mg/ml Cholesterol in ethanol
1 ml 1 M CaCl2
1 ml 1 M MgSO4
20 g Agar
Add distilled H2O up to 1 L
Mix and autoclave (50 min at 250 °F) to sterilize
Let cool down to a little above room temperature before adding 25 ml 1 M KH2PO4
Ensure mix is at pH 6
Pour into Petri dishes and store at 4 °C - M9 buffer
Add all the following ingredients together in 1 L vessel
3 g KH2PO4
6 g Na2HPO4
5 g NaCl
1 ml of 1 M MgSO4
Add water up to 1 L
Sterilize by autoclaving (50 min at 250 °F) and store at room temperature - 10% agarose pads
20 ml M9 buffer
2 g agarose- Stir 2 g of agarose into 20 ml of M9 to create a 10% agarose + M9 solution
- Microwave the solution, stirring every 5 s, until the solution is clear and runny. Stir continuously before using for pad making to avoid any solidification. This will also prevent excessive accumulation of bubbles
- Place two microscope slides with two layers of lab tape on top of each on either side of a clean microscope slide that does not have tape. Place a ½” glob of the agarose solution on the clear slide and immediately flatten with another clear microscope slide crosswise to the orientation of the bottom slide (see Figure 6)
Note: Move quickly to avoid cooling of the agarose solution. - Store in a humidity chamber to avoid drying out
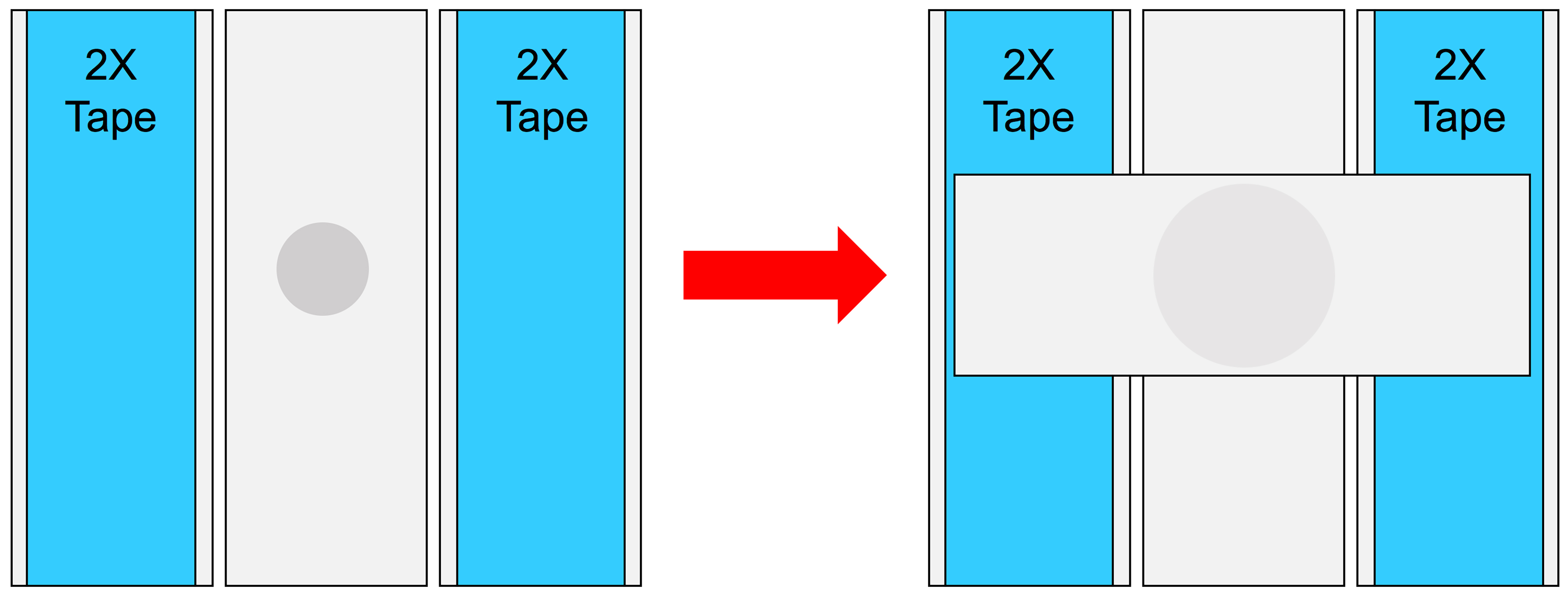
Figure 6. Agarose pad preparation. Illustration of the pressing of heated agarose to form agarose pads for live imaging sample preparation as detailed in Recipe 3.
Acknowledgments
This work was funded by the National Institutes of Health (NIH) [R01GM112657 to S.S.]. We thank Dylan Cooke for critically reading this manuscript. We are grateful to Yizhi Yin who helped in the initial stages of protocol development.
Competing interests
The authors have no conflicts of interest or competing interests.
References
- Aten, J. A., Stap, J., Krawczyk, P. M., van Oven, C. H., Hoebe, R. A., Essers, J. and Kanaar, R. (2004). Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science 303(5654): 92-95.
- Kong, X., Mohanty, S. K., Stephens, J., Heale, J. T., Gomez-Godinez, V., Shi, L. Z., Kim, J. S., Yokomori, K. and Berns, M. W. (2009). Comparative analysis of different laser systems to study cellular responses to DNA damage in mammalian cells. Nucleic Acids Res 37(9): e68.
- Koury, E., Harrell, K. and Smolikove, S. (2018). Differential RPA-1 and RAD-51 recruitment in vivo throughout the C. elegans germline, as revealed by laser microirradiation. Nucleic Acids Res 46(2): 748-764.s
- Porta-de-la-Riva, M., Fontrodona, L., Villanueva, A. and Ceron, J. (2012). Basic Caenorhabditis elegans methods: synchronization and observation. J Vis Exp(64): e4019.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Stiernagle, T. (2006). Maintenance of C. elegans. WormBook: 1-11.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Harrell, K. E., Koury, E. and Smolikove, S. (2018). Microirradiation for Precise, Double-strand Break Induction in vivo in Caenorhabditis elegans. Bio-protocol 8(24): e3130. DOI: 10.21769/BioProtoc.3130.
Category
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.