- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Method for Studying the Effect of Gene Silencing on Bacterial Infection-induced ERK1/2 Signaling in Bone-marrow Derived Macrophages


(*contributed equally to this work) Published: Vol 8, Iss 24, Dec 20, 2018 DOI: 10.21769/BioProtoc.3123 Views: 7249
Reviewed by: Shantibhusan SenapatiHaixia XuAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Macrophages are highly phagocytic cells that utilize various pathogen recognition receptors (PRRs) to recognize pathogen-associated molecular patterns (PAMPs). These PAMPs can be present within the microbe, such as bacterial CpG DNA, and are recognized by Toll-like receptor 9 (TLR9), a PRR present on the endosomal membrane of macrophages. PAMPs can also be present on the surface of microbes, such as Lipopolysaccharide (LPS), which decorates the outer membrane of gram-negative bacteria like Salmonella typhimurium and Escherichia coli. LPS is recognized by TLR4 present on the plasma membrane of macrophages, and LPS-TLR4 association leads to activation of signaling cascades including MAPK phosphorylation, which in turn promotes macrophage activation and microbial killing. This protocol describes the method for studying the role of a gene of interest in Extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) signaling, induced by bacterial infection in primary bone-marrow derived macrophages (BMDMs).
Keywords: Bone-marrow derived macrophagesBackground
Macrophages are phagocytic cells which can either be resident to specific tissues as Kupffer cells (in the liver) and peritoneal macrophages (in peritoneal cavity) or, can enter tissues in response to an infection. The primary function of macrophages involves phagocytosis and clearance of old damaged cells, and recycling of nutrients in the serum (recently reviewed in Shapouri-Moghaddam et al., 2018). However, for macrophages to clear microbes, there exists a need for their activation. Macrophages obtained from many tissues such as alveoli and peritoneal cavity includes high numbers of pre-activated macrophage population. However, macrophages derived from myeloid progenitor cells present in the bone marrow are comparatively naive and more responsive to activating stimulus (Epelman et al., 2014). Upon encounter of gram-negative bacteria by macrophages, LPS present on either the surface of a bacterium or shed by bacteria in the blood flow is captured by LBP (LPS-binding protein) and presented as a ligand to TLR4, a type I transmembrane protein present on the plasma membrane of macrophage that mediate the recognition of PAMPS such as LPS (Shimazu et al., 1999). The engagement of LPS with TLR4 (along with other co-stimulatory molecules) leads to recruitment of several adaptor proteins at the cytoplasmic tail of TLR4 followed by a cascade of intracellular events leading to activation of the nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) signaling cascades downstream to TLR4 including ERK1/2 (Buscher et al., 1995; Schaeffer and Weber, 1999; Gay et al., 2014; Platko et al., 2018; Arya et al., 2018). These signaling pathways directly or indirectly phosphorylate and activate various transcription factors that lead to the expression of genes involved in production and release of pro-inflammatory cytokines, reactive oxygen species, nitrosative burst and promotes macrophage activation (Satoh and Akira, 2016). Thus, ERK1/2 signaling plays an important role in augmenting macrophage response against intracellular pathogens. This protocol describes a method to investigate the role of a host factor of interest in modulating ERK1/2 activation in primary BMDMs in response to Salmonella typhimurium infection (see schematic shown in Figure 1 and Arya et al., 2018).
Materials and Reagents
- 5 ml syringe (Dispovan)
- 60-mm tissue culture dish (BD Falcon®, catalog number: 353002)
- 10-cm tissue culture dish (BD Falcon®, catalog number: 353003)
- 6-well tissue culture plate (BD Falcon®, catalog number: 353046)
- 50 ml Falcon tube (BD Falcon®, catalog number: 352070)
- 15 ml Falcon tube (BD Falcon®, catalog number: 352096)
- 14 ml round-bottom polypropylene tube (Corning, catalog number: 352006)
- 1.5 ml microcentrifuge tube (MCT) (Tarson, catalog number: 50010)
- 0.2 micron PVDF membrane (Bio-Rad, catalog number: 162-077)
- X-ray film (Carestream, catalog number: 6574958)
- Aluminum foil
- Cell scraper (HiMedia, catalog number: TCP104)
- Sterile 22 G needle (Dispovan)
- C57BL/6 male mice (typically 6-8 weeks old)
- Salmonella typhimurium strain SL1344
- RPMI medium 1640 (Lonza, catalog number: 12702F)
- Fetal bovine serum (FBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10082147)
- HEPES (1 M) (Thermo Fisher Scientific, GibcoTM, catalog number: 15630080)
- MEM Non-Essential Amino Acids Solution (100x) (Thermo Fisher Scientific, GibcoTM, catalog number: 11140-050)
- Antibiotic-Antimycotic (100x) (Thermo Fisher Scientific, GibcoTM, catalog number: 15240-062)
- Sodium Pyruvate (100 mM) (Thermo Fisher Scientific, GibcoTM, catalog number: 11360-070)
- GlutaMAXTM Supplement (Thermo Fisher Scientific, GibcoTM, catalog number: 35050-061)
- DPBS (Thermo Fisher Scientific, GibcoTM, catalog number: 14190-144)
- PBS, pH 7.4 (Thermo Fisher Scientific, GibcoTM, catalog number: 10010-023)
- Trypsin-EDTA (0.05%) (Thermo Fisher Scientific, GibcoTM, catalog number: 25300-054)
- Trypan Blue Solution (Thermo Fisher Scientific, GibcoTM, catalog number: 15250-061)
- Mouse M-CSF (macrophage-colony stimulating factor) (eBiosciences, catalog number: 34-8983)
- ACK (Ammonium-Chloride-Potassium) lysing buffer (Thermo Fisher Scientific, GibcoTM, catalog number: A10492-01)
- Protease inhibitor (Sigma-Aldrich, catalog number: P8340)
- Phosphatase inhibitor (Roche, catalog number: 4906837001)
- Bradford reagent (Sigma-Aldrich, catalog number: B6916)
- TWEEN® 20 (Sigma-Aldrich, catalog number: P9146)
- TritonTM X-100 (Sigma-Aldrich, catalog number: T8787)
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: L3771)
- Sodium deoxycholate (DOC) (Sigma-Aldrich, catalog number: D6750)
- Trizma® base (Tris base) (Sigma-Aldrich, catalog number: T6066)
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: 3014)
- Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA) (Sigma-Aldrich, catalog number: 3889)
- Ethylenediaminetetraacetic acid disodium salt dehydrate (EDTA-Na2) (Sigma-Aldrich, catalog number: E5134)
- Gentamicin solution (Sigma-Aldrich, catalog number: G1272)
- Streptomycin sulfate (Sigma-Aldrich, catalog number: S6501)
- Ethanol (Merck, catalog number: 108543)
- Skim milk (BD Difco, catalog number: 232100)
- LB broth (BD Difco, catalog number: 244620)
- SS agar (HiMedia, catalog number: M108)
- Laemmlisample buffer (Bio-Rad, catalog number: 161-0747)
- ON-TARGETplus Non-targeting Control siRNA Pool (Dharmacon, catalog number: D-001810-10)
- ON-TARGETplus Mouse Arl11 siRNA (Dharmacon, catalog number: L-055672-01-0005)
- Polyclonal anti-total ERK1/2 antibody (Cell Signaling Technology, catalog number: 4695)
- Polyclonal anti-phospho-ERK1/2 antibody (Cell Signaling Technology, catalog number: 4370)
- Peroxidase AffiniPure Goat Anti-Rabbit IgG (H+L) (Jackson ImmunoResearch, catalog number: 111-035-144)
- ECL Plus Western Blotting Substrate (Pierce, catalog number: 32132)
- 70% ethanol (see Recipes)
- Complete RPMI medium (see Recipes)
- Mouse M-CSF stock solution (0.4 mg/ml) (see Recipes)
- SS agar plate (see Recipes)
- Streptomycin stock solution (50 mg/ml) (see Recipes)
- LB broth (see Recipes)
- Opsonization buffer (see Recipes)
- Growth medium (GM) for infection (see Recipes)
- RIPA lysis buffer (see Recipes)
- Blocking buffer (see Recipes)
- PBST (see Recipes)
- Antibody dilution buffer (see Recipes)
Equipment
- Beaker
- 0.2 micron polyethersulfone filter
- Scissors and forceps (Fisher Scientific)
- Cell culture CO2 incubator (Thermo Scientific, model: 371)
- Biosafety cabinet class II (ESCO, model: AC2-4S8-NS)
- Inverted tissue culture microscope (Nikon, model: TS2)
- Hemocytometer (Sigma, catalog number: Z359629)
- Multimode reader (Tecan, model: Infinite M200)
- Incubator shaker (New Brunswick, model: Innova 42)
- Automatic X-ray Film Processor (Protec, model: OPTIMAX 2010)
- Flatbed scanner (Microtek, model: Bio-5000)
- Refrigerated centrifuge with plate centrifuge bucket big enough for a 6-well plate (Eppendorf, model: 5810 R)
- Semi-dry horizontal blotting system (Atto, model: WSE-4020)
- SDS-PAGE gel apparatus (Bio-Rad, model: Mini Protean Tetra Cell)
- Dancing shaker (Tarson, model: MC-02)
- Rocking shaker (Tarson, model: 4080)
- Flow cytometry
Software
- ImageJ software (NIH, https://imagej.nih.gov/ij/)
- Microsoft Excel software (MS Office)
Procedure
This protocol is divided into three parts: A) Generation of mouse primary BMDMs; B) Infection of BMDMs with Salmonella typhimurium; and C) Assessment of activated/phosphorylated levels ERK1/2 in Salmonella-infected macrophage cell lysates by Western Blotting. The readers are encouraged to see the schematic diagram shown in Figure 1 that describes the different steps for carrying out this protocol.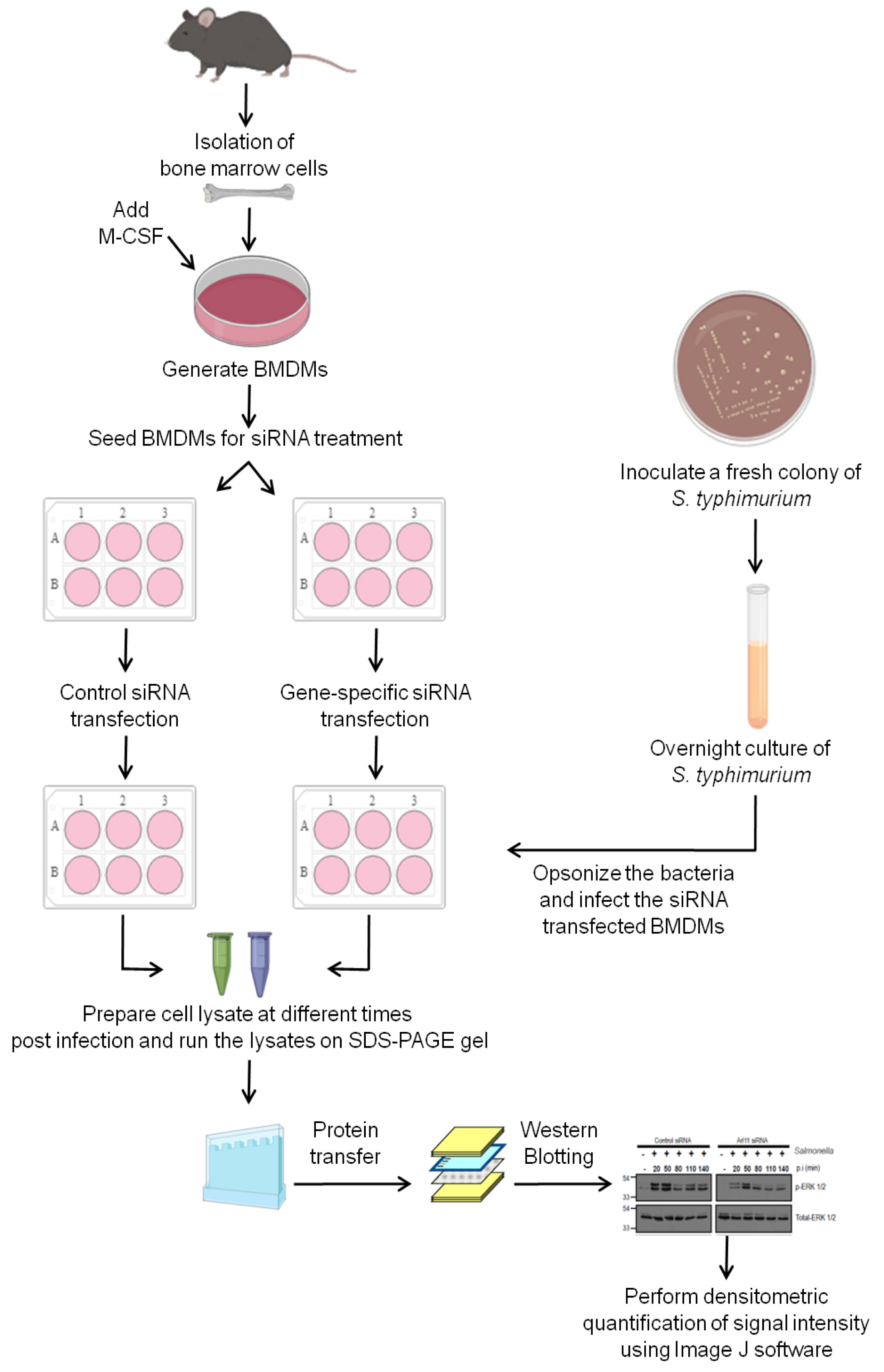
Figure 1. Schematic diagram showing the steps used to investigate the role of a host factor of interest in modulating ERK1/2 activation in primary bone marrow-derived macrophages (BMDMs) in response to Salmonella typhimurium infection. Isolate mouse bone marrow cells and culture in the presence of M-CSF to generate primary BMDMs. After generation of BMDMs transfect the cells with either control siRNA or siRNA against the gene of interest. Post-siRNA treatment, infect the BMDMs with opsonized stationary phase culture of Salmonella typhimurium. At the end of different times post infection, perform cell lysis and run the lysates on SDS-PAGE gel, perform the Western Blotting and develop the signal on X-ray films using chemiluminescent substrate. Quantify the signal intensity from the scanned X-ray films using ImageJ software.
- Generation of mouse primary BMDMs
- Prepare for mouse dissection by keeping three 60-mm tissue culture dishes filled with 70% ethanol (see Recipe 1), DPBS or complete RPMI media (see Recipe 2), and a pair of sterile (autoclaved) forceps and scissors over the clean area inside the biosafety cabinet (BSC). Also, place a square-shaped (12 x 12 inches) aluminum foil cleaned with 70% ethanol to be used as a platform for mouse dissection.
Note: Sterile conditions to be maintained all the time during the experiment. - Sacrifice mouse by cervical dislocation and immerse mouse in a beaker containing 70% ethanol. Using the tip of a small scissor make a cut near the thigh of the mouse and peel off the skin from the top of each hind limb down over the foot using scissors and forceps. Detach hind limb from the body by cutting the caudal bone along with the femur (cut along the bone that feels 90° to the femur). Cut off the foot (tarsus) along with the skin and discard. Keep the de-skinned limbs in the dish containing 70% ethanol for 5 min as this will make muscles dehydrate making them easier to detach from bone (see Figures 2A-2G).
Note: Remove muscles as aim of this step is to prevent contamination of BMDM cultures with muscle and fibroblast cells. - Remove excess muscles from the limbs using scissors while maintaining bone integrity. Detach the knee joint carefully by pressing softly with scissors to cut-off through the ligaments between femur and tibia (see Figures 2H-2I). Keep bones in 70% ethanol for 5 min and then transfer them to the dish containing DPBS for 5 min.
- Hold the bone at the center using a pair of forceps and cut both the ends (~3-6 mm from the end) of the tibia/femur using a scissor. Fill-up the 5 ml syringe with complete RPMI media and flush the bone marrow (BM) through one end of the bone into the 60-mm tissue culture dish containing complete RPMI media. Repeat this step through the other end of the bone to expel the BM from both ends of the bone (Figures 2J-2K).
- Using a 5 ml syringe, pass the collected BM through a 22 G needle 3-4 times to make single cell suspension and collect cells in a 50 ml Falcon tube (Figures 2L-2N).
- Centrifuge the cell suspension at 700 x g for 5 min at room temperature (RT), and decant the supernatant.
- Add 1 ml ACK lysing buffer to the cell pellet to lyse the RBCs and continue tap mixing for an incubation period of 1 min. Immediately add 10 ml DPBS and centrifuge the cell suspension at 700 x g for 5 min at RT (Figure 2O).
Note: Do not exceed the incubation time beyond 1 min as it might cause damage to cells other than RBC, and decrease overall yield of BMDMs. - Wash the cell pellet two times using 10 ml DPBS as described in Step A6.
Note: While adding DPBS observe for muscle contamination. If any muscle debris is present, take it out by using 1 ml pipette. - Following the last wash, resuspend the cell pellet in 15-20 ml of complete RPMI media and count the cells with a hemocytometer under an inverted tissue culture microscope using the trypan blue exclusion principle.
Note: The yield of BM cells varies a lot and accordingly different dilution of trypan blue should be used to get accurate cell count. Typically, one can use a starting dilution of 1:100 of trypan blue with an incubation period of 1 min for cell counting. - Seed ~4 million BM cells in a 10-cm tissue culture dish containing 8 ml of complete RMPI media supplemented with 30 ng/ml of M-CSF (see Recipe 3). Place the dishes in a cell culture incubator at 37 °C and 5% CO2. Consider this step as Day 1.
- Observe for the increase in the percentage of adhered macrophages on Day 2.
- On Day 3, replenish the BM culture with fresh media by adding half the volume of complete RPMI media that was initially used, i.e., 4 ml complete RPMI media supplemented with M-CSF (30 ng/ml).
- Observe for the increase in the percentage of adhered macrophages (around 60%) on Day 4.
- By Day 5, ~90% adherent macrophages will be observed (Figure 3). To use BMDMs in functional experiments, remove the media, and gently wash the cells with 10 ml DPBS. To detach the BMDMs, add 5 ml trypsin and place the dish inside the incubator for 5-10 min at 37 °C. After the incubation period, take a quick look at the cells under the microscope to confirm detachment. Next, perform trypsin neutralization by adding 5 ml complete RPMI media. Using a 1 ml pipette tip gently flush the cells and transfer the cell suspension into a 50 ml centrifuge tube.
Note: Perform step A14 very gently to avoid activation of macrophages. - Centrifuge the cell suspension at 700 x g for 5 min at RT, gently resuspend in 10 ml complete RPMI media and perform cell counting using a hemocytometer as described in Step A9.
- At this step, the collected cells can be analyzed for macrophage purity by flow cytometry and are ready for further characterization and functional experiments.
Note: For analyzing the purity of isolated macrophages by flow cytometry, readers can refer to Zhang et al., 2008.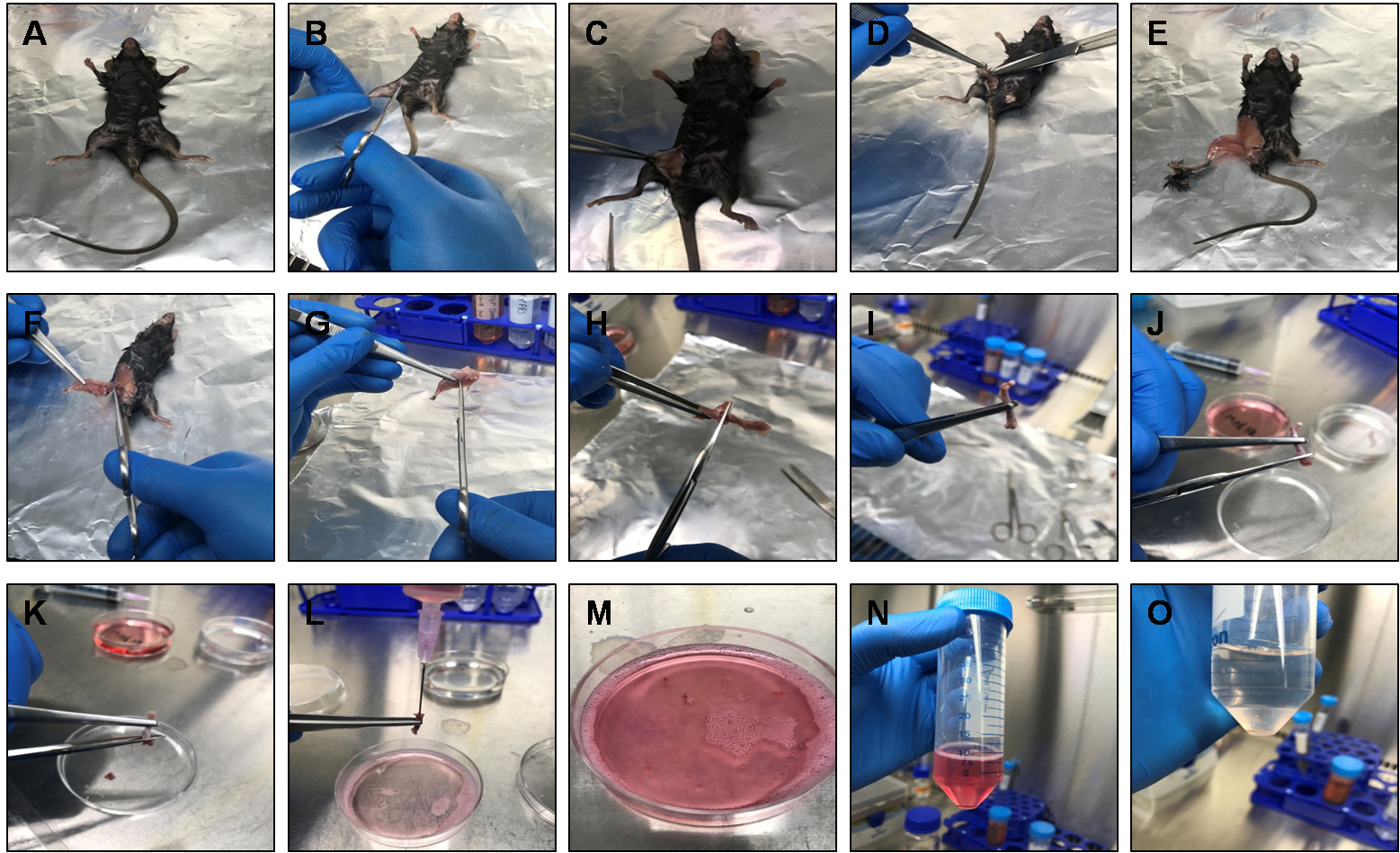
Figure 2. Dissection of mouse for the preparation of BMDMs. A. A 6-8 week-old C57BL/6 male mouse was sacrificed by cervical dislocation and sterilized in 70% ethanol and placed on a clean aluminum foil inside the biosafety cabinet. B-E. Using a small scissor make a cut near the thigh of the mouse and peel off the skin from the top of each hind limb down over the foot using scissors and forceps. F. Detach hind limb from the body by cutting the caudal bone along with the femur. G. Cut off the foot (tarsus) along with the skin. H and I. Remove excess muscles from the limbs using a scissor and detach the knee joint carefully by pressing softly with a scissor to cut-off through the ligaments between femur and tibia. J and K. Hold the bone at the center using forceps and cut both the ends of the tibia/femur using a scissor. L-N. Using a 5 ml syringe, pass the collected BM through a 22 G needle 3-4 times to make single cell suspension and collect cells in a 50 ml Falcon tube. O. Centrifuge and add ACK lysing buffer to the cell pellet to lyse the RBCs. Immediately DPBS and centrifuge the cell suspension to collect BM cells.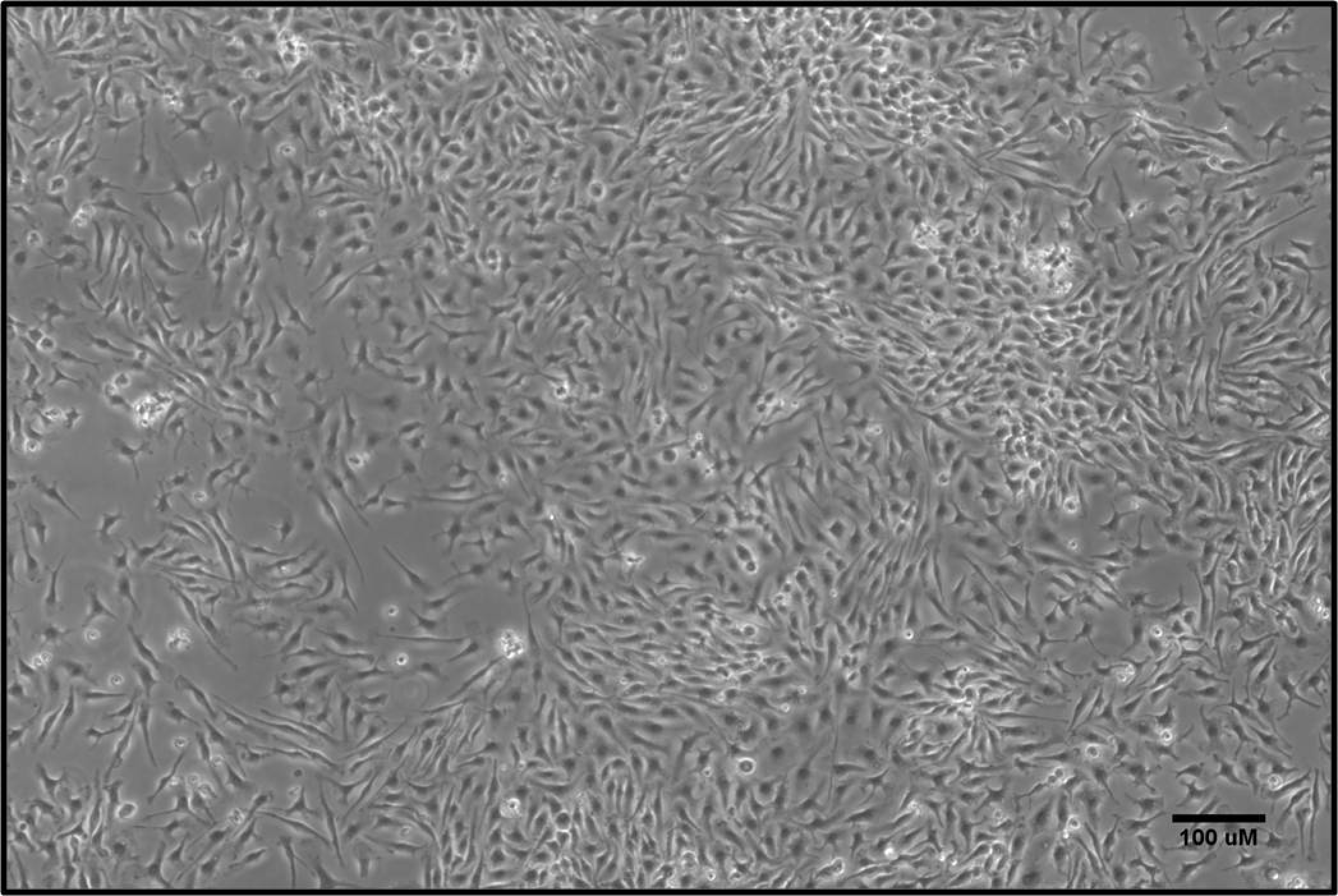
Figure 3. Characterization of mouse BMDMs. BM cells isolated from mouse hind limbs were cultured in the presence of M-CSF. On Day 5, the macrophage population was largely adherent. Scale bar = 100 µm.
- Prepare for mouse dissection by keeping three 60-mm tissue culture dishes filled with 70% ethanol (see Recipe 1), DPBS or complete RPMI media (see Recipe 2), and a pair of sterile (autoclaved) forceps and scissors over the clean area inside the biosafety cabinet (BSC). Also, place a square-shaped (12 x 12 inches) aluminum foil cleaned with 70% ethanol to be used as a platform for mouse dissection.
- Infection of BMDMs with Salmonella typhimurium
- Seed BMDMs at a density of 1 x 106 cells/well in a 6-well tissue culture plate in 2 ml complete RPMI media containing 30 ng/ml per well of mouse M-CSF. The number of 6-well tissue culture plates required will depend upon the number of siRNA treatments to be performed and different time points of infection to be analyzed.
- Next day, replace the media with fresh complete RPMI media containing 30 ng/ml of mouse M-CSF and transfect BMDMs with control siRNA and gene specific siRNA as per the manufacturer’s instructions. To illustrate this protocol, we have performed control siRNA or Arl11 siRNA as described in Arya et al., 2018.
Note: Readers are encouraged to standardize the conditions i.e., the amount of siRNA and treatment time in order to achieve the maximum silencing for the gene of their interest. - To prepare stationary culture for infection, inoculate an isolated colony of Salmonella typhimurium SL1344 strain from a freshly streaked SS agar plate containing 50 μg/ml streptomycin (see Recipes 4 and 5) in 3 ml LB broth (see Recipe 6) supplemented with streptomycin (50 μg/ml) in a 14 ml round-bottom polypropylene tube. Set up an overnight culture (~16 h) in a shaking incubator (200 rpm) at 37 °C. This step can be initiated at 56 h post siRNA treatment of BMDMs so that the duration of siRNA treatment (72 h) is completed by the time Salmonella culture is ready for infection.
Note: Salmonella typhimurium SL1344 strain is streptomycin resistant and streptomycin is added in the culture to prevent growth of any other contaminants. - Measure O.D. of overnight Salmonella culture (diluted to 1/10 in LB broth) at 600 nm wavelength using a spectrophotometer. Transfer the volume of bacterial culture equivalent to an O.D. = 1 (~109 live bacteria) in a MCT. Check the example calculation to estimate the volume required for an O.D. = 1.
Example calculation:
If O.D.600 of 1/10 dilution of overnight bacterial culture = 0.33
Thus, calculated O.D.600 of bacteria in 1 ml culture will be = 0.33 x 10 = 3.3
Number of bacteria equal 109 in 1 ml, i.e., O.D.600 = 1.0
Therefore, volume of bacterial culture required = (1/Actual O.D.600) x 1 ml= (1/3.3) x 1 ml = 0.303 ml
Transfer 0.303 ml of bacterial culture to a fresh MCT and make up the volume to 1 ml by adding 0.697 ml LB broth. - Centrifuge the diluted Salmonella culture at 8,600 x g for 3 min at RT, aspirate 0.9 ml LB broth and add equivalent volume of DPBS. Resuspend the bacterial cells by gentle vortexing and repeat the centrifugation step again to remove the LB broth.
Note: After centrifugation out of 1 ml only 0.9 ml volume is aspirated to prevent any loss of bacteria, and the centrifugation step is repeated twice in order to remove the LB broth completely. - Perform opsonization of bacteria. For efficient adherence of bacteria on the macrophage surface and phagocytosis, the bacterial surface is coated with IgG present in serum. Aspirate 0.9 ml DPBS after completion of Step B5 and resuspend the bacterial pellet in 900 μl opsonization buffer (see Recipe 7). Incubate the bacterial suspension at 37 °C for 20 min with intermittent invert mixing, followed by centrifugation at 8,600 x g for 3 min. Gently aspirate the supernatant (~0.9 ml) and add 0.9 ml DPBS to resuspend the bacterial cell pellet.
Note: After centrifugation out of 1 ml only 0.9 ml volume is aspirated to prevent any loss of opsonized bacteria. - Dilute the opsonized bacteria to 1/10 in GM (see Recipe 8), i.e., 4.5 ml GM (pre-warmed to 37 °C) + 0.5 ml of the opsonized bacteria. This implies, if O.D.600 = 1 means 1 x 109 bacteria/ml then 1/10 dilution will have 108 bacteria/ml.
Note: The Steps B6 and B7 should be performed in separate biosafety cabinets in order to minimize contamination. - Infect the plated BMDMs from Step B2 (i.e., post 72 h siRNA treatment) at a multiplicity of infection (MOI) 50:1. To perform this, remove the complete RPMI media and wash once with DPBS. Then quickly add 0.5 ml of diluted bacteria from Step B7 to each well. MOI is the number of bacteria added per cell during infection. See the example calculation to estimate the volume required to achieve an MOI of 50:1.
Example calculation:
BMDMs plated in a 6-well tissue culture dish at a density of 106 cells per well
So, for MOI = 50:1, number of bacteria required = 106 x 50 = 5 x 107
We have 108 bacteria in 1 ml (as in Step B7)
Therefore for 5 x 107, culture volume required = 1 ml x 5 x 107/108 = 0.5 ml bacterial dilution per well. - To avoid non-uniform phagocytosis of Salmonella by BMDMs synchronize the process by adding 1.5 ml GM to each well of a 6-well tissue culture plate, seal the plate with Parafilm to avoid spillage, and perform a centrifuge at 453 x g at 30 °C
for 5 min. - Remove the Parafilm and transfer the plate to the cell culture incubator at 37 °C and 5% CO2 for 20 min post infection (p.i.) sample.
- Assessment of activated/phosphorylated levels ERK1/2 in Salmonella-infected macrophage lysates by Western Blotting
- Aspirate the media from the wells to remove non-internalized bacteria and give one wash with DPBS (1 ml/well) and remove the DPBS completely without dislodging the cells.
- Preparation of cell lysates
Lyse the uninfected (control) and 20 min p.i. samples by adding ice-cold RIPA buffer supplemented with 1x protease inhibitors and 1x phosphatase inhibitors (0.1 ml per well of 6-well plate) (see Recipe 9). Immediately scrape the cells off the plate and transfer the extract to an MCT properly labeled with sample name. Keep on ice. - To the remaining sample wells, add 2 ml of GM (supplemented with 50 μg/ml gentamicin), and transfer the plate to a tissue culture incubator at 37 °C and 5% CO2 for the next time interval of infection as per your experiment requirement. For example, for the 50 min p.i. sample, incubate the sample well for 30 min. At the end of each time p.i., prepare the cell lysates as described in Step C2.
Note: Gentamicin is added to the media in order to prevent the growth of non-internalized bacteria which are left over even after washing, and also gentamicin exhibit minimal cytotoxicity to macrophages. - Keep the cell lysates on ice for up to 10 min (after scraping), followed by centrifugation at 16,600 x g at 4 °C for 10 min.
- Carefully collect the post nuclear supernatant (PNS) in a fresh MCT without disturbing the pellet. The PNS can be stored at -80 °C if SDS-PAGE is to be performed next day.
- Quantify the concentration of protein present in the PNS by Bradford assay using the manufacturer’s instructions. Normalize all the samples with respect to each other in order to load equal amount of protein (~15-20 μg per lane).
- After normalization calculate the volume required of each sample and transfer to fresh MCT and add Laemmli sample buffer to a final concentration of 1x, boil at 99 °C for 7 min followed by a short spin.
- Load samples onto 10% SDS-PAGE gels and follow the standard Western Blotting procedure.
Note: The SDS-PAGE gels need to be run in duplicate since antibodies against p-ERK1/2 and total-ERK1/2 detect band at the same size. - After performing the protein transfer step (30 Volts for 2 h in cold room), incubate PVDF membrane in blocking buffer (see Recipe 10) for overnight at 4 °C on a horizontal shaker.
- Remove the blocking buffer and wash the membrane three times for 5 min each with 0.05% PBST (see Recipe 11).
- Incubate membrane and primary antibody (at 1:1,000 dilution) in the antibody dilution buffer (0.05% PBST; see Recipe 12) with gentle agitation for 2 h at RT.
- Wash the membrane three times for 5 min each with 0.05% PBST on a dancing shaker.
- Incubate membrane with goat anti-rabbit IgG (HRP-conjugated) antibody (1:5,000 dilution) in 0.05% PBST with gentle agitation for 1 h at RT.
- Remove the secondary antibody solution and wash the membrane three times for 10 min each with 0.3% PBST on a dancing shaker.
- Prepare the ECL reagent as per the manufacturer’s instructions. Incubate the ECL substrate with membrane for 1 min, remove excess solution (membrane remains wet), wrap in plastic and expose to X-ray film and develop the signal in Automatic X-ray film processor (Figure 4).
- Scan the developed X-ray films using the flatbed scanner and perform the densiotometric quantification of phospho- and total-ERK1/2 band signal using ImageJ software as described in the data analysis section below.
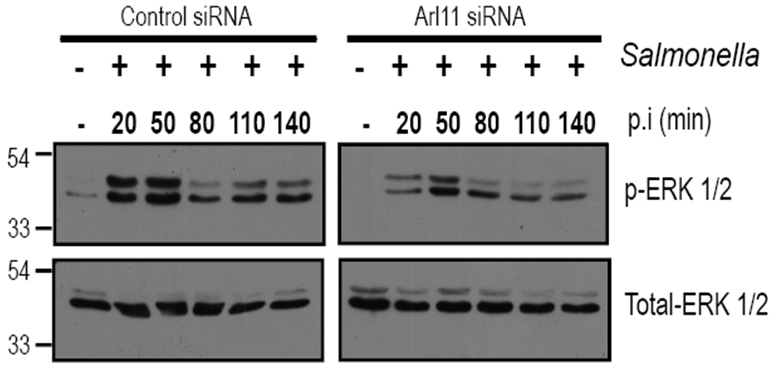
Figure 4. Arl11 depletion in BMDMs impairs ERK1/2 activation upon infection with Salmonella. Control- and Arl11-silenced BMDMs were infected with Salmonella typhimurium for different time periods, and lysates were prepared and blotted with the indicated anti-phospho-ERK1/2 and anti-total-ERK1/2 antibodies.
Data analysis
Densitometric quantification of phospho- and total-ERK1/2 signal using ImageJ software
- Launch ImageJ software and open the scanned images of phospho-ERK1/2 and total-ERK1/2 blots as shown in Figure 5A. Go to “File” → “Open” → “Browse” → “Select File” → “Open”.
- Select the “Rectangle Tool” from the ImageJ toolbar and draw a box which covers the band signal in lane 1 as shown in Figure 5B. Go to “Analyze” → “Gels” → “Select First Lane”.
- Drag and drop the selection from lane 1 to lane 2 as shown in Figure 5C. Then go to “Analyze” → “Gels” → “Select Next Lane”. Repeat this step until all the lanes are selected.
- To get the pixel intensity of each band, go to “Analyze” → “Gels” → “Plot Lanes”. Then select the “Wand tool” from the Image J toolbar and place the cursor (+) at the center of histogram for lane 1 and repeat this step for the next lanes and left click with the mouse to obtain area under the peak (pixel intensity) in a separate box as shown in Figure 5D. To perform this step, make sure “Area Dialog Box” is checked. To check for this, go to “Analyze” → “Set Measurements” → “Check Area Dialog Box” → Hit “OK”.
- Copy the measurements to MS Excel Sheet for both phospho-ERK1/2 and total-ERK1/2 blots for each time point of infection. Then calculate the ratio of phospho-ERK1/2 to total-ERK1/2 signal for each time p.i. and plot a clustered column graph. For more information, readers are encouraged to refer Figures 4a and 4b in Arya et al., 2018.

Figure 5. Densitometric quantification using ImageJ. Screenshots of different steps of densitometric quantification using ImageJ software are shown. For details refer to data analysis section of the protocol.
Recipes
- 70% ethanol
Mix 70 ml of absolute ethanol with 30 ml of sterile distilled water (DW) - Complete RPMI medium
RPMI 1640
10% Heat-inactivated FBS
10 mM HEPES
1 mM Sodium pyruvate
1x Antibiotic-Antimycotic
1x MEM Non-essential Amino acids
1x GlutaMAXTM Supplement
Store at 4 °C - Mouse M-CSF stock solution (0.4 mg/ml)
0.5 mg M-CSF
1.25 ml PBS (pH 7.4)
Filter sterilize using 0.2-micron polyethersulfone filter, aliquot and store at -80 °C
Note: To increase the shelf-life (upto1 year), use 0.1% BSA in PBS (pH 7.4) as solvent. - SS agar plate
6.3 g
100 ml DW
Heat to boiling with frequent agitation to dissolve the medium completely. Cool to about 45 °C, add the required concentration of antibiotic, mix well and pour into sterile Petri plates. Store the plates at 4 °C
Note: Do not autoclave or overheat SS-Agar media as it will destroy the selectivity of the medium. - Streptomycin stock solution (50 mg/ml)
0.5 g Streptomycin sulfate
10 ml DW
Filter sterilize, aliquot and store at -80 °C - LB broth
2.5 g LB mix
100 ml DW
Autoclave and store at RT in sterile conditions - Opsonization buffer
20% FBS in DPBS - GM for infection
RPMI 1640
10% Heat-inactivated FBS
10 mM HEPES
1 mM Sodium pyruvate
1x MEM Non-essential Amino acids
1x GlutaMAXTM Supplement
Store at 4 °C
Note: In the infection media no antibiotic is added in order to prevent arrest and killing of internalized bacteria. - RIPA lysis buffer
10 mM Tris-Cl (pH 8)
140 mM NaCl
1 mM EDTA
0.5 mM EGTA
1% TritonTM X-100
0.1% DOC
0.1% SDS
Store at RT
Note: Add protease and phosphatase inhibitors to RIPA lysis buffer before immediate use. - Blocking buffer
5% w/v non-fat dry milk in 0.05% PBST - PBST
Required percentage of TWEEN® 20 in PBS
Mix well by stirring and store at RT - Antibody dilution buffer
Primary and secondary antibodies dilution (as per the antibody datasheet) made in 0.05% PBST
Acknowledgments
This protocol was adapted and modified from previously published studies (Sindhwani et al., 2017; Arya et al., 2018). The authors acknowledge financial support from Council of Scientific & Industrial Research (CSIR), India. A. Tuli acknowledges financial support from the Wellcome Trust/Department of Biotechnology (DBT). This work was supported by the Wellcome Trust/DBT India Alliance Intermediate Fellowship awarded to A. Tuli (IA/I/14/2/501543). A. Tuli also acknowledges the infrastructure and financial support from CSIR-Institute of Microbial Technology (IMTECH, Chandigarh) (OLP-144, communication No. 038/2018).
Competing interests
The authors declare no competing financial interests.
Ethics
This study was carried out in strict accordance with the guidelines issued by the Committee for the Purpose of Supervision of Experiments on Animals (1094 55/1999/CPCSEA) under the Prevention of Cruelty to Animals Act 1960 and amendments introduced in 1982 by the Ministry of Environment and Forest, Government of India. All protocols involving mouse experiments were approved by the institutional animal ethics committee of the Council of Scientific and Industrial Research-Institute of Microbial Technology (approval IAEC/16/12).
References
- Arya, S. B., Kumar, G., Kaur, H., Kaur, A. and Tuli, A. (2018). ARL11 regulates lipopolysaccharide-stimulated macrophage activation by promoting mitogen-activated protein kinase (MAPK) signaling. J Biol Chem 293(25): 9892-9909.
- Buscher, D., Hipskind, R. A., Krautwald, S., Reimann, T. and Baccarini, M. (1995). Ras-dependent and -independent pathways target the mitogen-activated protein kinase network in macrophages. Mol Cell Biol 15(1): 466-475.
- Epelman, S., Lavine, K. J. and Randolph, G. J. (2014). Origin and functions of tissue macrophages. Immunity 41(1): 21-35.
- Gay, N. J., Symmons, M. F., Gangloff, M. and Bryant, C. E. (2014). Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol 14(8): 546-558.
- Platko, K., Lebeau, P. and Austin, R. C. (2018). MAPping the kinase landscape of macrophage activation. J Biol Chem 293(25): 9910-9911.
- Satoh, T. and Akira, S. (2016). Toll-like receptor signaling and its inducible proteins. Microbiol Spectr 4(6).
- Schaeffer, H. J. and Weber, M. J. (1999). Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol 19(4): 2435-2444.
- Shapouri-Moghaddam, A., Mohammadian, S., Vazini, H., Taghadosi, M., Esmaeili, S. A., Mardani, F., Seifi, B., Mohammadi, A., Afshari, J. T. and Sahebkar, A. (2018). Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol 233(9): 6425-6440.
- Shimazu, R., Akashi, S., Ogata, H., Nagai, Y., Fukudome, K., Miyake, K. and Kimoto, M. (1999). MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med 189(11): 1777-1782.
- Sindhwani, A., Arya, S. B., Kaur, H., Jagga, D., Tuli, A. and Sharma, M. (2017). Salmonella exploits the host endolysosomal tethering factor HOPS complex to promote its intravacuolar replication. PLoS Pathog 13(10): e1006700.
- Zhang, X., Goncalves, R. and Mosser, D. M. (2008). The isolation and characterization of murine macrophages. Curr Protoc Immunol Chapter 14: Unit 14 11.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Kumar, G., Arya, S. B. and Tuli, A. (2018). Method for Studying the Effect of Gene Silencing on Bacterial Infection-induced ERK1/2 Signaling in Bone-marrow Derived Macrophages. Bio-protocol 8(24): e3123. DOI: 10.21769/BioProtoc.3123.
- Arya, S. B., Kumar, G., Kaur, H., Kaur, A. and Tuli, A. (2018). ARL11 regulates lipopolysaccharide-stimulated macrophage activation by promoting mitogen-activated protein kinase (MAPK) signaling. J Biol Chem 293(25): 9892-9909.
Category
Immunology > Immune cell function > Macrophage
Biochemistry > Protein > Quantification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.