- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Systematic Quantification of GFP-tagged Protein Foci in Schizosaccharomyces pombe Nuclei

Published: Vol 8, Iss 24, Dec 20, 2018 DOI: 10.21769/BioProtoc.3117 Views: 6127
Reviewed by: Dennis NürnbergGunjan MehtaSamantha E. R. Dundon
Original research article
The authors used this protocol in:

Jun 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
DNA damage repair proteins form foci in response to DNA damaging agents. The efficiency and integrity of the DNA repair pathway of a particular eukaryotic (mutant) strain is usually determined by the number of foci formed compared with their wild-type counterpart. Conventionally, focus number is determined visually, and this low accuracy may obscure the identification of a weaker phenotype, particularly when the output is low. Here, using the homologous recombination protein Rhp54 as an example, we present a protocol that can increase the consistency of foci identification among samples and can significantly improve the efficiency of foci quantification for large sample sizes. A similar method can be applied to other foci-forming proteins.
Keywords: Fission yeastBackground
Eukaryotic cells are equipped with a set of DNA damage response mechanisms to defend themselves against internal and external stresses. External stresses, such as chemical agents cause damage or mutation to the DNA and, if left unrepaired, will propagate to the offspring and lead to genomic instability. Methyl methanesulfonate (MMS) is one example of a chemical (alkylating) agent that methylates DNA bases and stalls the replication fork, thereby generating double-strand breaks (DSBs) when the replication fork collapses (Arnaudeau et al., 2001). In Schizosaccharomyces pombe, DSBs are often repaired by the error-free homologous recombination (HR) pathway (Ferreira and Cooper, 2004). HR proteins, such as Rhp54, lead to foci formation in response to MMS treatment, with the number of foci formed increasing proportionately to the level of DNA damage (Maki et al., 2011). Recent work has shown that a mutation in the histone H3 methyltransferase Set2 can affect Rhp54 foci formation during MMS treatment, thereby suggesting a role for Set2 in MMS resistance (Lim et al., 2018).
Quantification of foci number can reflect the robustness of DNA repair mechanisms in cells. However, measuring foci formation efficiency using fluorescently tagged DNA repair proteins often involves manual counting, which is susceptible to human error and is time-consuming (Rübe et al., 2008; Lorenz et al., 2009). There are several free or commercially available software on the market that afford better accuracy in foci determination and increase the throughput; however, there are several problems with such software (Herbert et al., 2014; Ledesma-Fernández and Thorpe, 2015; Oeck et al., 2015; Wiesmann et al., 2015). First, there is lack of a fully systematic and user-friendly way to move from image acquisition to image processing to foci analysis (counting); indeed, a user-friendly program must allow the user to have more control during each step of the process without requiring a background in computer programming (scripts and macros). Second, most of the available software or plugins were developed based on foci formation in mammalian cells, particularly targeting γH2AX foci, and thus might not be suitable for determining foci formation in yeast cells, which are much smaller in size (Böcker and Iliakis, 2006; Jucha et al., 2010; Anderson et al., 2013). The FociQuant software has been used for foci quantification in yeast with few complications; this is because the protocol focuses on the kinetochore protein, which forms a single distinct focus due to the clustering of centromeres (Ledesma-Fernández and Thorpe, 2015). However, difficulties arise in the quantification of foci for HR proteins; because DSBs can occur anywhere in the genome, multiple foci can be scattered in the nucleus and in different focal planes. Therefore, we are unable to use pre-existing methods of foci quantification to determine the foci of HR proteins accurately.
To overcome these problems, we employed the Nikon microscope system for image acquisition and NIS-Elements Advance Research software (v5.02) with the Extended Depth of Focus (EDF) plugin and “General Analysis” for image processing and foci analysis, respectively. Similar to other advanced microscope systems, DAPI and GFP Z-stack images are captured in a single file with one mouse-click. Because the cells exist in a 3D conformation, the EDF function enables the focused image of each plane captured by the Z-stack to be combined into a single, focused image. In this way, foci at different focal planes are included, thereby increasing the accuracy of the quantification. Furthermore, the software enables automated foci quantification by first determining the nucleus via DAPI staining, followed by counting the number of GFP foci in it; this prevents the inclusion of other non-nucleus, GFP noise. Although the analysis process is automated, the user can manually deselect regions or noise that are incorrectly determined by the software. The control and commands on the software are user-friendly, and the user does not need to have programming knowledge to modify certain quantification steps. In addition, the software is not only suitable for the analysis of images captured by a Nikon microscope but can be used to analyze images taken from other sources through additional steps.
Here, using MMS-dependent activation and foci formation of GFP-tagged Rhp54 as an example, we present an unbiased and precise method for the quantification of foci number formed by Rhp54-GFP in yeast. We have applied this method in recent work (Lim et al., 2018) to count foci number in wild-type and set2-mutant cells following MMS treatment.
Materials and Reagents
- Pipette tips
- Microcentrifuge tubes (1.5 ml) (Eppendorf, catalog number: 0030120086)
- Centrifuge tubes (Sigma-Aldrich, model: Greiner 15 ml, catalog number: T1818)
- Microscope slides (Continental Lab Products, catalog number: 7105)
- Microscope cover glasses 22 x 22 mm (Paul Marienfeld GmbH & Co, Marienfeld SUPERIOR, catalog number: 0101050)
- Grade GF/C Glass microfiber filters, 25 mm circle, 1.2 μm pore size (Whatman, catalog number: 1822-025)
- C-terminal GFP-tagged Rhp54 S. pombe strain (Tang et al., 2015; Lim et al., 2018)
- 972, S. pombe wild-type (WT) strain (without any GFP tagging) (Lim et al., 2018)
- Methyl methanesulfonate (MMS) in 99% stock (Sigma-Aldrich, catalog number: 129925)
CAUTION: MMS is a toxic substance and a potential carcinogen. Users should wear all protective wears while working with it. MMS-containing waste media should be placed in a container for cytotoxic waste before disposal by a licensed chemical waste disposal company. - Methanol (AR-grade) in 100% (Fisher Scientific, catalog number: 10284580)
- 4',6-diamidino-2-phenylindole (DAPI) for nucleus staining (Sigma-Aldrich, catalog number: D8417)
- NaCl (Sigma-Aldrich, catalog number: S9888)
- KCl (Sigma-Aldrich, catalog number: P5405)
- Na2HPO4 (Sigma-Aldrich, catalog number: S5136)
- KH2PO4 (Sigma-Aldrich, catalog number: P9791)
- Hydrochloric acid 37% (Sigma-Aldrich, catalog number: 320331)
- Yeast extract (BD Biosciences, BactoTM, catalog number: 212750)
- Adenine (Sigma-Aldrich, catalog number: A8626)
- Glucose (Sigma-Aldrich, catalog number: G8270)
- Immersion Oil for microscopy (Nikon, catalog number: MXA20235)
- 1x phosphate-buffered saline (PBS) (see Recipes)
- 2 N Hydrochloric acid (HCl) (see Recipes)
- Yeast extract adenine (YEA) media for cell growth (see Recipes)
Equipment
- P20 Pipetman (Gilson, catalog number: F123600)
- P200 Pipetman (Gilson, catalog number: F123601)
- P1000 Pipetman (Gilson, catalog number: F123602)
- Conical flask, 250 ml (Kartell, catalog number: 1462)
- Shaker incubator (Eppendorf, model: Innova® 44, catalog number: M1282-0002)
- Inverted fluorescence microscope (e.g., Nikon; model: Eclipse Ti-E) with:
- Oil immersion objective lens (Nikon; model: CFI PLAN APO VC 100XH 1.4 NA)
- DAPI filter (excitation λ: 350 nm; emission λ: 460 nm) (Chroma; model: ET-DAPI; catalog number: 49000)
- FITC filter (excitation λ: 480 nm; emission λ: 535 nm) (Chroma; model: FITC /EGFP /Bodipy FI /Fluo3 /DiO; catalog number: 41001)
- Digital camera (Hamamatsu; model: ORCA-Flash2.8; catalog number: C11440-10C)
- Low-temperature freezer (-80 °C) (Thermo Fisher Scientific, model: FormaTM 700 Series)
- Refrigerated centrifuge (4 °C) (TOMY DIGITAL BIOLOGY, model: MX-305)
- Forceps (Sigma-Aldrich, catalog number: F4267)
- Spectrophotometer (Biochrom, ULTRASPEC, catalog number: 80-2116-30)
- Vacuum filtration holder set, consisting of a conical collection flask and a filter holder (Fisherbrand, catalog number: 09-753G)
- Vacuum pump (Rocker Scientific, model: Rocker 400, catalog number: 167400-11(22))
- Autoclave (ALP, model: CL-40M)
Software
- Nikon NIS Elements AR (v5.02) with Extended Depth of Focus (EDF) and General Analysis plugin (purchased from Nikon Co.)
- Microsoft Office 365–Excel
Procedure
- Sample preparation
- For each sample, prepare 3 ml of 100% methanol in a 15 ml centrifuge tube and pre-chill at -80 °C for at least 1 h before fixation.
- For each strain, inoculate at least 10 ml of cells per time point and grow overnight (~16 h) in YEA to an optical density (OD600nm) of 0.25 at 30 °C in a shaking incubator at 110 rpm. OD is determined by spectrophotometer.
Note: For example, if there will be 5 time points (0, 1, 2, 3 and 4 h, for instance), then grow 50 ml (5 x 10 ml) of cell culture in a conical flask. - Add 0.01% of a 99% MMS stock into the cell culture and start timing.
Note: MMS can be substituted with other drugs or treatments that will initiate foci formation. - As a negative control for GFP signal, grow a 10 ml wild-type strain without any GFP tagging (WT) to OD600nm 0.5 using the conditions in Step A2.
- Cell harvesting and fixation
- Collect 10 ml of cells by passing the culture through the vacuum filtration holder set with glass microfiber filters.
Note: Leave the vacuum pump on for an extra few seconds to ensure most of the liquid is removed. - Immediately submerge the cell-containing filter into pre-chilled methanol using forceps. Repeat the process for the other time points
- Fix the cells in methanol by leaving them at -80 °C for at least 15 min or up to 2 to 3 weeks.
- Collect 10 ml of cells by passing the culture through the vacuum filtration holder set with glass microfiber filters.
- Rehydration of cells
Note: Keep the tubes on ice throughout the whole process of rehydration.- Add 1 ml of ice-cold 1x PBS to the 15 ml tube, invert to mix. Dislodge the cells from the filter paper by repeatedly pipetting the PBS-methanol mixture inside the 15 ml tube over the surface of the filter and then remove the filter disc from the solution with forceps.
Note: Dislodging the cells gently will prevent the generation of excessive fibers, which will cause unevenness when mounting on a glass slide. - Rehydrate the cells by adding 6 ml of ice-cold 1x PBS gradually, i.e., add 1 ml, invert to mix, add another 1 ml and mix.
Note: Do not add all 6 ml at the same time as this may burst the cells. - Centrifuge at 1,600 x g for 1 min to collect the cell pellet.
- Remove the supernatant and transfer the cells to a 1.5 ml microcentrifuge tube with 1 ml of 1x PBS.
- Wash thrice each with 1 ml ice-cold 1x PBS.
- Resuspend the cells in 20 µl of ice-cold 1x PBS and keep on ice until observation.
Note: Observations must be done on the same day the samples are rehydrated.
- Add 1 ml of ice-cold 1x PBS to the 15 ml tube, invert to mix. Dislodge the cells from the filter paper by repeatedly pipetting the PBS-methanol mixture inside the 15 ml tube over the surface of the filter and then remove the filter disc from the solution with forceps.
- Image acquisition
- To view the Rhp54-GFP signals, mount 1 µl of cells with an equal volume of 1 µg/ml DAPI onto a glass slide and cover with a microscope cover glass.
Note: Coverslip with thickness No. 1 is used in the current experimental procedures. For optimal results, use coverslips with a thickness that is recommended for each individual type of microscope lens. No sealing of the slides is required as the sample is visualized immediately after mounting. - Observe the S. pombe cells with a 100x magnification objective lens using a fluorescence microscope.
Note: We use the Nikon Eclipse Ti-E microscope system with NIS-Elements Advance Research software (v5.02) as an example. - Set up an automated image acquisition parameter (ND Acquisition): first capturing a still image of DAPI then taking a Z-stack for GFP.
Note: Acquisition of the Z-stack images is important, as the foci may only be focused in certain focal planes. The number of images captured defines the resolution. For accurate quantification and reconstruction of nucleus image perfectly, according to Nyquist-Shannon sampling theorem, Z-slice of 285 nm per individual steps is necessary (based on objective oil lens of 100x magnification and 1.4 NA) (Shannon, 1949). This is equal to ~14 Z-steps over a total depth of 4 µm. - To avoid photobleaching when running Z-scan, the cells are first focused by viewing the DAPI-stained nuclei. Once ready, click the “Run now” button for automated image acquisition. The captured image will be saved to a pre-destined folder automatically.
Note: A new field of view should be observed as the focus plane moves away from the previous photobleached area during the scan in the Z-axis. - Capture a sufficient number of images with a total number of cells > 500 per sample.
Note: All strains in the same experiment need to have images taken on the same day and continuously (without turning the microscope on/off or adjusting the light source) to ensure consistency and inter-strain comparability. - Combined images of DAPI and Z-stack GFP will be saved as .nd2 file format.
- To view the Rhp54-GFP signals, mount 1 µl of cells with an equal volume of 1 µg/ml DAPI onto a glass slide and cover with a microscope cover glass.
- Image processing
- To combine the GFP images from the different Z-axes into a single, focused frame, open a .nd2 image, click on the “Create EDF Focused Document” button (Figure 1A).
Note: The EDF module functions by combining the focused portion in each plane into a single focused image. - To standardize the variation in background intensity on the post-EDF image, select “Draw Square Background ROI” and box-up an area without any cells (Figure 1B). Select “Subtract Background Using ROI” to remove the background.
- To enhance the intensity of the GFP foci for easier detection by the software, select from the menu bar: Image > Local Contrast > set to 3 Degrees and with a radius of 0.65 µm.
Note: Local contrast is a digital image processing step to increase the contrast of the image with respect to local surroundings, aiming to emphasize the details or sharpen an image. The “degree” is the contrast multiplicator or magnitude of contrast amplification. “Radius” is the size of the adjacent area (pixel) to be processed. When the radius value is small, it will make the bright pixels become brighter while dimming the darker adjacent pixels to enhance the smaller-scale details. Increasing the radius value makes a wider adjacent area visible but results in the loss of details, the GFP foci in this case. Hence, “degree” and “radius” have to be set such that the fine details of GFP can be enhanced while at the same time not exaggerated. - Save the .nd2 file.
- Repeat Steps E1-E4 for other images.
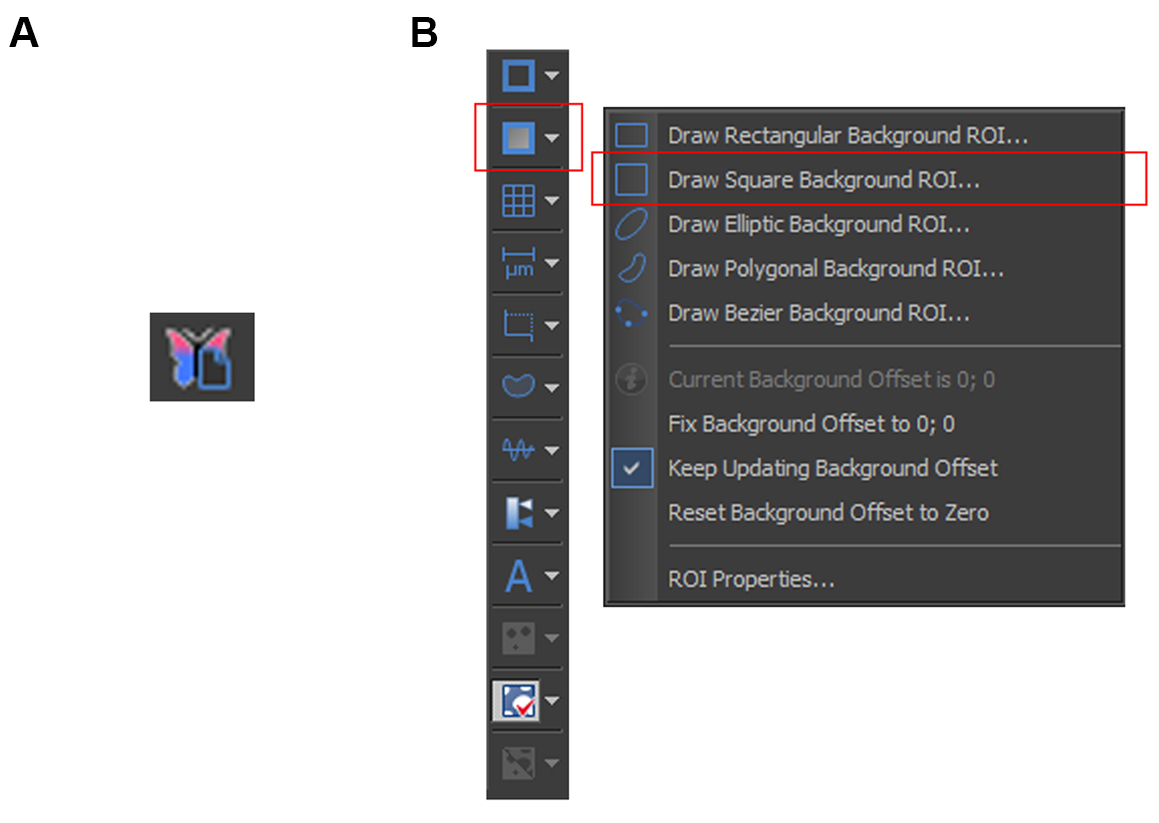
Figure 1. Creating a focused image and removing the background noise. A. Button to create an EDF-focused image. B. Red boxes indicate the selection required for defining the background area on an image.
- To combine the GFP images from the different Z-axes into a single, focused frame, open a .nd2 image, click on the “Create EDF Focused Document” button (Figure 1A).
- Optimizing and determination of GFP foci (Batch process)
Images of the positive control strain should be used as a reference to determine the parameters for GFP foci quantification. In the case of Lim et al. (2018), wild-type Rhp54-GFP cells that were treated with MMS for 4 h were used to set up the parameters. Example of images taken for DAPI and GFP signal is shown in Figure 2.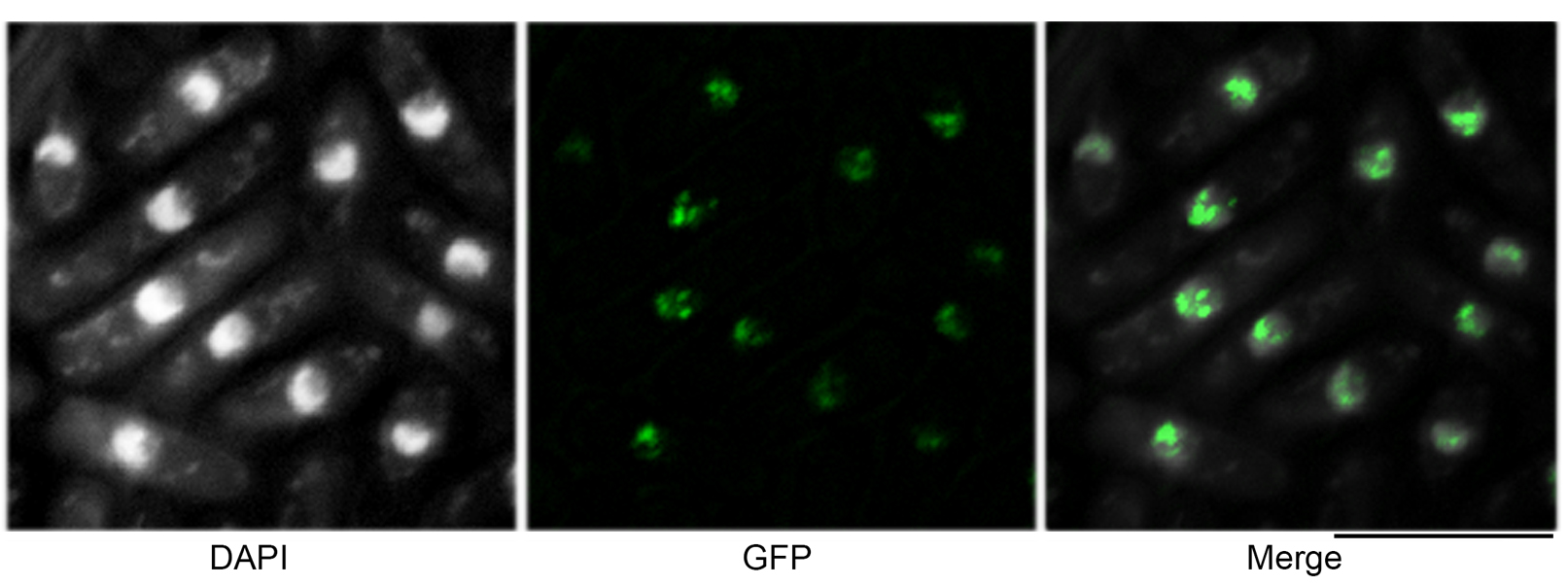
Figure 2. Example of DAPI and GFP images. A single .nd2 file consists of images of DAPI and GFP channel. The merged image of both channels shows that Rhp54-GFP signal localized to the nucleus. Scale bar: 10 μm.- Open a processed .nd2 image (EDF and background subtraction done, after Step E4); at the menu bar, select Image > General Analysis.
- Under the “Analysis Palette” of the General Analysis window (Figure 3A), select “Add Channel” to add both the DAPI and FITC (GFP) channels (Figure 3B).
- To define a parameter for DAPI detection, select DAPI on the left and threshold on the right, and adjust accordingly (Figures 3A and 3B). The changes made will show up in real time on the image, as in Figure 3C; blue signals indicate the DAPI area defined after adjustment of the threshold.
Note: Ensure the “Preview” box is ticked at the bottom of the window to view changes in real-time. See Note 1. - For FITC (GFP), select “Bright Spot Detection” (Figure 3D, red box on the right; Figure 3E) and adjust other parameters accordingly until the number of pink spots appearing on the image matches the number of GFP foci observed by eye (Figure 3F). See Note 2.
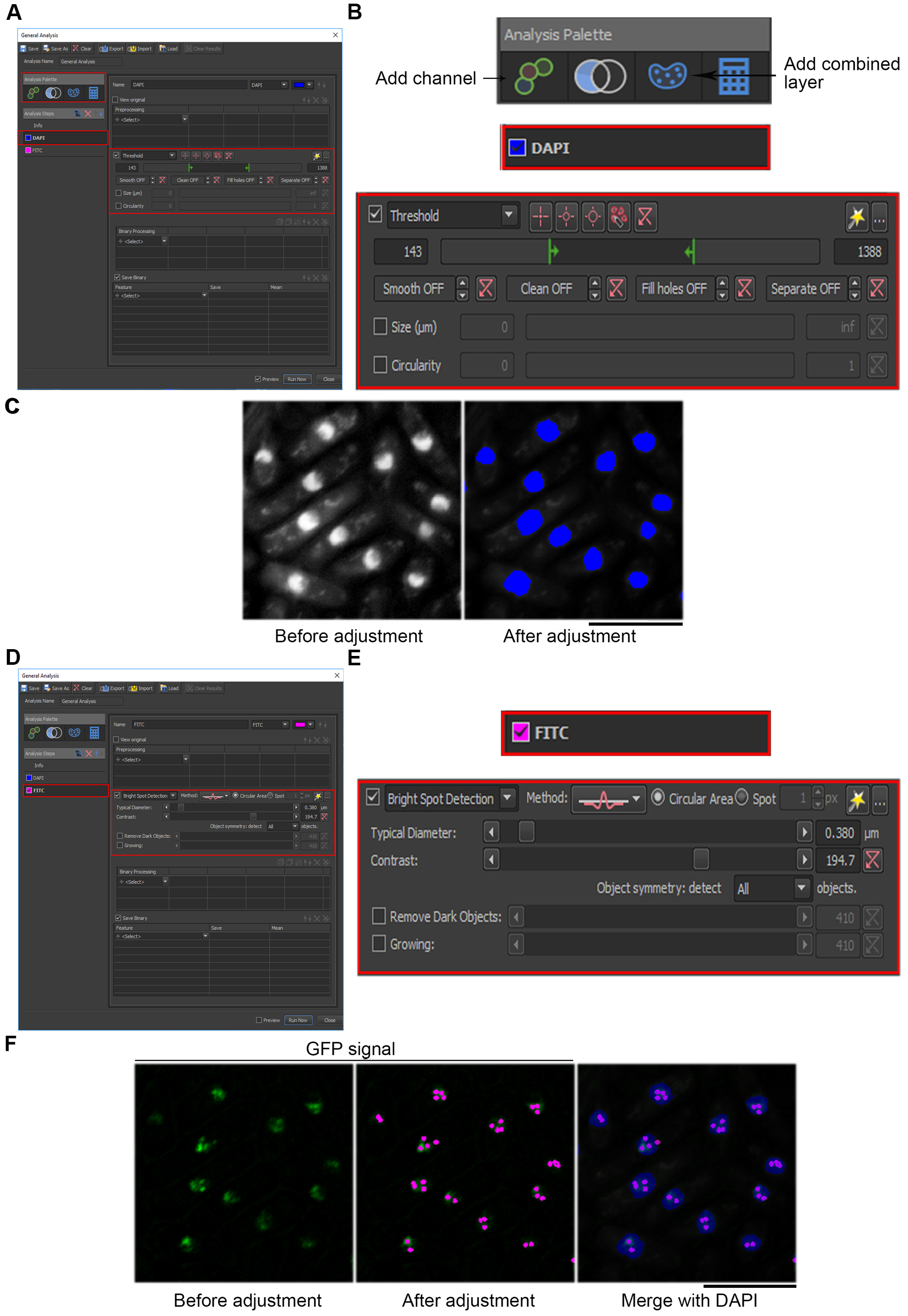
Figure 3. Adjusting the parameters for detecting DAPI and GFP signals. A. Select the DAPI channel (red box, left) and adjust the threshold and other parameters (red box, right). The changes made will be reflected in real time on the image as shown in C. B. The zoom-in view of the area boxed with red in A. C. Images of DAPI before and after adjustment of the threshold. Blue signals indicate the DAPI area. Scale bar: 10 μm. D. Select the FITC channel (red box, left) and adjust the parameters for Bright Spot Detection (red box, right). The changes made will be reflected in real time on the image as shown in F. E. The zoom-in view of the area boxed with red in D. F. Images of GFP before and after the adjustment of parameters, and a merged view for both DAPI and GFP foci detected. Pink signals indicate GFP foci. Scale bar: 10 μm. - To count the GFP foci number in the nucleus, under “Analysis Palette” select “Add Combined Layer” (Figure 3B) and adjust the following criteria: ROI = “DAPI”, Feature = “Count”, Measured On = “FITC” (Figures 4A and 4B). Save the final parameter in .ga format.
- Press “Run Now” and the counting results will be shown in the “Automated Measurement Results” table (Figure 4C), with “Source” indicating the image file name, “RoiID” indicating each of the individual DAPI-positive foci identified, and “NumberObjects” indicating the number of GFP-positive foci identified.
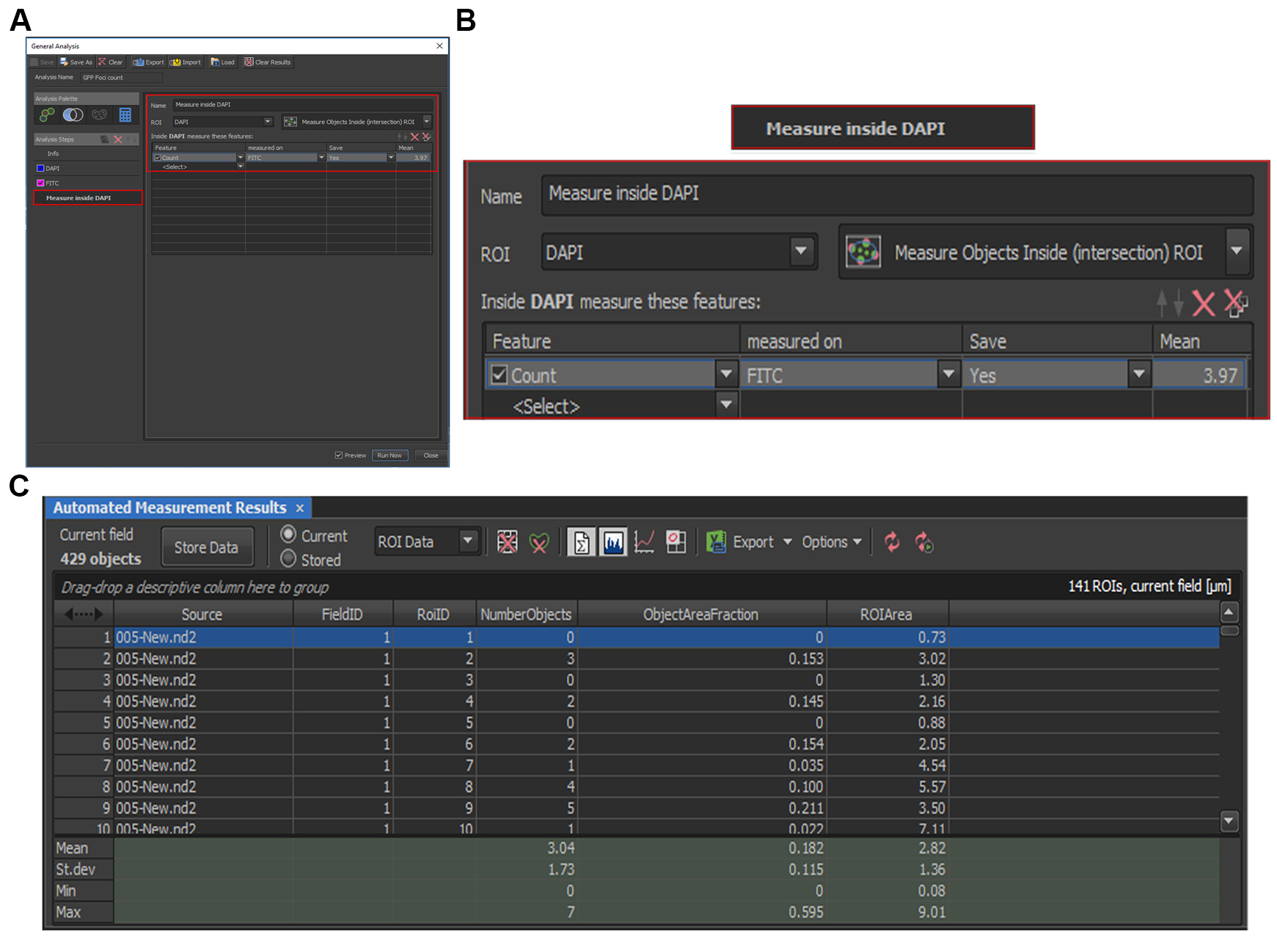
Figure 4. Quantification of GFP foci inside the DAPI signal. A. “Measure inside DAPI” is added by selecting “Analysis Palette > Add Combined Layer” and with the following criteria: ROI = “DAPI”, Feature = “Count”, Measured On = “FITC”. B. Zoom-in view for area boxed red in A. C. Automated Measurement Results table, with “Source” indicating the image file name, “RoiID” indicating each of the individual DAPI identified, and “NumberObjects” indicating the number of GFP foci identified in the particular DAPI signal. - To batch process all the images, select at the menu bar: Image > Analysis Explorer. Select the method or criteria that will be used for the batch analysis (Figure 5A). In this case, select “General Analysis”; as in Figure 4A, the “Analysis Name” is “General Analysis”. Alternatively, the previously set parameter (.ga file) can also be loaded. Then click on “Batch”.
- In the “Batch Analysis” window, select the location of the folder and the image files to be processed and then press “Run” (Figure 5B). After the images are processed, the results will be shown in the “Automated Measurement Results” window, as in Figure 4C. Export the raw data to Microsoft Excel for data analysis.
Note: The threshold or parameters set can only be used on samples taken on the same day.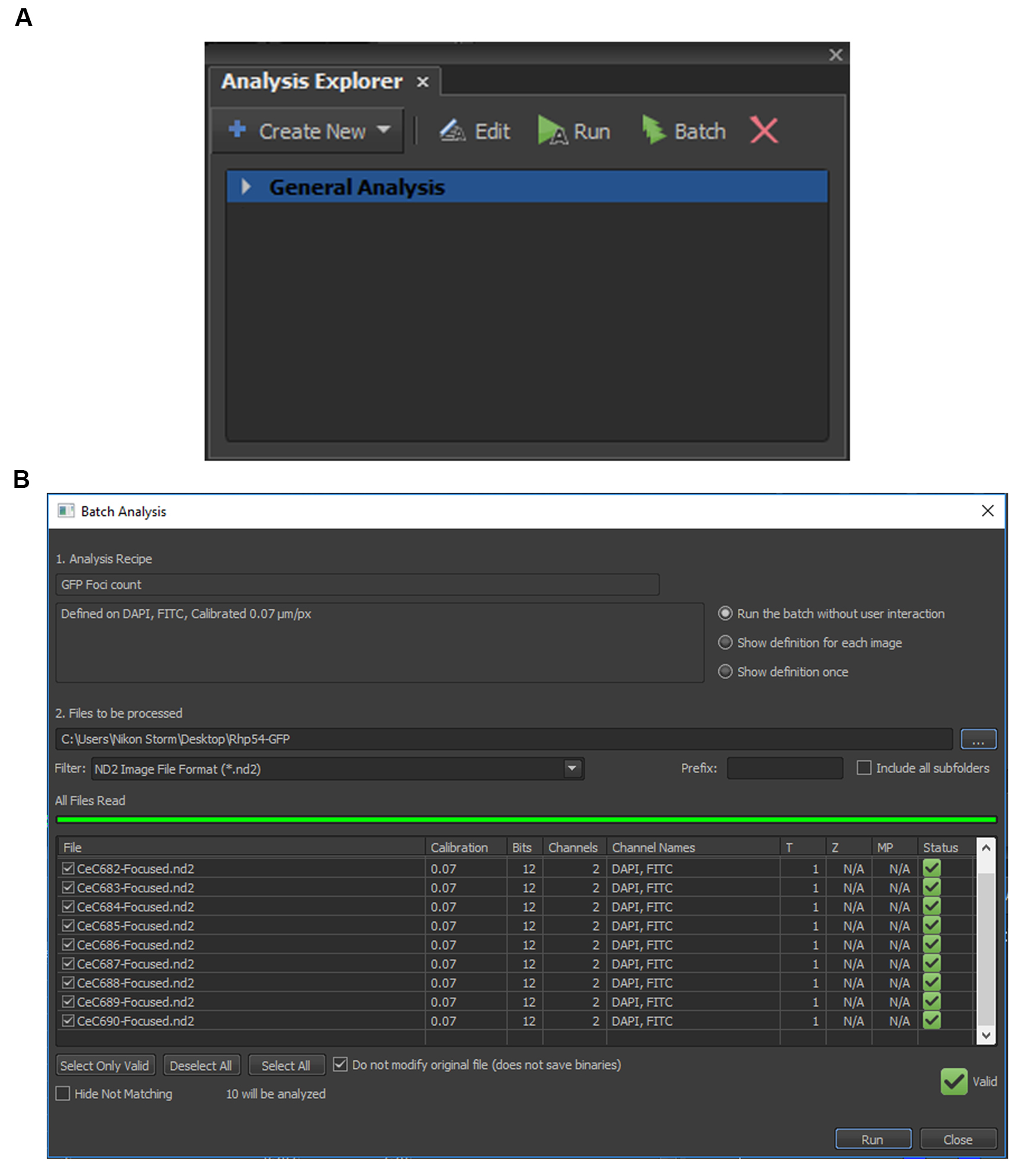
Figure 5. Batch processing of images. A. Analysis Explorer showing previous processed files and parameters used. B. “Batch Analysis” is used to select the .nd2 files to be selected for batch process using the same parameters.
- Analysis of images obtained from other microscope systems
DAPI and GFP Z-stack images obtained from other microscope systems can also be analyzed using the Nikon NIS Elements software, with some additional steps.- Z-stack images must be exported into individual .tif format images at the original microscope system.
Note: If there are 7 steps for a Z-stack, 7 individual images will be attained after exporting. - Ensure that the file name is in sequence. For example D1.tif, D2.tif, D3.tif … D7.tif.
- At the menu bar, select: File > Import/Export > Create ND File from File Sequence.
- Select the folder containing the individual Z-stack images, and choose “Z Series” (red box, Figure 6A) and press “Convert”. This will convert the GFP images into a single .nd2 file.
- If the images are in RGB, after Step G4, at the menu bar select: Image > Convert > Convert to grey (Figure 6B). This step should be performed on the DAPI image.
- On the GFP Z-stack image, create a single, focused image by selecting “Create EDF Focused Document” button (Figure 1A).
- To overlap DAPI and GFP images, drag the window with the post-EDF GFP image into the DAPI image to create a single overlap .nd2 file.
- Continue with Steps E2 to F8.
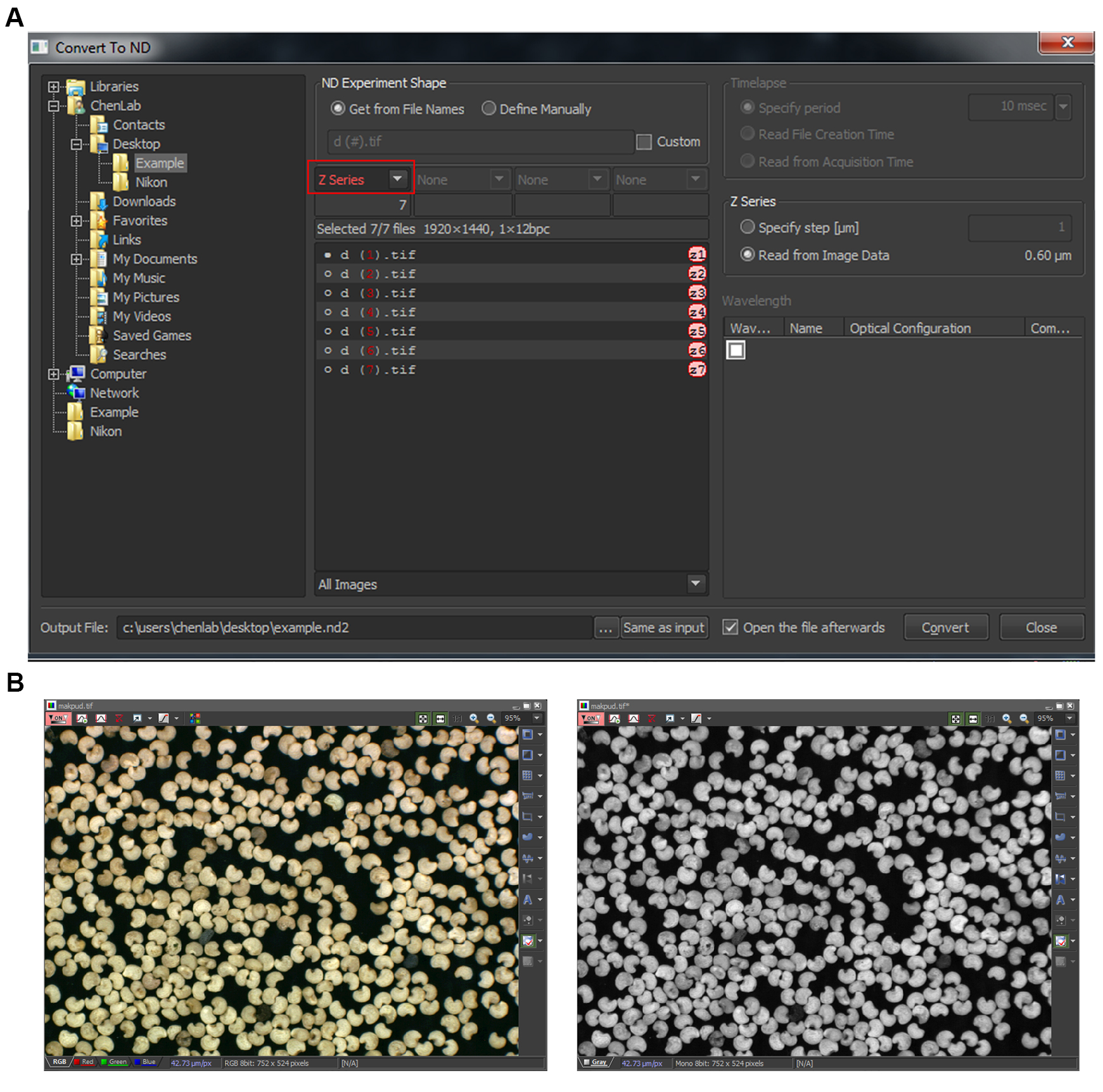
Figure 6. Conversion of images obtained from other microscopy systems. A. Individual Z-stack images will be combined into a single Z-stack .nd2 file. Red box: select “Z series” to create a Z-stack file. B. Conversion of a random RGB image (left) to a greyscale image (right). Notice that at the bottom of the window it changes from three colors (Red, Green, Blue) to one (Grey). Image shown is the sample image obtained inside NIS-Elements Advance Research software.
- Z-stack images must be exported into individual .tif format images at the original microscope system.
Data analysis
- In the exported Excel sheet, quantify the number of cells according to the number of foci.
- Cells that have more than 4 GFP foci (> 4) are counted and expressed as a percentage of the total number of cells counted.
- Apply similar procedures for the other time points and samples so that they can be compared with each other.
- Graph of the percentage of cells with more than 4 GFP foci (> 4) against time.
Note: An example of different number of GFP foci identified by the software in cells (Figure 7). Only cells contained more than 4 GFP foci were included in the graph (Figure 9A). See Note 3.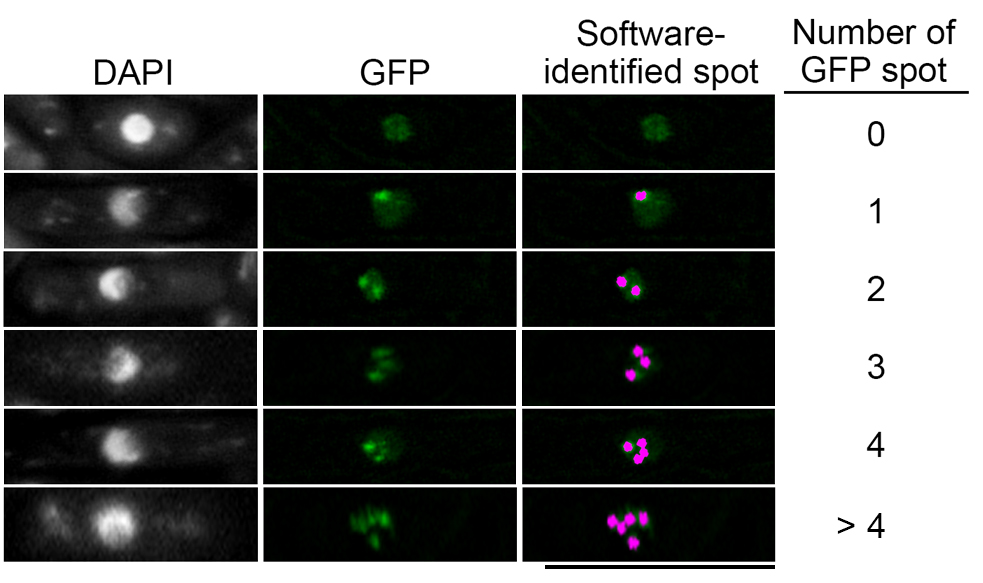
Figure 7. Cells with different number of GFP foci identified by software. Based on the parameter set earlier in Step F4, the identified GFP foci were shown as pink dots, and the actual count will be recorded as in Figure 4C. Scale bar: 10 μm.
Notes
- The inclusion of non-nuclear DAPI-stained cells in the detection (Step F3).
Suggestion: To ensure higher accuracy and decrease non-specific counting of GFP foci, after the batch run, manually remove the non-specific DAPI-stained area from the counting. A nucleus is defined by the round shape of the DAPI stained area and is usually located in the middle of G2 phase cells or appeared in two, one at each end for mitotic cells. Figure 8A shows an example of nuclei stained with DAPI and other non-specific staining (noise).- At the Binary Toolbar, select “delete object”, click on DAPI signals that are defined incorrectly by the software (noise) and remove them (Figure 8B).
- Select “auto detect” and click on the area where the nucleus is to be selected (Figure 8B). After modification, at Figure 4B, select “update measurement”.
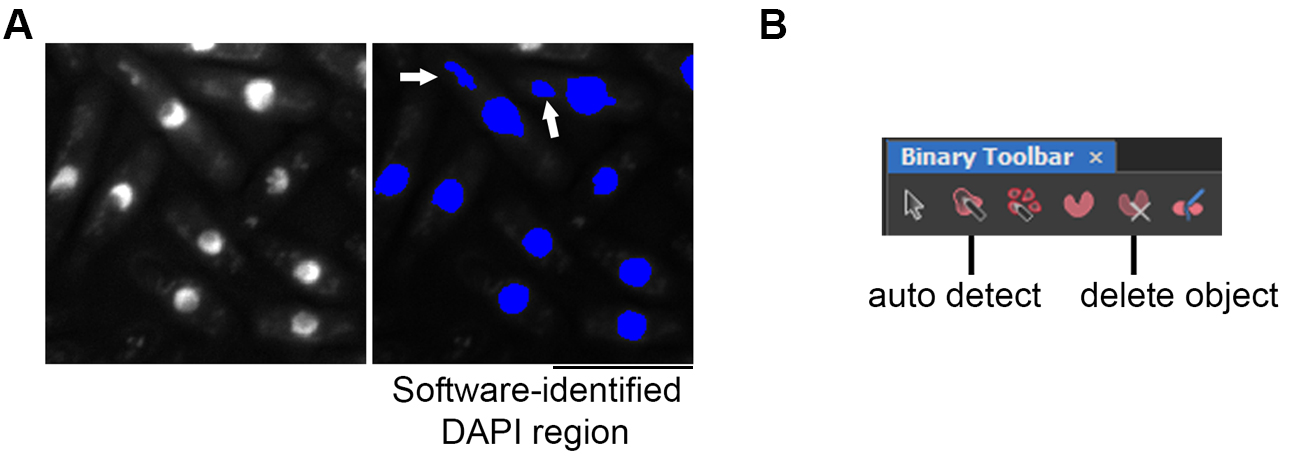
Figure 8. Binary toolbar to manually define the nucleus region and remove non-specific DAPI staining. A. Original image of DAPI-stained cells (left) and nucleus region identified by the software that appeared in blue (right). The arrow highlights a non-nucleus staining noise that was removed prior to analysis. Scale bar: 10 μm. B. The signal identified by the software using set parameters can be manually edited using the Binary Toolbar. “Delete object” is used to deselect noise and any other incorrectly defined regions. “Auto detect” is to redefine the area of the nucleus (DAPI signal).
- The foci determined by the software are lower in number than visually observed (Step F4).
Suggestion: Because GFP intensity varies between sample batches, similar parameters cannot be applied to images taken on different days, even if a similar strain is used. Hence, for every batch of samples, the wild-type is needed for the initial determination of parameters. If the initial GFP intensity is too low, the GFP signal needs to be enhanced further by adjusting the Local Contrast (Step E3) until the software can identify the number of spots seen with the naked eye. - Standard deviation (SD) is large when combining the triplicates of the experiment thus masking the difference (if any) between wild type and mutant strains.
Suggestion: It is not recommended to combine all biological repeats for calculations of average and SD. Instead, it is better to present data in separate sets to show repeatability of the trend. Alternatively, the results can be shown as relative values, such that all values from different strains and time points are normalized to the value of the wild type at the final time point (e.g., wild type 4 h after the addition of MMS). For example, the value at individual time points expressed in percentage (Figures 9A and 9B) was divided by that of wild-type at 4 h (46.09%) to give a relative value (Figures 9B and 9C). Through this approach, variation among different experimental repeats could be minimized to allow statistical comparison when data from different repeats were to combine.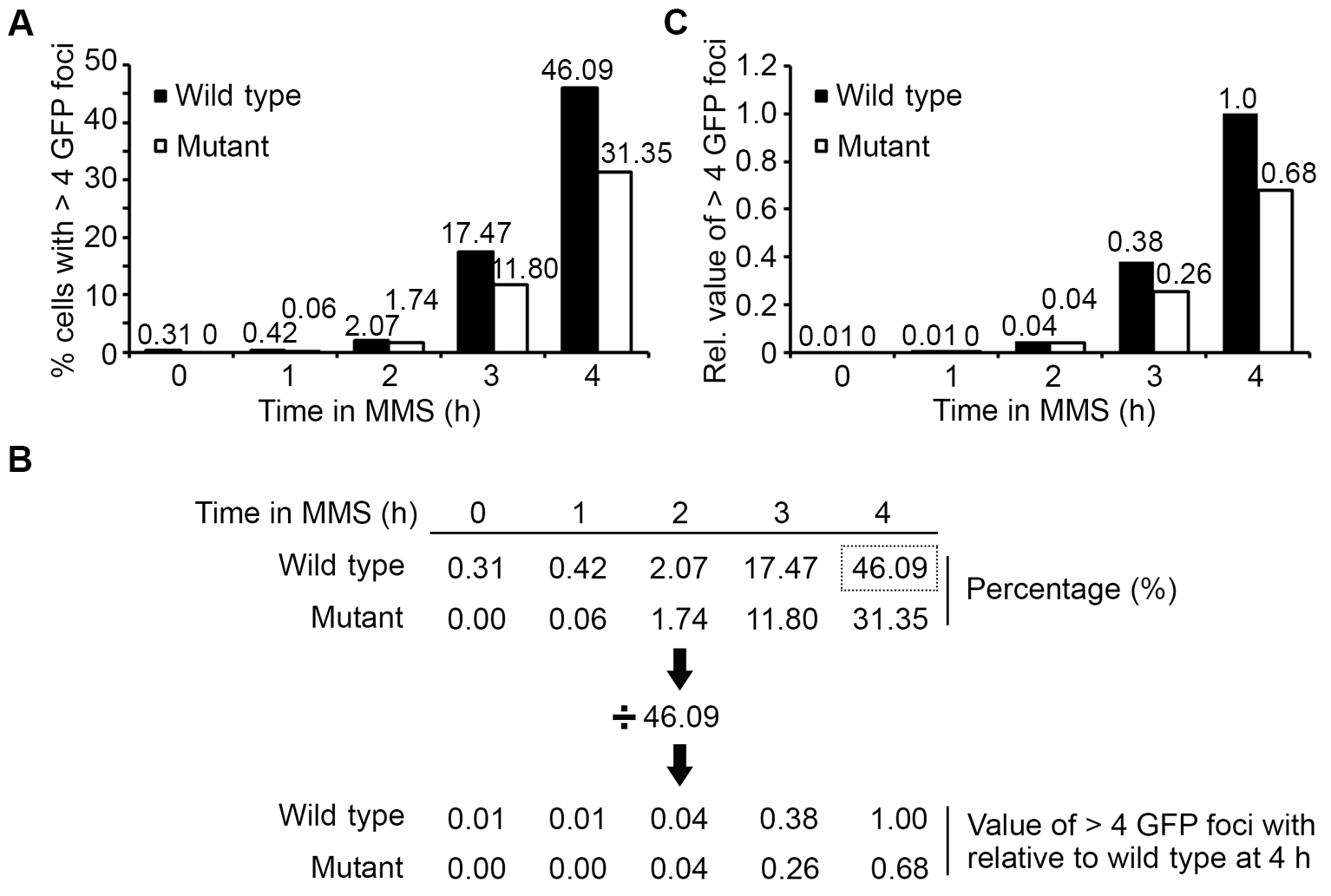
Figure 9. Example of data processing. A. Cells with more than four GFP foci are quantified and plotted against the time in MMS treatment. Black bar: wild-type; White bar: mutant. B. Readings obtained from (A) are recalculated relative to the wild type at 4 h (boxed with dotted line). C. Representation of data in B. - Further application: The protocol described herein can be further adapted to quantify fluorescent focus signals in the whole cell (inclusive of cytosol) instead of simply in the nucleus with the following modifications.
Adjust the parameters accordingly in Step F3 (Figure 3A), such that the blue signal encompasses the whole cell instead of simply the nucleus. In doing so, the software will quantify the number of foci that are present in the whole cell instead of those in the nucleus only, in subsequent Steps F4-F8.
Recipes
- 1x phosphate-buffered saline (PBS)
137 mM NaCl
2.7 mM KCl
10 mM Na2HPO4
1.8 mM KH2PO4
Adjust to pH 7.4 using 2 N HCl
Autoclave and store at room temperature - 2 N Hydrochloric acid (HCl)
41.788 ml distilled water
8.212 ml stock Hydrochloric acid 37% - Yeast extract adenine (YEA) media for cell growth
In the fume hood, add HCl to the water slowly (caution: not vice versa)
5 g yeast extract
75 mg adenine
30 g glucose
1 L of distilled water
Adjust the pH to 5.5 using 2 N HCl
Autoclave and store at room temperature
Acknowledgments
We thank Rebecca Jackson for critically editing this manuscript and Nikon Imaging Centre for the use of analysis software. This work was supported by a Singapore Ministry of Education MOE2016-T2-2-063 awarded to E.S.C.
Competing interests
The authors declare no conflict of interest.
References
- Anderson, D., Andrais, B., Mirzayans, R., Siegbahn, E. A., Fallone, B. G. and Warkentin, B. (2013). Comparison of two methods for measuring γ-H2AX nuclear fluorescence as a marker of DNA damage in cultured human cells: applications for microbeam radiation therapy. J Inst 8(6): C06008.
- Arnaudeau, C., Lundin, C. and Helleday, T. (2001). DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol 307(5): 1235-1245.
- Böcker, W. and Iliakis, G. (2006). Computational methods for analysis of foci: validation for radiation-induced γ-H2AX foci in human cells. Radiat Res 165(1): 113-124.
- Ferreira, M. G. and Cooper, J. P. (2004). Two modes of DNA double-strand break repair are reciprocally regulated through the fission yeast cell cycle. Genes Dev 18(18): 2249-2254.
- Herbert, A. D., Carr, A. M. and Hoffmann, E. (2014). FindFoci: a focus detection algorithm with automated parameter training that closely matches human assignments, reduces human inconsistencies and increases speed of analysis. PLoS One 9(12): e114749.
- Jucha, A., Wegierek-Ciuk, A., Koza, Z., Lisowska, H., Wojcik, A., Wojewodzka, M. and Lankoff, A. (2010). FociCounter: a freely available PC programme for quantitative and qualitative analysis of γ-H2AX foci. Mutat Res 696(1): 16-20.
- Ledesma-Fernández, E. and Thorpe, P. H. (2015). Fluorescent foci quantitation for high-throughput analysis. J Biol Methods 2(2).
- Lim, K. K., Nguyen, T. T. T., Li, A. Y., Yeo, Y. P. and Chen, E. S. (2018). Histone H3 lysine 36 methyltransferase mobilizes NER factors to regulate tolerance against alkylation damage in fission yeast. Nucleic Acids Res 46(10): 5061-5074.
- Lorenz, A., Osman, F., Folkyte, V., Sofueva, S. and Whitby, M. C. (2009). Fbh1 limits Rad51-dependent recombination at blocked replication forks. Mol Cell Biol 29(17): 4742-4756.
- Maki, K., Inoue, T., Onaka, A., Hashizume, H., Somete, N., Kobayashi, Y., Murakami, S., Shigaki, C., Takahashi, T. S., Masukata, H. and Nakagawa, T. (2011). Abundance of prereplicative complexes (Pre-RCs) facilitates recombinational repair under replication stress in fission yeast. J Biol Chem 286(48): 41701-41710.
- Oeck, S., Malewicz, N. M., Hurst, S., Rudner, J. and Jendrossek, V. (2015). The Focinator - a new open-source tool for high-throughput foci evaluation of DNA damage. Radiat Oncol 10: 163.
- Rübe, C. E., Grudzenski, S., Kühne, M., Dong, X., Rief, N., Löbrich, M. and Rübe, C. (2008). DNA double-strand break repair of blood lymphocytes and normal tissues analysed in a preclinical mouse model: implications for radiosensitivity testing. Clin Cancer Res 14(20): 6546-6555.
- Shannon, C. E. (1949). Communication in the presence of noise. Proc. IRE, 37:10-21.
- Tang, M. Y., Guo, H., Nguyen, T. T., Low, L. S., Jackson, R. A., Yamada, T. and Chen, E. S. (2015). Two fission yeast high mobility group box proteins in the maintenance of genomic integrity following doxorubicin insult. Gene 562(1): 70-75.
- Wiesmann, V., Franz, D., Held, C., Münzenmayer, C., Palmisano, R. and Wittenberg, T. (2015). Review of free software tools for image analysis of fluorescence cell micrographs. J Microsc 257(1): 39-53.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Lim, K. K. and Chen, E. S. (2018). Systematic Quantification of GFP-tagged Protein Foci in Schizosaccharomyces pombe Nuclei. Bio-protocol 8(24): e3117. DOI: 10.21769/BioProtoc.3117.
Category
Microbiology > Microbial cell biology > Cell imaging
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.