- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantitation of Regulatory Activity for the Complement Alternative Pathway Using an Adaptation of the AP50 in vitro Assay
Published: Vol 8, Iss 24, Dec 20, 2018 DOI: 10.21769/BioProtoc.3116 Views: 6350
Reviewed by: Vamseedhar RayaproluPaul JordanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Complement pathways function to identify and remove pathogens and infected cells. There are three complement pathways: the classical, lectin and alternative pathway (AP). While all pathways are activated following pathogen stimuli, the AP is constitutively active and tightly controlled by activators (e.g., Factor B, Factor D) and negative regulators (e.g., Factor H). Complement activity can be measured by well-established methods that are often used in a diagnostic setting to determine the CH50 (50% complement hemolytic activity) or AP50, specifically to measure AP activity. The protocol here has adapted the traditional AP50 method designed to measure AP activity in human sera, to measure the positive or negative AP regulatory activity within a given test sample. The assay relies on the ability of AP components in human serum to lyse rabbit erythrocytes under in vitro conditions specific for the AP with subsequent release of hemoglobin that is quantitated by measurement of optical density. Our method has added test substances, such as cell culture media with defined changes in individual complement components and determined the ability to either promote or inhibit AP activity in vitro. Thus, this protocol reflects the overall functional ability of a sample to effect AP activity and can be used in the research laboratory to determine AP regulatory activity in a complex biological sample, or to test the ability of drugs or novel biomolecules to regulate AP activity.
Keywords: ComplementBackground
Complement is part of the innate immune system and acts to recognize and clear pathogens and stimulate adaptive immune responses. The three complement pathways, the classical (CP), lectin (LP) and alternative pathways (AP), constitute a series of cleavage and activation of complement proteins, generating enzymatically active protein complexes, such as convertase complexes, and bioactive split products, such as C3a. The three pathways converge on cleavage of the complement component C3 and one common downstream outcome is the generation of a membrane attack complex (MAC) and subsequent lysis of target cells (Ricklin et al., 2010). Functional activity of complement can be measured by assays such as the 50% complement hemolytic activity (CH50 assay), where a stimulus (antibody coated red blood cells), is provided in vitro to the complement components in a biological sample. The subsequent lysis of the target red blood cells (RBC) is quantitated by release of hemoglobin and measured by an increase in optical density. If target complement components such as C3 are missing from the test biological sample, either genetically or due to complement consumption resulting from activation of complement in vivo, then CH50 will be low. Similarly, the AP50, specifically measures the activity of the AP where rabbit RBC spontaneously activate the human AP resulting in RBC lysis (Figure 1A). The presence of EGTA chelates calcium to prevent CP activation and the addition of Mg2+, restores this requirement for the AP, yielding buffer conditions specific to determine AP activity (Joiner et al., 1983). CH50 and AP50 assays are routinely used in many diagnostic laboratories to quantitate complement activity in human blood (Prohaszka et al., 2016). In the traditional application of the AP50, a high AP activity is reflected by low availability of complement components due to complement ‘consumption’ and thus low lysis of rabbit RBC. The lysis of rabbit RBC is quantitated by detecting the release of free hemoglobin by an increase in optical density (Figure 1A). This traditional assay has been adapted here, to keep the starting complement components in the assay constant by use of one source of human serum and measure the ability of factors added in test substances to promote the cleavage of the complement components from human serum, under conditions specific for the AP (Figure 1B). In this case, with constant input of complement components, adding a sample containing a stimulator, such as factor D or B will result in increased RBC lysis, while adding a negative regulator, such as factor H (FH) will result in decreased RBC lysis. A previous study has applied a similar approach to measure complement convertase activity in human serum (Blom et al., 2014) and the method here has adapted this to measure the net stimulatory or inhibitory ability of cell culture supernatants for the AP (Cabezas et al., 2018).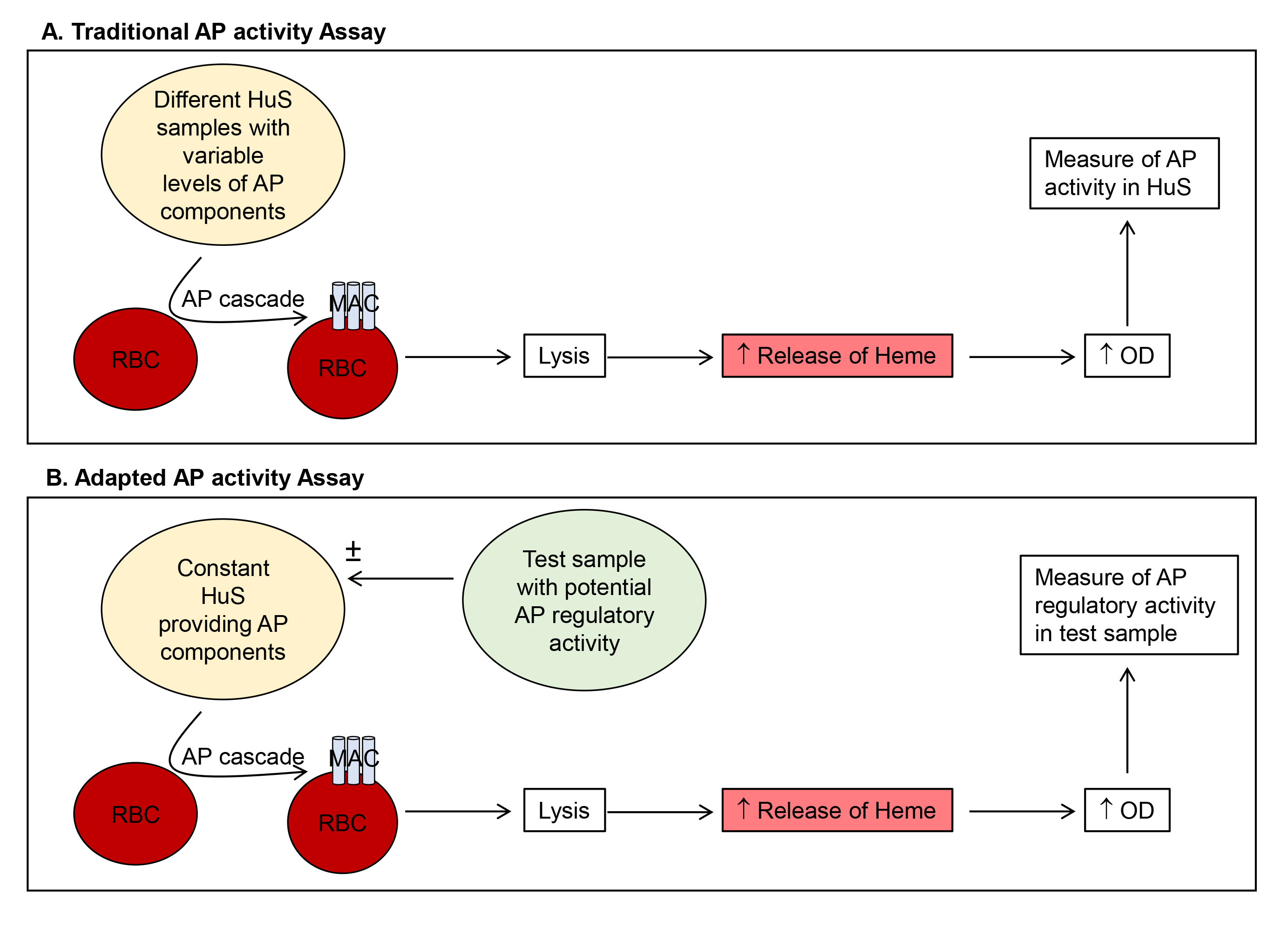
Figure 1. Comparison of traditional (A) and adapted (B) AP activity assays. Method (A) uses variable levels of HuS to quantitate AP activity in the test HuS samples. Our adapted method (B) utilizes a constant HuS sample and quantitates the AP regulatory activity of added test samples. HuS = human serum; OD = optical density.
Materials and Reagents
- Pipette tips (Axygen Scientific, catalog number: T-200-Y)
- 1.5 ml flip-topped tubes (Axygen Scientific, catalog number: MCT-150-C)
- Plastic film (Greiner multiwell plate sealer, Sigma-Aldrich, catalog number: A5596)
- Borosilicate glass beads, 5 mm (Sigma-Aldrich, catalog number: Z143944)
- 96-well flat bottomed plates (Costar, catalog number: 3596)
- Barbitone complement diluent tablets (Oxoid, catalog number: BR0016)
- NaOH pellets (UNIVAR, catalog number: A482)
- Concentrated HCl (ANALAR, catalog number: 101256J)
- Human serum (HuS), non-heat inactivated, store at -20 °C (collected from discard blood following routine clinical venesection in accordance with Southern Adelaide Local Health Network, Human Ethics Research Committee approval #343.16-HREC/16/SAC/306)
- Rabbit blood, freshly collected (under Flinders University Animal Welfare Committee approval to use scavenge material)
- Purified FH, generated in-house from human blood and stored at -20 °C [Diluted to 1 µg/µl in vehicle prior to use (Cabezas et al., 2018)]
- Vehicle = corresponding carrier for the test samples for example DMEM (Hyclone, catalog number: SH30022.01)
- Sodium chloride (NaCl) (Santa Cruz, catalog number: sc-203274A)
- Gelatin solution (2% solution) (Sigma-Aldrich, catalog number: G1393)
- EGTA (Sigma-Aldrich, catalog number: E3889)
- Veronal buffer (see Recipes)
- AP buffer (see Recipes)
- Saline (see Recipes)
- 10 N NaOH (see Recipes)
Equipment
- Pipettes (Thermo Fisher Scientific, model: Finnpipette F1)
- 50-100 ml conical flask/Erlenmeyer flask (Thermo Fisher Scientific, catalog number: KIM 265520-1)
- Oven (Labnet mini incubator)
- Hemocytometer (Counting chamber) (Hawksley, catalog number: AC1000)
- Plate centrifuge (Heraeus Sepatech Omnifuge 2.0 RS)
- Microplate reader (multimode detector) (Beckman Coulter, model: DTX880)
Procedure
- Preparation of rabbit RBC
Note: All procedures are performed on a standard laboratory bench.- Collect rabbit blood into a 50-100 ml flask containing a layer of glass beads and gently swirl to allow a clot to form. This should occur within 10 min and be complete by approximately 20 min.
- Decant the defibrinated blood. Allow the cells to settle under gravity for 2-5 min. Discard supernatant and wash in approximately 25 ml AP buffer. The starting solution will be red, then becomes clear upon washing. Once the final cell-free supernatant is clear (usually following 3-4 washes) the cells are ready for use. Store at 4 °C until use (maximum 1-2 months).
- Prior to use, re-wash RBC in approximately 25 ml AP buffer.
- Enumerate the cells using a hemocytometer and adjust to 5 x 107 cells/ml with AP buffer.
- Assay method
- Dilute HuS, 2 in 5 with AP buffer to chelate calcium and inactivate the CP and LP, resulting in a 40% HuS stock. For instance; add 720 μl of AP buffer to 480 μl of HuS in a 1.5 ml tube.
- Incubate on ice, 15 min.
- In 1.5 ml flip-topped tubes, mix 40% HuS with the test substance and AP buffer in a total volume of 100 μl. Include final concentrations of HuS at 0, 5% and 10% HuS for each test sample. Include 0, 5% and 10% HuS alone, controls. Include 0, 5% and 10% HuS with 50 µg purified FH, as a negative regulator (inhibition) control. Volume should be reconstituted to 100 μl with an appropriate vehicle, i.e., the media corresponding to the test sample, such as DMEM or RPMI. Sample set up is shown in Table 1.
Note: Each test sample is assayed with a final concentration of HuS at 5% and 10% to ensure activity is neither saturated nor at the limit of detection so that both activators and inhibitors of the AP can be measured.
Table 1. Tube components for test samples and controls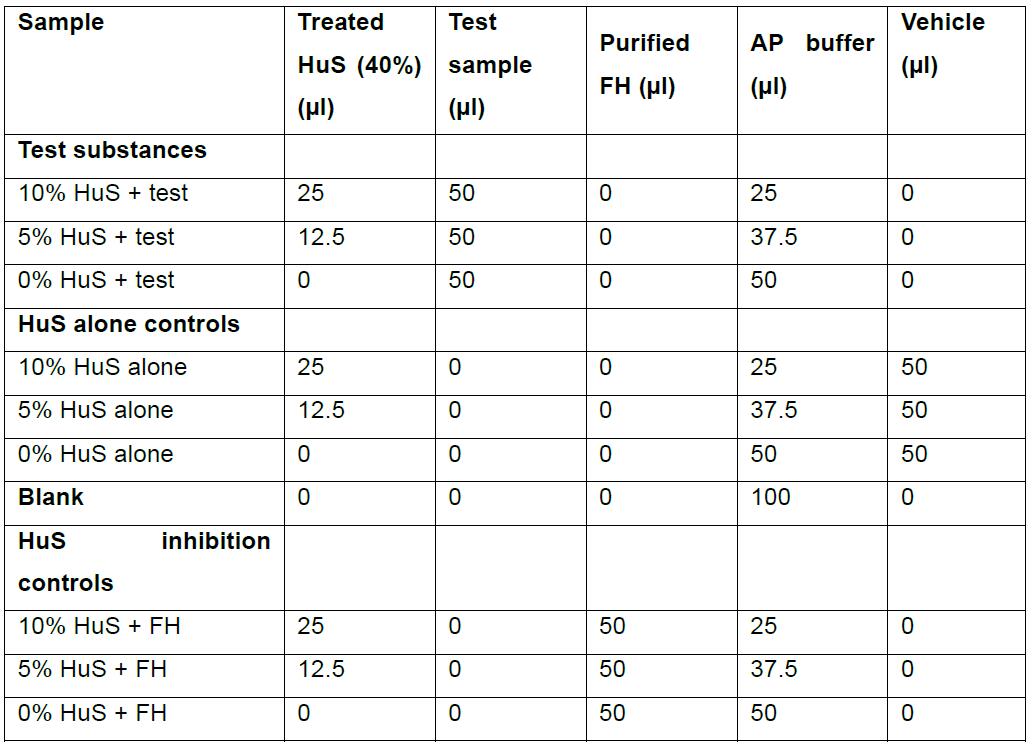
- Add 50 μl of each sample mix from Step B3 into a 96-well plate, in duplicate or triplicate if desired.
- Add 50 μl of the RBC suspension prepared as above in Procedure A. Include a total hemolysis control (50 μl cells + 50 μl of water). An example of sample setup is shown in Figure 2A.
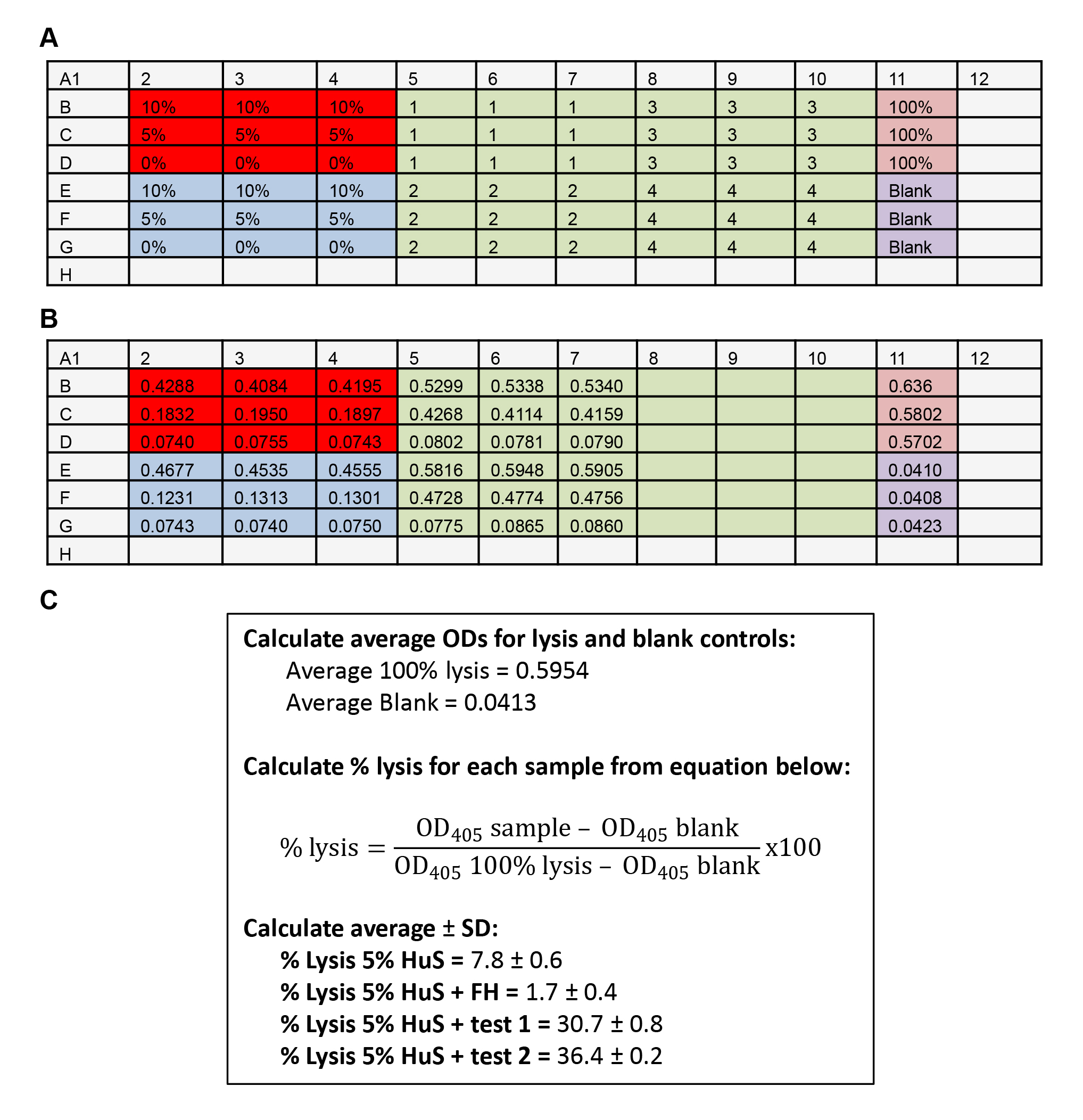
Figure 2. Example data. A. Assay plate setup. Red = HuS alone controls; Blue = HS + FH inhibition controls; Green = test samples 1-8; Pink = 100% hemolysis; Purple = Blank control Grey = wells left empty. B. OD measurements. C. Example calculations. - Seal plate with plastic film and incubate for 30 min in a 37 °C oven with gentle rocking every 10 min.
- Add 150 μl of ice-cold saline buffer to all wells except the 100% hemolysis control.
- Spin the plate, 1,000 x g, 5 min.
- Carefully remove 100 μl of the clarified supernatant with a pipette and transfer to a new flat-bottomed 96-well plate, taking care not to disturb the pelleted RBC.
- Read absorbance at 405 nm, yielding an example range of OD values shown in Figure 2B.
Data analysis
- Average the OD405 values for 100% lysis and blank.
- For each well, calculate % lysis using the equation shown in Figure 2C.
- Calculate average % lysis ± SD of test triplicates.
- Confirm controls are acting appropriately: 0, 5% and 10% HuS alone (should show increasing lytic ability); HuS + FH (inhibition); 0% HuS + test (lytic ability of test sample alone).
- Determine significance P < 0.05 (e.g., by Student's t-test [one variable] or one-way ANOVA/Tukey's test [multiple variables]), compared to the appropriate %HuS alone control using GraphPad Prism.
Recipes
- Veronal buffer
One barbitone complement dilutent tablet is dissolved in 100 ml of warm water and stored at room temperature
Note: Veronal buffer, used to make AP buffer contains barbitone, which is a narcotic drug. Ensure buffers and reagents are stored in accordance with regulations. - AP buffer
Veronal buffer plus 0.01 M EGTA and 0.1% (w/v) gelatin, pH 7.5Final con. For 1 L 0.01 M EGTA 3.80 g 0.1% (w/v) gelatin 200 ml 2% gelatin solution - Boil solution, by microwaving to solubilize gelatin
- Add EGTA and adjust the pH to 8-8.5 by dropwise addition of 10 N NaOH to dissolve EGTA. Once the solution is clear then add concentrated HCl to adjust final pH to 7.5
- Filter sterilize to prevent microbial growth
- Saline
Final con. For 1 L 0.15 M NaCl 8.76 g - 10 N NaOH
Final con. For 1 L 10 N 400 g
Note: All buffers are made up in MilliQ water.
Acknowledgments
Ms Cabezas-Falcon is supported by an Australian International Post-graduate Research Scholarship.
Competing interests
Authors have no conflicts of interest or competing interests.
Ethics
Human sera was collected in accordance with approval from the Southern Adelaide Clinical Human Research Ethics Committee (SA HREC EC00188), ORF number 343.16. Sheep blood was collected in accordance with the Flinders University Animal Welfare Committee approval to use scavenge material.
References
- Blom, A. M., Volokhina, E. B., Fransson, V., Stromberg, P., Berghard, L., Viktorelius, M., Mollnes, T. E., Lopez-Trascasa, M., van den Heuvel, L. P., Goodship, T. H., Marchbank, K. J. and Okroj, M. (2014). A novel method for direct measurement of complement convertases activity in human serum. Clin Exp Immunol 178(1): 142-153.
- Cabezas, S., Bracho, G., Aloia, A. L., Adamson, P. J., Bonder, C. S., Smith, J. R., Gordon, D. L. and Carr, J. M. (2018). Dengue virus induces increased activity of the complement alternative pathway in infected cells. J Virol 92(14).
- Joiner, K. A., Hawiger, A. and Gelfand, J. A. (1983). A study of optimal reaction conditions for an assay of the human alternative complement pathway. Am J Clin Pathol 79(1): 65-72.
- Prohaszka, Z., Nilsson, B., Frazer-Abel, A. and Kirschfink, M. (2016). Complement analysis 2016: Clinical indications, laboratory diagnostics and quality control. Immunobiology 221(11): 1247-1258.
- Ricklin, D., Hajishengallis, G., Yang, K. and Lambris, J. D. (2010). Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11(9): 785-797.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Falcon, S. C., Gordon, D. L. and Carr, J. M. (2018). Quantitation of Regulatory Activity for the Complement Alternative Pathway Using an Adaptation of the AP50 in vitro Assay. Bio-protocol 8(24): e3116. DOI: 10.21769/BioProtoc.3116.
Category
Immunology > Complement analysis > Virus
Microbiology > Microbe-host interactions > Virus
Biochemistry > Protein > Activity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.