- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Visualization and Quantification of Cell-to-cell Movement of Proteins in Nicotiana benthamiana


Published: Vol 8, Iss 24, Dec 20, 2018 DOI: 10.21769/BioProtoc.3114 Views: 7652
Reviewed by: Zhibing LaiChao-Jan LiaoJuan Du
Original research article
The authors used this protocol in:

May 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Cell-to-cell movement of proteins through plasmodesmata is a widely-established mechanism for intercellular signaling in plants. Current techniques to study intercellular protein translocation rely on single-cell transformation using particle bombardment or transgenic lines expressing photo-inducible fluorophores. The method presented here allows visualization and objective quantification of (effector) protein movement between N. benthamiana leaf cells. Agroinfiltration is performed using a single binary vector encoding a GFP-tagged protein of interest that is either mobile or non-mobile (MP; non-MP), together with an ER-anchored mCherry. Upon creation of mosaic-like transformation patterns, cell-to-cell movement of the MP can be followed by monitoring translocation of the GFP signal from mCherry labeled transformed cells into neighboring non-transformed cells. This process can be visualized using confocal microscopy and quantified following protoplast isolation and flow cytometric cell analysis. This method overcomes the limitations of existing methods as it allows rapid and objective quantification of protein translocation without the need of creating transgenic plants.
Keywords: Protein translocationBackground
Many proteins use plasmodesmata as channels to move from one cell to another. The aperture of plasmodesmata determines the size of molecules able to pass. To visualize whether a protein is able to move from cell to cell, current techniques include particle bombardment and photo-inducible fluorophores (e.g., DRONPA) (Gerlitz et al., 2018). A drawback of DRONPA is that it is highly time consuming since transgenic lines have to be created (Gerlitz et al., 2018). Transient transformation using particle bombardment overcomes this limitation. However, since it involves single cell transformation the fluorescent signal fades out rapidly when distributed over neighboring cells (Figure 1A). Fading is a limitation of both techniques and hampers detection of movement of poorly expressed proteins (Cao et al., 2018). Moreover, quantification of protein translocation with these methods is laborious and subjective, as only relative low numbers of translocation events can be monitored using a confocal microscope.
Our newly developed method allows both visualization and objective quantification of intercellular protein translocation in N. benthamiana leaves. The method is derived from a protocol for analyzing cell-to-cell movement of the Non-Structural Movement (NSm) protein from Tomato Spotted Wilt Virus (Feng et al., 2016). By transiently transforming multiple cells in a mosaic-like pattern movement of green fluorescently tagged proteins from transformed (labeled red) to non-transformed cells (non-fluorescent) can be monitored (Figure 1B). We adjusted this method to allow monitoring of effector translocation and provide an unbiased quantification procedure to record cell-to-cell movement. The effector proteins Avr2 and Six5 from the fungus Fusarium oxysporum were employed to test this system. This effector pair manipulates plasmodesmata to facilitate cell-to-cell movement of Avr2 in a Six5 dependent manner (Cao et al., 2018). In the absence of Six5 the Avr2 protein behaves as a non-mobile protein (Non-MP) and is retained in the transformed cell. The dependency of one protein on another one for cell-to-cell movement represents an anomaly, as typically mobile proteins (MP) have intrinsic properties to translocate themselves as exemplified by the NSm protein (Feng et al., 2016). 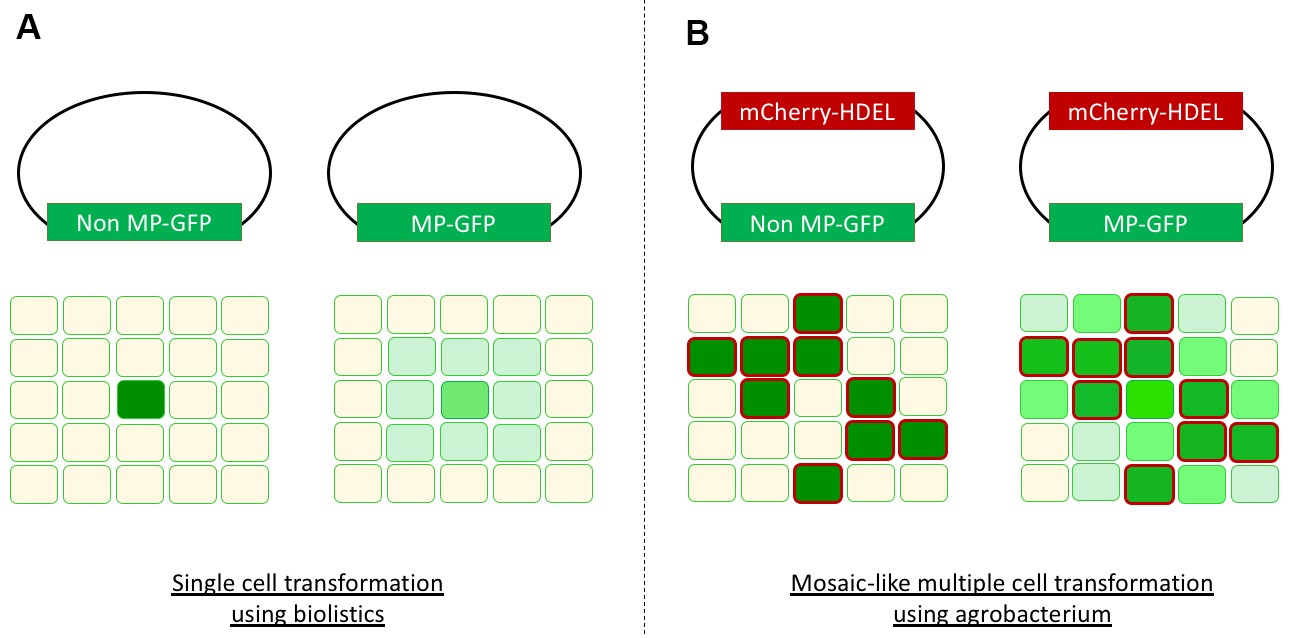
Figure 1. Overview cell-to-cell movement visualization. A. Single cell transformation using biolistics with a non-mobile protein (Non-MP-GFP) will result in a single green cell (left). A mobile protein (MP-GFP) will spread to neighboring cells, resulting in a green halo (right). B. Agrobacterium-mediated transient transformation of multiple cells in a mosaic-like pattern. In case of a non-mobile protein the transformed cells are marked both red (mCherry-HDEL) and green (Non-MP-GFP), and surround non-fluorescent untransformed cells (left). A mobile protein (MP-GFP) will be translocated from transformed cells (mCherry-HDEL and MP-GFP) to the neighboring untransformed cells resulting in an increased GFP signal in these cells (right). The intensity of this GFP signal in the untransformed cells varies depending on the amount of neighboring transformed cells, an artist’s impression is depicted here.
For our study a single binary vector carrying both Avr2-GFP and mCherry-HDEL was used for agroinfiltration in N. benthamiana (Ma et al., 2012). The mCherry-HDEL protein is ER-localized and cannot translocate to neighboring cells, thereby serving as a marker for transformed cells. Transformed cells show both green GFP and red mCherry signals, while untransformed cells lack both signals. However, upon co-infiltration with a second binary vector encoding SIX5 these neighboring untransformed cells will become green fluorescent as they acquire the AVR2-GFP protein from neighboring transformed cells. To quantify the intercellular translocation of Avr2-GFP, protoplasts can be isolated from infiltrated leaf sectors and subjected to flow cytometric cell analysis (Figure 2). Flow cytometry allows determination of the number of GFP + mCherry and GFP-only cells in presence- and absence of Six5, thereby enabling quantification of AVR2-GFP effector translocation. Potential false positive GFP-only events will be recorded in the AVR2-GFP control lacking SIX5, allowing determination of the base line level of GFP-only cells. Use of flow cytometry to quantify cell-to-cell movement provides an objective and unbiased way to assess intercellular translocation in a large number of cells. Other possible applications for this method include, among others, plasmodesmata aperture measurement by using single, double or triple GFP fusions following treatment of the plant with various (a)biotic stresses and/or pharmaceutical compounds. Furthermore, mutants and derivatives of proteins-of-interest can be screened for their altered translocation properties. 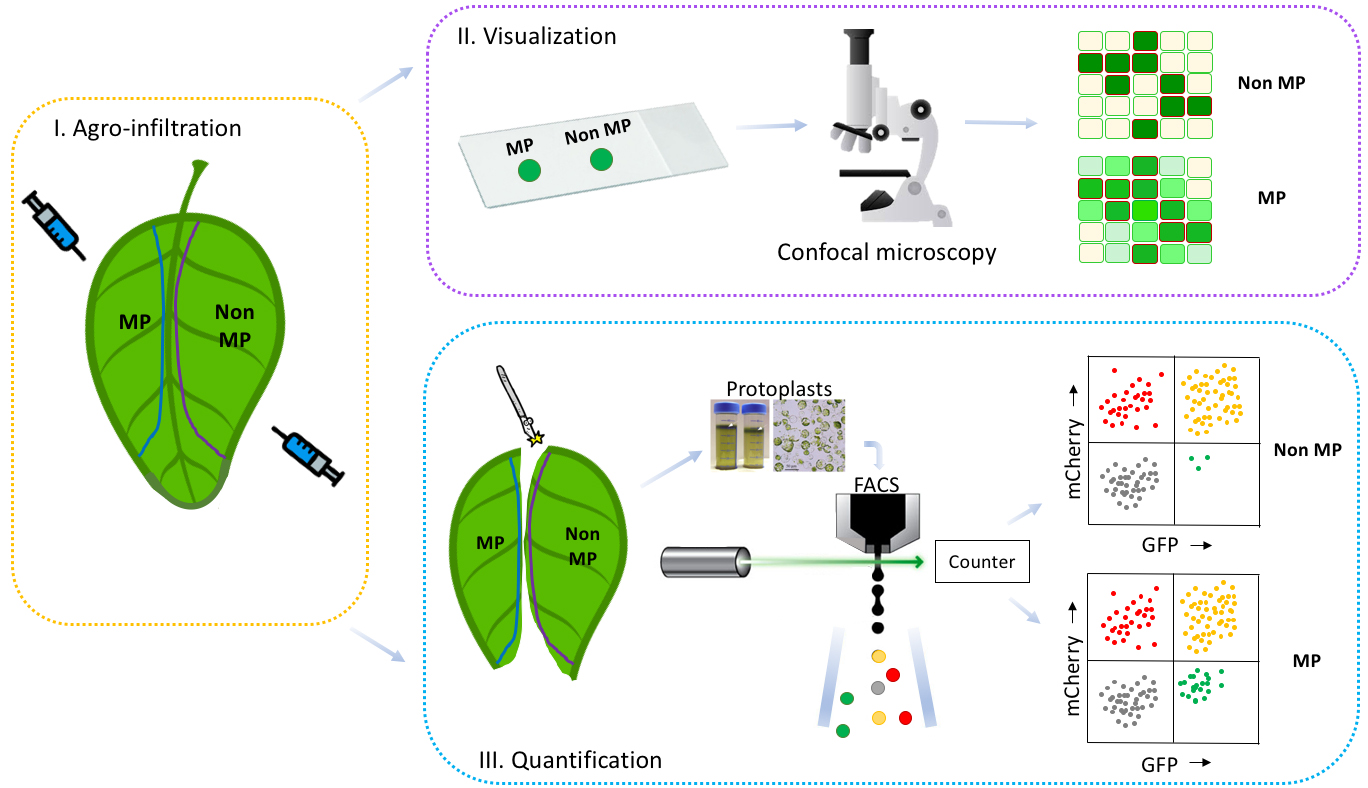
Figure 2. Workflow of the method to detect and quantify cell-to-cell movement of proteins. A schematic representation of the workflow (I, Agro-infiltration; II, Visualization; III, Quantification) to determine cell-to-cell movement of proteins is shown. By using this method to study the mobility of a protein of interest, one of two potential patterns will be obtained: either a mobile protein pattern (II, III MP) or a non-mobile protein pattern (II, III Non-MP). By comparing the protein of interest’s pattern with a positive MP control and a negative Non-MP control, a conclusion can be drawn regarding the protein of interest mobility. Our workflow consists of the following three steps: I. Infiltrate sectors of N. benthamiana leaves with Agrobacterium harboring constructs containing genes encoding either mobile (MP) or non-mobile proteins (Non-MP). II. Harvest leaf disks from the infiltrated leaf sectors and analyze the mosaic-like transformation patterns for MP and Non-MP with confocal microscopy to detect potential cell-to-cell movement. III. Isolate protoplasts from the infiltrated leaf sectors and count the number of red, yellow or green colored cells with flow cytometry (FACS) to quantify cell-to-cell movement.
Materials and Reagents
- Syringes 1 ml (BD Plastikpak, catalog number: 303172)
- Centrifuge tube 50 ml, conical bottom (Greiner Bio One, catalog number: 210261)
- Plastic pipette 25 ml (Greiner Bio One, catalog number: 760180)
- Microscope slides (Thermo Scientific, Menzel-Glazer, catalog number: 631-0098)
- Cover slides, 24 x 60 mm (Thermo Scientific, Menzel-Glazer, catalog number: 15747592)
- Square plates, 120 mm (Greiner Bio One, catalog number: 688102)
- Petri dishes, 94 mm (Greiner Bio One, catalog number: 632102)
- Non-sterile scalpel blades, No. 21 curved cutting edge (Swann-Morton, catalog number: 0107)
- Aluminum foil, thin
- Glass Pasteur pipettes (long) (WU Mainz, catalog number: 10216234)
- Steel mesh 80 (177 μm) (cell culture reagents) (Sigma, catalog number: S3770)
- 5 ml Polypropylene round bottom tubes (Falcon, catalog number: 352063)
- Membrane filter, 0.45 μm pore size (Millipore, Milex-HV, catalog number: SLHV033RS)
- Edding 400 permanent marker, 1 mm round point (Edding)
- E. coli (DH5α)
- Agrobacterium (GV3101)
- Nicotiana benthamiana
- pZK538 vector (Addgene, catalog number: 118012)
- pDGB3alpha2_35S:P19:Tnos (GB1203) (p19) (Addgene, catalog number: 68214)
- Avr2-GFP pZK538 (Addgene, catalog number: 118011) (Cao et al., 2018)
- NSm pZK538 (Addgene, catalog number: 118011-13) (Feng et al., 2016)
- Phusion polymerase
- DMSO
- Gamborg B5 medium (Duchefa, catalog number: G0209)
- MES sodium salt (Duchefa, catalog number: M1503)
- Calcium chloride dihydrate (CaCl2•2H2O) (Merck, catalog number: 102382)
- Ammonium nitrate (NH4NO3) (Merck, catalog number: 101187)
- Sucrose (Duchefa, catalog number: S0809)
- Macerozyme R-10 (Duchefa, catalog number: M8002)
- Cellulase onozuka RS (Duchefa, catalog number: C8003)
- Ethanol (96%) (VRW chemicals, catalog number: 83804)
- Commercial bleach (< 5% Hypochlorite)
- Sterile MilliQ water
- Potassium hydroxide (KOH) (Merck, catalog number: 105033)
- DpnI restriction enzyme (Thermo Scientific, catalog number: ER1701)
- GeneJet PCR purification kit (Thermo Scientific, catalog number: K0702)
- GeneJet Gel extraction kit (Thermo Scientific, catalog number: K0692)
- NEBuilder Hifi DNA assembly cloning kit (NEB, catalog number: E5520S)
- Agarose (Sphaero Q, catalog number: s103a)
- MS basal salt mixture (Duchefa, catalog number: M0221)
- Acetosyringone (C10H12O10) (Merck, catalog number: D134406)
- TEX buffer (see Recipes)
- Leaf digestion solution (see Recipes)
- MMAi medium (see Recipes)
Equipment
- Scalpel holder
- Scale
- Burner
- Flow hood
- Forceps
- Pipettes
- Leaf puncher 5 mm (Well Tech, Rapid core, catalog number: E69039-50)
- Confocal microscope LSM 510 (Zeiss, model: LSM510)
- EVOS FL cell imaging system (Thermo Fisher, model: EVOS FL, catalog number: AMF4300)
- Haemocytometer 0.100 mm; 0.0025 m2 (Merck, Blaubrand, catalog number: BR719520)
- Rotina 420R Centrifuge (Hettich Zentrifugen, model: Rotina 420R, catalog number: 4701)
- PCR machine (Bio metra, TAdvanced)
- Gel electrophoresis machine (Bio-Rad)
- Pipetus pipetting pump (Hirschmann Laborgerate, Pipetus, catalog number: 9907200)
- Heat block (Grant Instruments, model: QBD2, catalog number: 11470958)
- FACS Aria III BD (BD Biosciences, model: FACS Aria III)
Software
- Kaluza Flow Cytometry Analysis software version 1.2 (Beckman Coulter, US)
- Fiji (Fiji is just ImageJ) version 1 (https://fiji.sc/) (Schindelin et al., 2012)
Procedure
- Construct generation
- Linearize the pZK538 vector (Addgene #118012) using inversion PCR with Phusion polymerase and primer set FP7939 (GGC TAT CGT TCG TAA ATG GTG) and FP7940 (GGA TCC ATG GTG AGC AAG GGC GAG GAG CTG TTC ACC GGG GTG GT).
Note: Add 3% DMSO to the standard Phusion PCR mix, use a total volume of 20 μl. - Use the following PCR program:
- 98 °C 30 s
- 98 °C 10 s
- 57 °C 30 s
- 72 °C 15 min
- Repeat steps b-d 30 times
- 72 °C 10 min
Note: After 15 cycles add extra 0.1 μl of Phusion to the reaction.
- Digest linearized PCR product with DpnI restriction enzyme to remove original template.
- Clean up the digested PCR product with GeneJet PCR purification kit.
- Electrophorese 1 μl of linearized pZK538 vector on a 1% agarose gel for size and concentration estimation.
- Design primers to amplify your gene of interest [HiFi DNA assembly cloning protocol (NEB)]:
Fw primer: CACCATTTACGAACGATAGCCATGCCTGTGGAAGATGCCGATTC
Rv primer: ACCACCCCGGTGAACAGCTCCTCGCCCTTGCTCACCATGGATCCATCCTCTGAGATAGTAAGATAGTAG
Note: Replace the underlined section with gene of interest specific sequence. Remove the stop codon for your reverse primer, since GFP will be fused C-terminally. - Amplify your MP and non-MP of interest using PCR with Phusion polymerase (standard Phusion PCR mix, with a total volume of 20 μl).
- Separate the PCR products on a 1% agarose gel and isolate the fragment using a GeneJet gel extraction kit.
- Estimate the DNA concentration by loading 1 μl on a 1% agarose gel.
- Clone your gene of interest into the linearized cell-to-cell movement vector pZK538, according to HiFi DNA assembly cloning protocol (NEB).
- Use the following reaction mix (vector:insert ratio 1:2):
100 ng of linearized pZK538 vector (14 kb)
7 ng of MP/non-MP insert (500 bp)
10 μl NEBuilder HiFi DNA Assembly Master Mix
Fill up to 20 μl with MilliQ water
Note: Adjust the amount of insert DNA according to its size in base pairs (bp). - Incubate the reaction mix at 50 °C in a heat block for 45 min.
- Transform 2 μl of reaction mix to E. coli (DH5α) (Green and Sambrook, 2012).
- Check your final construct by sequencing the insert (FP7939: CACCATTTACGAACGATAGCCATGCCTGTGGAAGATGCCGATTC).
Note: Replace the underlined section with gene of interest specific sequence. - If correct, proceed to transform 25 ng of your final construct to Agrobacterium (GV3101) using electroporation (1.25 kV,
25 μF, 400 Ω) (Mersereau et al., 1990).
- Linearize the pZK538 vector (Addgene #118012) using inversion PCR with Phusion polymerase and primer set FP7939 (GGC TAT CGT TCG TAA ATG GTG) and FP7940 (GGA TCC ATG GTG AGC AAG GGC GAG GAG CTG TTC ACC GGG GTG GT).
- Nicotiana benthamiana plant growth and agroinfiltration
- Grow plants according to the protocol described by Ma et al., 2012.
Note: Growing plants in a growth chamber instead of a greenhouse provides better quality protoplasts. - In short: Germinate 10-15 seeds in a growth chamber (22.0 °C, 70% humidity, short day: 13 h dark; 11 h light) and transfer the seedlings seven days after sowing to separate 10 cm pots.
- Proceed with agroinfiltration when plants are five-week-old, this stage is optimal for agroinfiltration (Ma et al., 2012) and also gives sizable leaves for later protoplast isolation.
Note: Counting from the top of the plant, use leaves number two, three and four for infiltration (see Figure 4 from Ma et al., 2012). - Use the protocol for agroinfiltration as described by Ma et al., 2012 (but use different OD600 bacterial titers, see below).
- Incubate the agrobacteria in MMAi medium (see Recipes) for at least two hours prior to agroinfiltration.
- Try a range of different bacterial titers (e.g., OD600 of 0.5, 0.1, 0.05 etc.) to determine the optimal concentration for a mosaic-like transformation pattern for your MP and non-MP of interest. As a control (non-MP) Avr2-GFP can be used. In our hands, a final OD600 of 0.05 is best used for Avr2-GFP (Figure 3). Six5 and GUS can be infiltrated at OD600 of 0.5.
Note: If a low intensity of your GFP signal is encountered, the silencing suppressor p19 (see Addgene #68214 for similar construct) can be co-infiltrated (mix cultures before infiltration) using an OD600 of 0.5. - Infiltrate one leaf half (up to the main vein), with your MP-GFP and non-MP-GFP in the other half, mark the infiltrated sectors with an Edding 400 permanent marker (1 mm).
- Analyze leaf disks of the transformed areas 72 h after infiltration with a confocal microscope (see Procedure C).
Note: Timing could differ based on the expression level of the protein of interest. In our set-up imaging 72 h after infiltration resulted in the highest fluorescence levels.
- Grow plants according to the protocol described by Ma et al., 2012.
- Imaging cell-to-cell movement with confocal microscopy
- Punch leaf disks of the infiltrated areas with a 5 mm leaf puncher and place with bottom side up on microscope slide. Place a cover slide (24 x 60 mm) on top of your samples. Prevent dehydration of the sample by adding MilliQ water around the leaf disks.
- Set up the confocal microscope LSM 510: For GFP excitation use an Ar-ion laser to excite at 488 nm and record emission at 505-550 nm. For mCherry excitation use an Argon-2 laser to excite at 568 nm and record emission at 585-615 nm. For both GFP and mCherry use a laser strength of 14% and a pinhole of 1 airy unit.
- Select the pallete tab and use the range indicator to fine-tune the fluorescent image settings. Adjust the maximum and minimum intensity of both your GFP and mCherry signal, by changing the detector gain and amplifier offset.
- If proper GFP and mCherry fluorescent signals are observed, proceed to look for “gaps” (2 to 3 untransformed non-fluorescent cells) in the mCherry channel that are surrounded by 4 to 5 mCherry fluorescent cells. If cell-to-cell movement is occurring in non-fluorescent areas in the mCherry channel, it will display a GFP signal in the GFP channel (Figure 3. bottom panels). This mobile GFP signal can be relatively weak compared to surrounding transformed cells, but can be distinguished from aspecific background signals by its clean and specific localization to nuclei and cytosol (Figure 3. bottom panels).
Note: Ideally, most cells will be transformed resulting in occasional gaps in a confluent fluorescent surrounding. Increasing the number of gaps in our hands led to a decreased signal intensity.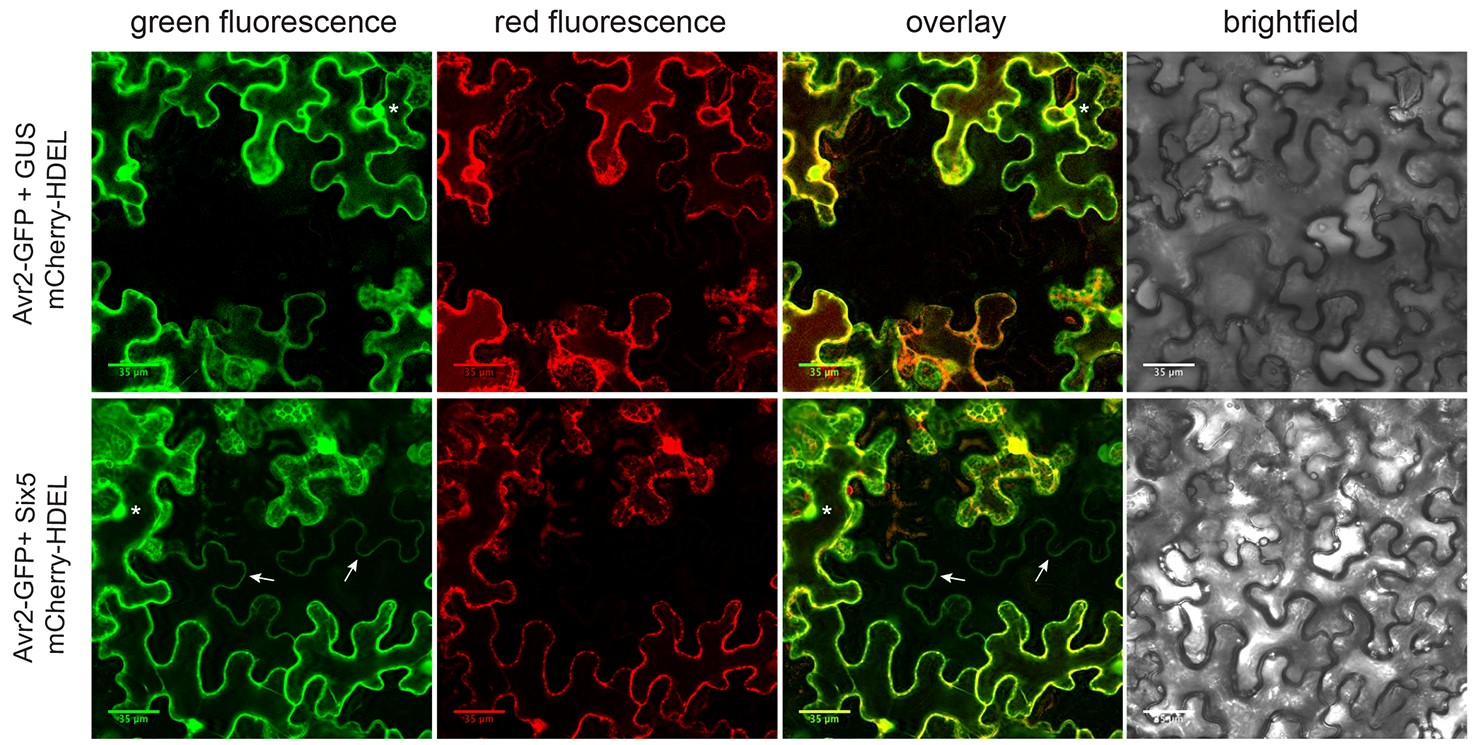
Figure 3. Mosaic-like transformation pattern showing cell-to-cell movement of Avr2-GFP. Co-agroinfiltration of Agrobacterium strains carrying a pZK538 vector encompassing genes encoding mCherry-HDEL and Avr2-GFP with either an Agrobacterium strain carrying a SIX5 or GUS-containing binary into N. benthamiana leaves. Transformed cells show both a red ER-localized signal and a green Avr2-GFP signal localized to cytoplasm and nucleus (indicated by an asterisk). The ER-localized mCherry is unable to move to neighboring untransformed cells and serves as a transformation marker. In the absence of Six5 (non-mobile Avr2-GFP), no red or green fluorescence was observed in untransformed cells adjacent to transformed cells (top panels). In the presence of Six5 (mobile Avr2-GFP), untransformed cells that flank a transformed cell show green fluorescence in cytoplasm and occasionally in the nucleus, indicating cell-to-cell translocation of Avr2-GFP (indicated by arrows, bottom panels). The above figure is adapted from Cao et al. (2018). Scale bars = 35 μm.
- Protoplast isolation
- Our protoplast isolation protocol is based on the method described by Faraco et al. (2011), with minor modifications (Figure 4).
- Collect the infiltrated leaves and include an un-infiltrated mock control (second, third or fourth leaf of one of the plants used for infiltration).
Note: Harvest one leaf at a time to prevent wilting of the other leaves during sample preparation.
Note: Perform all following steps in a flow hood. TEX buffer can be easily contaminated, leading to dead protoplasts.- Prepare five beakers, one with 250 ml 70% ethanol, one with 100 ml 1% hypochlorite solution (a 5-fold dilution of commercial bleach showed to be equivalent) and three with 250 ml sterile MilliQ water. Also prepare leaf digestion solution by adding 45 ml TEX buffer to 5 ml leaf digestion solution aliquot (10x) (previously prepared, see Recipes).
Note: One leaf digestion solution aliquot is good for five samples, 10 ml per sample. Thaw the appropriate number of aliquots for your experiment during the aforementioned preparations. - Sterilize leaves (one at a time) by dipping each of them in 70% ethanol for a few seconds and immersion in 1% hypochlorite solution for 30 s. Next, rinse the leaves three times by immersion in sterile H2O.
Note: Hold the leaf with sterile forceps by the stem to avoid tearing of the leaf. - Place the sterilized leaf in a sterile square Petri dish and use a sterilized scalpel to cut the infiltrated leaf area along the markings.
- Place the leaf half in a sterile round Petri dish containing 2 ml leaf digestion solution (1x). Use a sharp scalpel to slice the leaf into 2-5 mm strips.
Note: Use sterile forceps to gently hold the leaf in place and replace your scalpel blade regularly. - Add 8 ml of leaf digestion solution to the leaf strips and make sure to submerge each strip well by gently pushing them under.
- Close the Petri dish and wrap in aluminum foil.
- Incubate the plates overnight (approximately 15 h) at room temperature without shaking.
- Next morning, unpack the plates in a flow hood.
Note: Properly digested leaf strips will look almost transparent. Green protoplasts should be floating in the medium. - Use sterilized forceps to remove all leaf strips and gently move them up and down in the solution to release remaining protoplasts.
- Create a V-shaped opening in the Petri dish by heating your scalpel blade in a gas burner for a few seconds and use it to make a small incision at the side of the Petri dish. While cutting slightly turn the blade to one side to create a V-shape. This opening can be used to pour your protoplast suspension.
- Place the autoclaved steel mesh 80 (177 μm) over a clean sterile Petri dish and wet the mesh with a little TEX buffer. Gently pour the protoplast solution, using the V-shaped opening, through the mesh. Add approximately 5 ml TEX buffer to the original Petri dish to collect remaining protoplasts and also pour them through the mesh into the new Petri dish. Create another V-shaped opening in this new Petri dish.
- Decant the protoplast solution, via the V-shaped opening, in 50 ml Greiner tubes and adjust the volume to 25 ml with TEX buffer.
Note: Hold the 50 ml Greiner tube at a 45° angle to allow a gentle flow preventing shearing forces to the protoplasts. - Spin for 10 min using a Rotina 420R Centrifuge at 100 x g at room temperature, with acceleration 2 and deceleration 0.
Note: Be careful when retrieving samples, living protoplasts will form a ring at the top and are fragile! - Use long sterilized Pasteur pipettes connected to a Pipetus pipetting pump to remove the buffer and as much debris as possible.
Note: Slow down the speed of the pump when removing the debris and when the liquid reaches the bottom to avoid losing the floating protoplast cells. - Resuspend the protoplast suspension in 25 ml TEX buffer.
Note: Hold the 50 ml Greiner tube at a 45° angle, to avoid shearing. - Repeat the washing steps (D15-D16) once more.
- After the second spin, resuspend the protoplast suspension in approximately 3-4 ml TEX buffer.
- Determine the quality (intact; round cells) and concentration of the protoplasts by counting the cells with bright field microscopy, using a hemocytometer. A concentration of ca. 3 x 107 cells/ml is sufficient.
- Proceed with flow cytometry analysis of the protoplast samples.
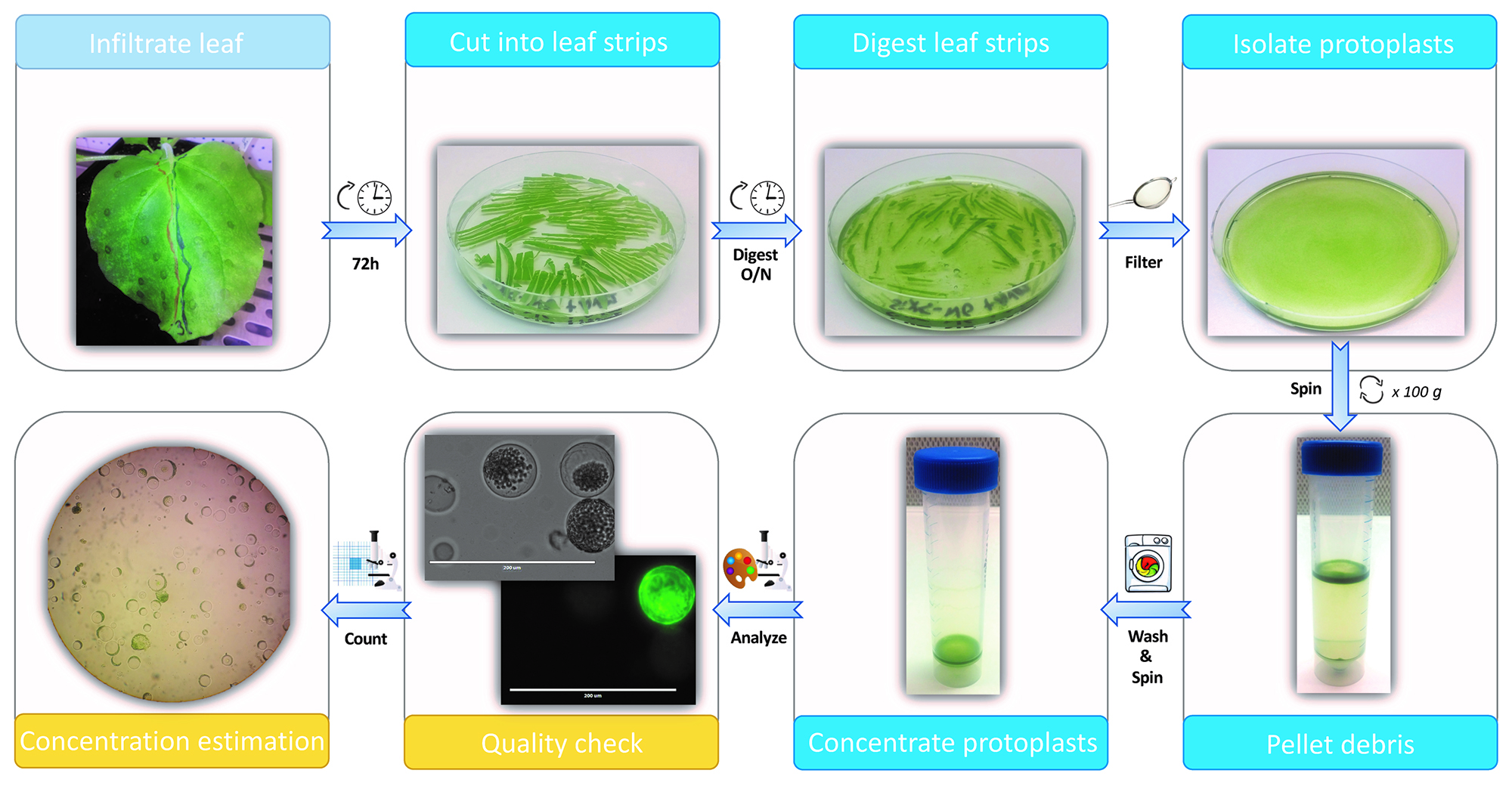
Figure 4. Protoplast isolation method. A flow scheme for protoplast isolation is shown. A N. benthamiana leaf is infiltrated with MP-GFP and Non-MP-GFP construct containing Agrobacterium strains 72 h prior to protoplast isolation (light blue). Infiltrated leaf sectors are cut into leaf strips, digested overnight in leaf digestion solution and filtered over a mesh (blue, top panels). Isolated protoplasts are centrifuged (100 x g) to pellet the debris. The protoplasts are then washed by removing the debris, along with most of the buffer and resuspended in fresh TEX buffer. Following centrifugation and removing the buffer, the protoplasts are concentrated in the few milliliters remaining (blue, bottom panels). Quality (intact; round cells) and fluorescence of the protoplasts is checked using brightfield- or fluorescence- microscopy. Subsequently, concentration of the protoplast suspension is estimated by counting cells using a hemocytometer (yellow).
- Flow cytometry
- Mix 500 μl protoplast suspension and 500 μl TEX buffer and pipette them over a 50 μm filter into 5 ml Polypropylene round bottom tubes.
- Place the tube into a FACSAria III (BD Biosciences, US) loading chamber and have the sampled loaded by the machine.
- Record the red and green channel, using the following laser settings: excitation of GFP at 488 nm and emission capture at 530/30 nm. mCherry excitation at 561 nm and emission capture at 610/20 nm.
- Adjust the voltage of the red and green channel to spread the population on scale.
- Count 50,000 cells per sample, with a Neutral density filter 2.0 to keep forward scatter (FSC) protoplasts on scale.
Data analysis
- Perform 3 independent biological replicates, each consisting of 3 infiltrated leaves per sample and subsequent cell counts.
Note: All following steps are performed with Kaluza Flow Cytometry Analysis software version 1.2 (Beckman Coulter, US).
- Make a dot plot with GFP on x-axis and mCherry on y-axis for each sample and use the mock sample to set the background fluorescence parameters, by dragging the quadrant dividers around the mock cell population (Figure 5A. Mock).
- Copy the mock settings to each sample and calculate the percentage of cells in each quadrant (Q1, Q2, Q3, Q4) (Figure 5A).
- Calculate the number of GFP-only cells within the total GFP population [(Q4/(Q3 + Q4)] and compare this for your MP and non-MP. These final percentages can be visually represented in a clustered column graph (Figure 5B).
Note: From the dot plots (Figure 5A) it is apparent that the mCherry-only population (Q2) also changes in the presence of Six5. This is due to the movement of Avr2-GFP to not only untransformed cells, but also to mCherry-only cells, shifting them towards the mCherry + GFP population (Q3). - Calculate the average for the replicates and use a paired t-test to determine if there is a significant difference between your MP and non-MP (Figure 5C).
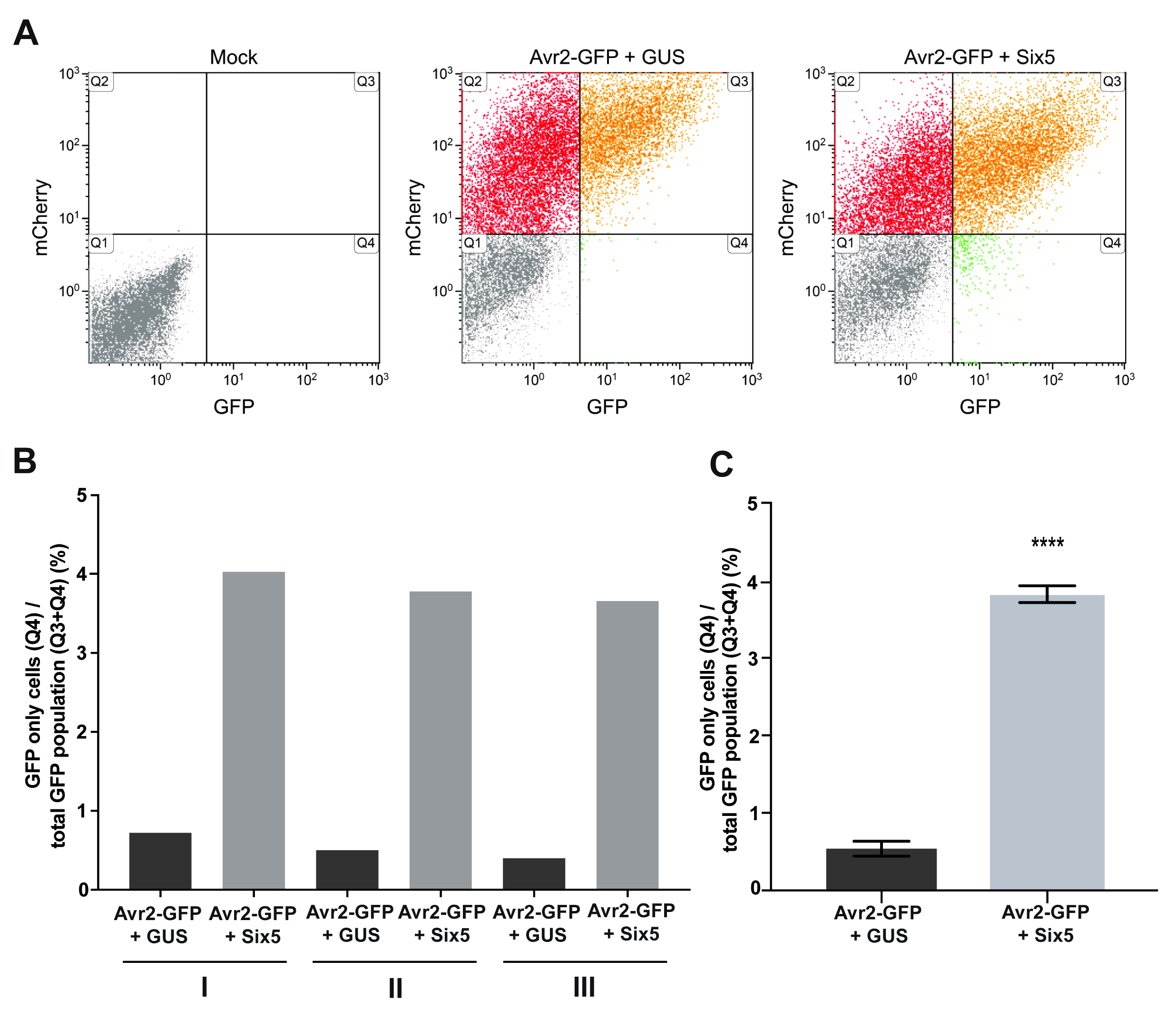
Figure 5. Quantification of Avr2-GFP translocation by fluorescence assisted cell counting. A. To assess Six5-mediated translocation of Avr2-GFP, protoplasts were isolated from Avr2-GFP/mCherry-HDEL agro-infiltrated leaves and the number of cells showing only green, only red, or both green and red fluorescence was counted using flow cytometry. Background autofluorescence levels were determined using untransformed cells (mock) as reference. Based on the mock, four quadrants were established following expression of the Avr2-GFP/mCherry-HDEL construct: Q1, non-transformed cells; Q2, cells expressing mCherry only; Q3, cells expressing mCherry + GFP; and Q4, accumulating GFP only. In the presence of Six5 (mobile Avr2-GFP), a higher number of GFP-only cells was observed in Q4 as compared to that of the GUS infiltrations (non-mobile Avr2-GFP). In the case of a normal (singular) MP, Avr2-GFP can be used as a non-MP control. B. Example graph of three quantification replicates. C. Aggregate data comprising quantification measurements I-III and statistical analysis. Paired t-test shows a significant difference with a P value of < 0.0001 (****). Error bars indicate standard error. The above figure is adapted and modified from Cao et al. (2018).
Notes
As a Non-MP control Avr2-GFP (Cao et al., 2018) (Addgene #118011) can be used. As a MP control the Avr2-GFP/Six5 combination (Cao et al., 2018) or the NSm protein (Feng et al., 2016) (Addgene #118011-13) can be used.
Recipes
- MMAi (made fresh)
5 g/L MS basal salt mixture (Murashige and Skoog medium without gamborg B5 vitamins)
20 g/L Sucrose
10 mM MES
200 μM Acetosyringone - TEX buffer (1 L)
3.1 g/L Gamborg B5 medium
500 mg/L MES
750 mg/L CaCl2•2H2O
250 mg/L NH4NO3
136.9 g/L sucrose
Dissolve in 1 L of MilliQ water, pH 5.7 (with 1 M KOH), filter sterilize (0.45 μm pore), store at 4 °C - 10x leaf digestion solution
2% w/v macerozyme R-10
4% w/v cellulase Onozuka RS
Dissolve in TEX buffer (100 ml is enough for 100 digestions)
Mix for at least 30 min, then centrifuge at 3,000 x g for 15 min
Filter sterilize supernatant with 0.45 μm pore filter, make 5 ml aliquots in 50 ml tubes and store at -20 °C
Dilute to a total volume of 50 ml with TEX buffer before use
Acknowledgments
We are grateful to LCAM-FNWI for confocal microscopy support and Ben Cornelissen and Nico Tintor for their feedback on the manuscript. Part of the presented protocol was based on previous studies by Ma et al., 2012 and Faraco et al., 2011. This work was supported by the NWO-Earth and Life Sciences funded VICI project No. 865.14.003.
Competing interests
The authors declare that there are no conflicts of interest.
References
- Cao, L., Blekemolen, M. C., Tintor, N., Cornelissen, B. J. C. and Takken, F. L. W. (2018). The Fusarium oxysporum Avr2-Six5 effector pair alters plasmodesmatal exclusion selectivity to facilitate cell-to-cell movement of Avr2. Mol Plant 11(5): 691-705.
- Faraco, M., Di Sansebastiano, G. P., Spelt, K., Koes, R. E. and Quattrocchio, F. M. (2011). One protoplast is not the other! Plant Physiol 156(2): 474-478.
- Feng, Z., Xue, F., Xu, M., Chen, X., Zhao, W., Garcia-Murria, M. J., Mingarro, I., Liu, Y., Huang, Y., Jiang, L., Zhu, M. and Tao, X. (2016). The ER-membrane transport system is critical for intercellular trafficking of the NSm movement protein and tomato spotted wilt tospovirus. PLoS Pathog 12(2): e1005443.
- Gerlitz, N., Gerum, R., Sauer, N. and Stadler, R. (2018). Photoinducible DRONPA-s: a new tool for investigating cell-cell connectivity. Plant J 94(5): 751-766.
- Green, M. R. and Sambrook, J. (2012). Molecular Cloning: A Laboratory Manual (4th edition). Cold Spring Harbor Laboratory Press.
- Ma, L., Lukasik, E., Gawehns, F. and Takken, F. L. W. (2012). The use of agroinfiltration for transient expression of plant resistance and fungal effector proteins in Nicotiana benthamiana leaves. Methods Mol Biol 835: 61-74.
- Mersereau, M., Pazour, G. J. and Das, A. (1990). Efficient transformation of Agrobacterium tumefaciens by electroporation. Gene 90(1): 149-151.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Blekemolen, M. C., Tark-Dame, M. and Takken, F. L. (2018). Visualization and Quantification of Cell-to-cell Movement of Proteins in Nicotiana benthamiana. Bio-protocol 8(24): e3114. DOI: 10.21769/BioProtoc.3114.
Category
Plant Science > Plant cell biology > Intercellular communication
Plant Science > Plant transformation > Agrobacterium
Molecular Biology > Protein > Intercellular translocation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.