- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

An in vitro Microscopy-based Assay for Microtubule-binding and Microtubule-crosslinking by Budding Yeast Microtubule-associated Protein

Published: Vol 8, Iss 23, Dec 5, 2018 DOI: 10.21769/BioProtoc.3110 Views: 7544
Reviewed by: Pengpeng LiFarah HaqueJer-Sheng LIN
Original research article
The authors used this protocol in:

Sep 2017
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
In this protocol, we describe a simple microscopy-based method to assess the interaction of a microtubule-associated protein (MAP) with microtubules. The interaction between MAP and microtubules is typically assessed by a co-sedimentation assay, which measures the amount of MAP that co-pellets with microtubules by centrifugation, followed by SDS-PAGE analysis of the supernatant and pellet fractions. However, MAPs that form large oligomers tend to pellet on their own during the centrifugation step, making it difficult to assess co-sedimentation. Here we describe a microscopy-based assay that measures microtubule binding by direct visualization using fluorescently-labeled MAP, solving the limitations of the co-sedimentation assay. Additionally, we recently reported quantification of microtubule bundling by measuring the thickness of individual microtubule structures observed in the microscopy-based assay, making the protocol more advantageous than the traditional microtubule co-pelleting assay.
Keywords: MicrotubuleBackground
Microtubules are dynamic polar filaments made up of polymerized tubulin subunits. Cells use microtubules to organize the cytosol and to build complex architectures necessary for cell growth and division. For instance, during interphase, microtubules act as molecular highways for motor proteins to manipulate the position of various cargoes in the cell. During mitosis, microtubules assemble the mitotic spindle to accomplish the critical task of chromosome segregation (Wadsworth and Khodjakov, 2004; Wadsworth et al., 2011). To date, we know that numerous regulators, including motor and non-motor microtubule-associated proteins (MAPs), interact with microtubules and participate in a variety of motile activities carried out by microtubules (Goshima and Scholey, 2010). In particular, many of these factors ensure that cells undergo proper mitotic progression, by helping to generate pulling and pushing forces on the microtubules for centrosome separation (Gonczy et al., 1999; Tanenbaum et al., 2008), nuclear envelope breakdown (Salina et al., 2002), mitotic spindle assembly (Rusan et al., 2002), chromosome capture and congression (Schmidt et al., 2005), spindle centering and positioning (Omer et al., 2018; Zulkipli et al., 2018), spindle checkpoint inactivation (Howell et al., 2001), and spindle elongation (Sharp et al., 1999; Khmelinskii et al., 2009).
We recently showed that She1, a MAP that regulates dynein motility along microtubule tracks (Markus et al., 2012), also crosslinks spindle microtubules to help maintain spindle integrity during spindle positioning in budding yeast (Zhu et al., 2017). In this bio-protocol, we describe a sensitive fluorescence-based assay that enables visualization and quantification of microtubule binding and crosslinking activities exhibited by a MAP. Because this assay does not need biochemical quantities of purified MAPs, we believe that it represents a significant advantage over the traditional microtubule co-sedimentation assay.
Materials and Reagents
- Avant 1.7 ml microcentrifuge tubes (MIDSCI, catalog number: AVSS1700)
- Pipette tips, 20 µl (Rainin Instrument, LLC, catalog number: 17005091)
- Pipette tips, 250 µl (Rainin Instrument, LLC, catalog number: 17005093)
- Razor blades (Fisher Scientific, catalog number: 12640)
- Microcentrifuge tube rack (Fisher Scientific, catalog number: 22-313630)
- Scotch double-sided tape, ¾ in. x 400 in. (3M)
- Microscope slides, 3 in. x 1 in. (Fisher Scientific, catalog number: 12518100A3)
- Cover glass, No. 1.5, 30 mm x 22 mm (Fisher Scientific, catalog number: 12544A)
- Filter forcep (EMD Millipore, catalog number: XX6200006P)
- Kimwipe (Fisher Scientific, catalog number: 06666A)
- Microscope lens paper, 4 in. x 6 in. sheets (Fisher Scientific, catalog number: 11996)
- Unlabeled porcine tubulin, > 99% pure, 1 mg (Cytoskeleton, Inc., catalog number: T240) (store at -80 °C)
- HiLyte Fluor 488 labeled porcine tubulin, 20 µg (Cytoskeleton, Inc., catalog number: TL488M) (store at -80 °C)
- Rhodamine labeled porcine tubulin, 20 µg (Cytoskeleton, Inc., catalog number: TL590M) (store at -80 °C)
- 100 mM GTP (Cytoskeleton, Inc., catalog number: BST06-010) (store at -20 °C)
- 10 mM Taxol (Cytoskeleton, Inc., catalog number: TXD01) (store at -20 °C)
- Precision Red Advanced Protein Assay Reagent (Cytoskeleton, Inc., catalog number: ADV02) (store at room temperature)
- PIPES (Sigma-Aldrich, catalog number: P1851) (store at room temperature)
- EGTA (Fisher Scientific, catalog number: O2783-100) (store at room temperature)
- Magnesium Chloride Hexahydrate (Fisher Scientific, catalog number: BP214-500)
- Ultrapure water from Milli-Q® Advantage A10 Water Purification System (Millipore Sigma)
- THETM alpha Tubulin Antibody, mAb, mouse (GenScript, catalog number: A01410) (store at -20 °C)
- Pluronic F-127, 100 g (Anatrace, catalog number: P305) (store at room temperature)
- Carl Zeiss ImmersolTM 518 F Immersion Oil (Fisher Scientific, catalog number: 1262466A) (store at room temperature)
- PEM Buffer (see Recipes)
- Blocking buffer (see Recipes)
Equipment
- Ice bucket (Fisher Scientific, catalog number: 07210123)
- Milli-Q® Advantage A10 Water Purification System (Millipore Sigma)
- Sorvall centrifuge equipped with S120-AT2 rotor (Thermo Fisher Scientific, model: Discovery)
- 37 °C incubator (Fisher Scientific, catalog number: 11690525D)
- Microcentrifuge (Eppendorf, model: 5424)
- Inverted fluorescence microscope equipped with Perfect Focus System and a 100x/1.49 NA objective (Nikon, model: Ti-E)
- Laser launch system with 405/488/561/640 nm wavelengths at 15 mW per line (Nikon, model: LUN4)
- Electron multiplying CCD camera (Andor Technology, model: iXon 888)
- GFP filter cube set for imaging HiLyte Fluor 488 or Alexa Fluor 488 fluorescence (Chroma Technology Corp., catalog number: 49002)
- TRITC filter cube set for imaging rhodamine or TMR fluorescence (Chroma Technology Corp., catalog number: 49008)
- HP workstation with 3.5 GHz 6 core Xeon processor, 64 GB memory, 1 TB hard drive, and 64bit Windows 7 (Micro Video Instruments, Inc., catalog number: 99909)
- Pipet-Lite LTS pipette L-20XLS+ (Rainin Instrument, LLC, catalog number: 17014392)
- Pipet-Lite LTS pipette L-200XLS+ (Rainin Instrument, LLC, catalog number: 17014391)
Software
- NIS-Elements Advanced Research (Nikon) (https://www.nikoninstruments.com/Products/Software/NIS-Elements-Advanced-Research)
- ImageJ (NIH, https://imagej.nih.gov/ij/download.html)
- KaleidaGraph (http://www.synergy.com/wordpress_650164087/kaleidagraph/)
Procedure
- Prepare fluorescently-labeled microtubules
- Dissolve unlabeled porcine tubulin (Cytoskeleton, Inc.) on ice in PEM buffer (Recipe 1) to a final concentration of 5 mg/ml (e.g., add 200 µl of PEM buffer to 1 mg of unlabeled lyophilized tubulin).
- Dissolve HiLyte Fluor 488 or rhodamine-labeled tubulin (Cytoskeleton, Inc.) on ice in PEM buffer to a final concentration of 5 mg/ml (e.g., add 4 µl of PEM buffer to 20 µg of fluorescently-labeled lyophilized tubulin).
- Mix 50 µl of unlabeled tubulin with 4 µl of fluorescently-labeled tubulin in a clean 1.7 ml microfuge tube. This step and subsequent steps may be performed under normal room light. It is not necessary to pipet in the dark.
- Incubate the tubulin mixture on ice for 10 min without shaking.
- Centrifuge the tubulin mixture for 10 min at 298,000 x g and 4 °C (in Sorvall Discovery centrifuge with S120-AT2 rotor) to remove unwanted tubulin aggregates. A pellet is usually not visible after centrifugation in this step, since the lyophilized tubulin would normally dissolve completely in Steps A1 and A2 above.
- Transfer the supernatant containing the pre-cleared tubulin into a new 1.7 ml microfuge tube.
- Add 0.54 μl of 100 mM GTP (i.e., final GTP concentration at 1 mM) to polymerize the pre-cleared tubulin. Mix the solution well by pipetting up and down three times using the L-200XLS+ pipette. Do not over-pipet, as pipetting three times is sufficient to mix the solution and initiate the polymerization of tubulin subunits.
- Incubate the GTP/tubulin mixture in a 37 °C incubator for 20 min without shaking.
- Take the tube out of the 37 °C incubator and briefly spin it at 21,130 x g for 10 sec in an Eppendorf 5424 microcentrifuge to pellet any condensation that might have formed in the lid of the microfuge tube. Next, add 0.27 μl of 10 mM Taxol (i.e., final Taxol concentration at 50 μM) and mix well by pipetting up and down three times using the L-200XLS+ pipette.
- Incubate the tubulin/GTP/Taxol mixture at 37 °C for another 20 min without shaking.
- Centrifuge the tube containing the Taxol-stabilized microtubules at 20,156 x g for 10 min at room temperature in an Eppendorf 5424 microcentrifuge. This step will separate the polymerized microtubules from the free, unpolymerized tubulin.
- Discard the supernatant and resuspend the pellet in 50 μl of PEM buffer and 0.25 µl of 10 mM Taxol. To avoid shearing the polymerized microtubules, resuspend the pellet gently by pipetting up and down using the L-200XLS+ pipette and a pipet tip that has been shaved off with a razor blade.
- Once the pellet is completely dissolved, the concentration of the microtubules in the solution will be at ~50 μM. Alternatively, the concentration can be measured and determined by the Precision Red Advanced Protein Assay Reagent (Cytoskeleton, Inc.), following the manufacturer’s protocol.
- Store the microtubules at room temperature in the dark (e.g., in a drawer). This microtubule preparation is good for up to one month at the room temperature.
- Assemble the imaging chamber
- Cut two pieces of double-sided tape and stick them down on a clean microscope slide using filter forceps as shown in Part 1 of Video 1 and Figure 1A. The two pieces of tape should be placed in parallel and at 1.5 mm apart from each other in the middle of the microscope slide. No specific procedure to clean the surface of the microscope slide is required.
 Video 1. Preparing imaging chamber for microscopy. Part 1, a video clip showing Steps 1-3 of Procedure B. Part 2, a video clip showing how to pipet solution into the imaging chamber. Part 3, a video clip demonstrating how to replace the solution in the imaging chamber. For visualization purpose, PEM buffer spiked with bromophenol blue was used in the demonstration shown in the clips for Parts 2 and 3.
Video 1. Preparing imaging chamber for microscopy. Part 1, a video clip showing Steps 1-3 of Procedure B. Part 2, a video clip showing how to pipet solution into the imaging chamber. Part 3, a video clip demonstrating how to replace the solution in the imaging chamber. For visualization purpose, PEM buffer spiked with bromophenol blue was used in the demonstration shown in the clips for Parts 2 and 3. - Gently put down a clean cover glass on top of the two pieces of double-sided tape. Press down on the cover glass along the double sided tape regions, creating a strip of chamber sandwiched between the cover glass and the microscope slide (i.e., the imaging chamber) as shown in Figure 1B. No specific treatment of the cover glass surface is required before it is pressed down on the microscope slide.
- Flip the cover glass/slide assembly over, so that the cover glass is now at the bottom relative to the microscope slide.
- Prepare the anti-tubulin antibody solution by diluting THETM alpha Tubulin Antibody (GenScript) from 500 µg/ml to 33.3 µg/ml using PEM buffer. Store unused antibody solution at 4 °C for further use.
- Pipet 7 µl of the anti-tubulin antibody into the imaging chamber. Fill the chamber with the antibody solution by injecting slowly at an angle from one side of the chamber using an L-20XLS+ pipette (see Figure 1C). Also see Part 2 of Video 1 for a demonstration of the technique. Consider rehearsing this step on a practice chamber with only PEM buffer because the anti-tubulin antibody solution might not flow into the chamber on the first try.
- Allow the anti-tubulin antibody to stick to the chamber by incubating at room temperature for 5 min.
- Replace the anti-tubulin antibody solution in the imaging chamber with blocking buffer (Recipe 2). To do so, cut a piece of Kimwipe into triangular shape and hold the pointed end against one side of the imaging chamber. While the antibody solution is being drawn out of the chamber by the Kimwipe, pipet 7 µl of blocking buffer solution into the other side of the chamber (see Figure 1D). Also see Part 3 of Video 1 for a demonstration of the technique to replace the solution in the imaging chamber.
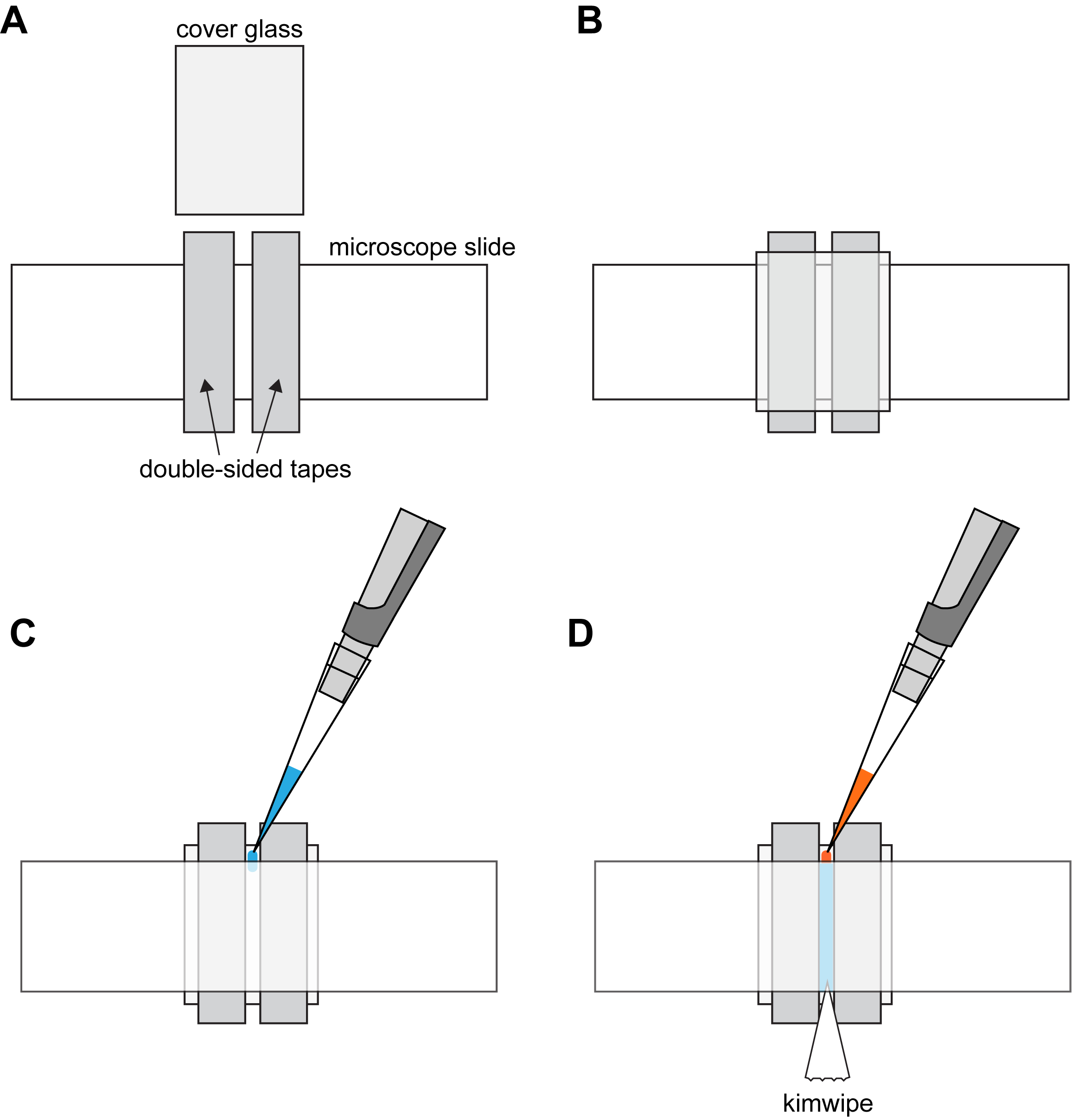
Figure 1. Chamber assembly. A. Adhere double-sided tapes to microscope slide. B. Apply cover glass onto the double-sided tapes. C. Turn the cover glass and slide over. Fill the chamber with anti-tubulin antibody solution. D. Flow-in blocking buffer by injecting on one side while drawing out with a Kimwipe on the other side. - Allow the blocking buffer to incubate at room temperature for 5 min.
- Prepare the microtubule solution by diluting the microtubule stock (prepared as described in Procedure A above) from 50 µM to 0.125 µM using PEM buffer. To avoid shearing the microtubules, use a cut pipet tip when pipetting the microtubules, as described in Step 12 of Procedure A.
- Replace the blocking buffer in the imaging chamber with the diluted microtubule solution. Similar to Step B7 above, use a piece of Kimwipe to draw the blocking buffer out of the chamber from one side while pipetting the microtubule solution into the chamber on the other side.
- Allow the microtubules to attach to the anti-tubulin antibody by incubating at room temperature for 5 min.
- Bring the chamber to an inverted fluorescence microscope for imaging.
- Cut two pieces of double-sided tape and stick them down on a clean microscope slide using filter forceps as shown in Part 1 of Video 1 and Figure 1A. The two pieces of tape should be placed in parallel and at 1.5 mm apart from each other in the middle of the microscope slide. No specific procedure to clean the surface of the microscope slide is required.
- Visualize microtubule binding and crosslinking
The following steps require the experimenter to have in hand a small quantity of test microtubule-binding protein purified and labeled with a fluorescent probe that is different from that used for labeling the microtubules. We refer the experimenter to other studies (Markus et al., 2012; Zhu et al., 2017) for protocols involving purification and labeling of HALO-tagged fusion proteins with TMR or Alexa Fluor 488, which works well for the following steps.- Mix 10 µl of 125 nM Taxol-stabilized microtubules (labeled with HiLyte Fluor 488 as described in Procedure A) with 1 μl of
10 µM MAP (labeled with TMR) in PEM buffer. Add 9 µl of PEM buffer to bring the total reaction volume to 20 μl. Alternatively, the same volume and concentration of Taxol-stabilized microtubules labeled with rhodamine (prepared as described in Procedure A) can be mixed with MAP labeled with Alexa Fluor 488 in this step. Microtubules mixed with buffer alone should be performed separately as a control. - Incubate the mixture at room temperature for 10 min.
- Flow the microtubules/MAP mixture into an imaging chamber that has been treated with anti-tubulin antibody and blocking buffer (Steps 1-8 in Procedure B). As described for Step 10 in Procedure B, use a piece of Kimwipe to draw the blocking buffer out of the chamber from one side while pipetting the microtubules/MAP solution into the chamber on the other side. To avoid shearing the microtubules, use a cut pipet tip when pipetting the microtubules into the imaging chamber.
- Allow the microtubules/MAP to attach to the anti-tubulin antibody by incubating at room temperature for 5 min.
- To wash away unattached microtubules/MAP, flow-in fresh blocking buffer once into the imaging chamber.
- Incubate at room temperature for 5 min. Bring the chamber to a microscope room for imaging.
- To visualize the microtubules/MAP and microtubules/buffer samples, use a Nikon Ti-E inverted microscope equipped with Perfect Focus System and a 100x/1.49 NA objective.
- Place a drop of immersion oil on the cover glass side of the imaging chamber and mount the slide on the microscope stage.
- Use a LUN4 laser launch system (Nikon) to illuminate the fluorophores and an iXon 888 electron multiplying CCD camera (Andor Technology) to image the fluorescently-labeled microtubules and MAP.
- Use a GFP filter cube set (Chroma Technology Corp.) to visualize Alexa Fluor 488 or HiLyte Fluor 488 fluorescence. Use a TRITC filter cube set (Chroma Technology Corp.) to visualize rhodamine or TMR fluorescence.
- Bring the microtubules into focus relative to the 100x/1.49 NA objective. Adjust focusing in the GFP channel for HiLyte Fluor 488-labeled microtubules. Alternatively, use the TRITC channel if Step C1 uses rhodamine-labeled microtubules. An example of an image of HiLyte Fluor 488-labeled microtubules is illustrated in Figure 2.
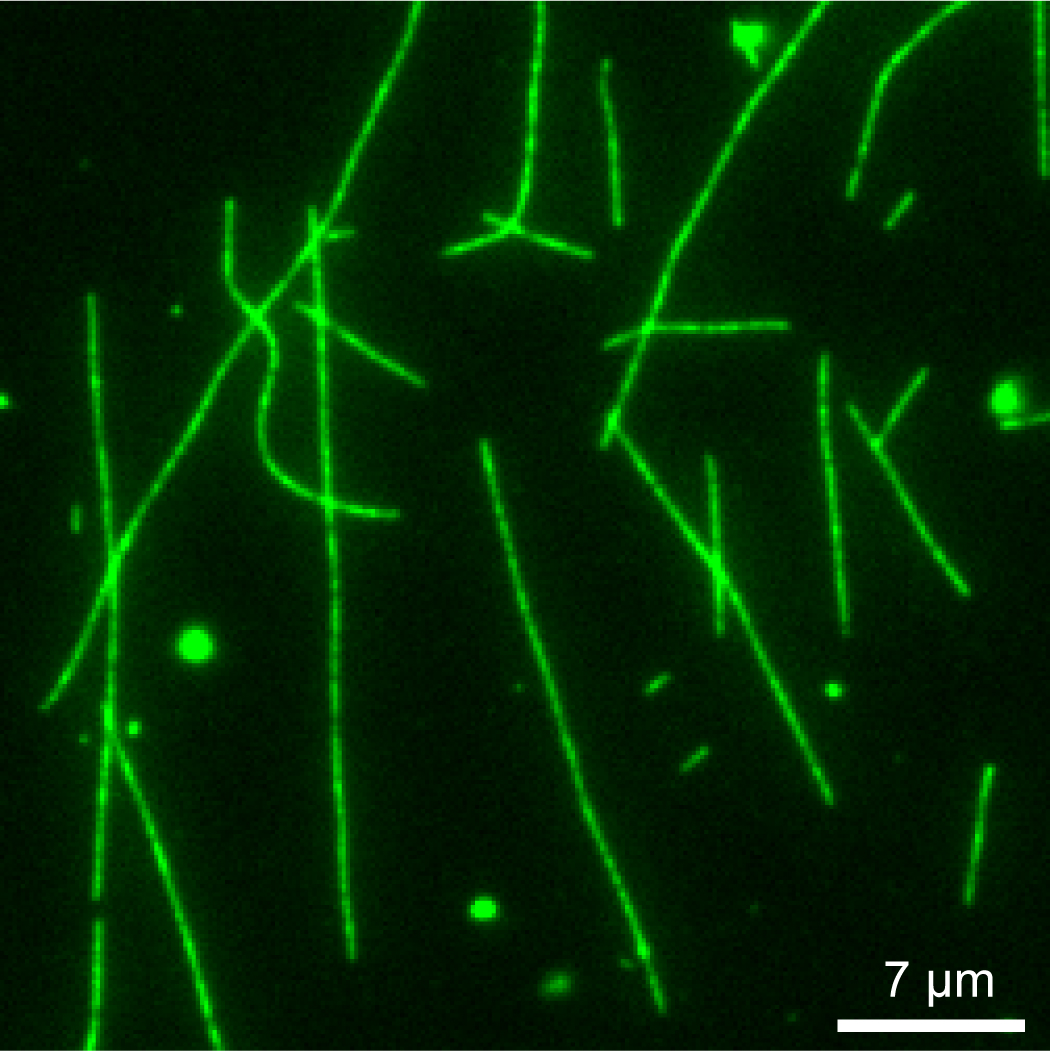
Figure 2. Example of HiLyte Fluor 488-labeled microtubules. Microtubules bound to anti-tubulin antibody in the imaging chamber were visualized in a Nikon Ti-E inverted microscope in this picture. Scale bar = 7 µm. - Switch to the TRITC channel to visualize the bound MAP on the microtubules. Alternatively, switch to GFP channel if Step C1 uses Alexa Fluor 488-labeled MAP.
- To acquire two-color pictures, use a PC workstation with NIS-Elements software (Nikon), which controls channel switching, laser intensity, and camera settings. An example of two-color images of HiLyte Fluor 488-labeled microtubules and TMR-labeled MAP is illustrated in Figure 3.
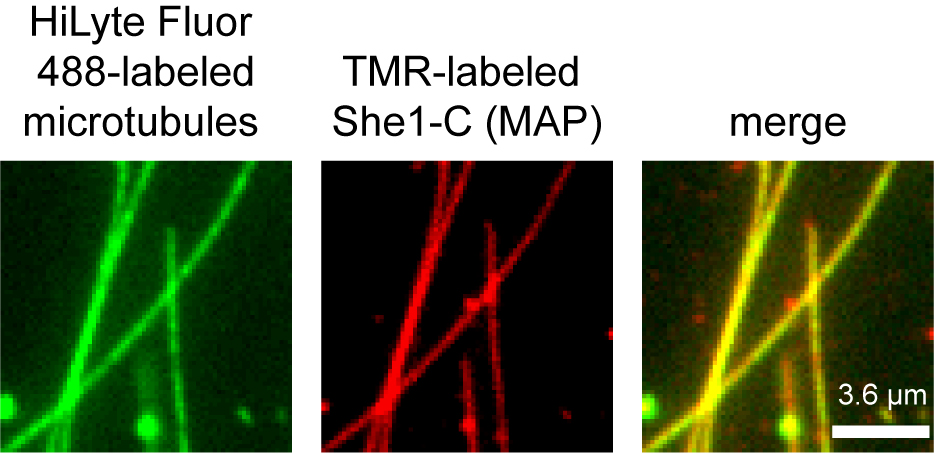
Figure 3. Example of two-color images of fluorescently-labeled microtubules and MAP. HiLyte Fluor 488-labeled microtubules mixed with a TMR-labeled recombinant MAP, She1-C-TMR, purified as described in Zhu et al. (2017), were visualized in the imaging chamber in this picture. Scale bar = 3.6 µm. - To collect enough data for subsequent analysis, capture at least 20 different fields of two-color microtubules/MAP images per chamber. Repeat the experiment with at least three independent preparations of MAP.
- Use identical camera settings to compare the amount of MAP bound to the microtubules from chamber to chamber or when different MAP concentrations are used (if needed).
- Some MAPs might crosslink or bundle microtubules. Image microtubules in the presence and absence of MAP to determine the frequency of observing microtubule bundles. Bundles can be distinguished from individual microtubules by measuring the microtubule cross-sectional intensity (as described in Data Analysis).
- Mix 10 µl of 125 nM Taxol-stabilized microtubules (labeled with HiLyte Fluor 488 as described in Procedure A) with 1 μl of
Data analysis
- To quantify the relative amount of bound MAP on microtubules, use the segmented line tool in ImageJ software (NIH). Trace a line along individual microtubules observed in the two-color images (as illustrated in Figure 4, left panel). Measure the mean fluorescence intensity of MAP per unit length for at least 50 individual microtubules (see Figure 4). Subtract the background of each measurement by moving the line tool to an adjacent area without microtubules (as illustrated in Figure 4, right panel). Calculate the average value of the corrected mean intensity per unit length.
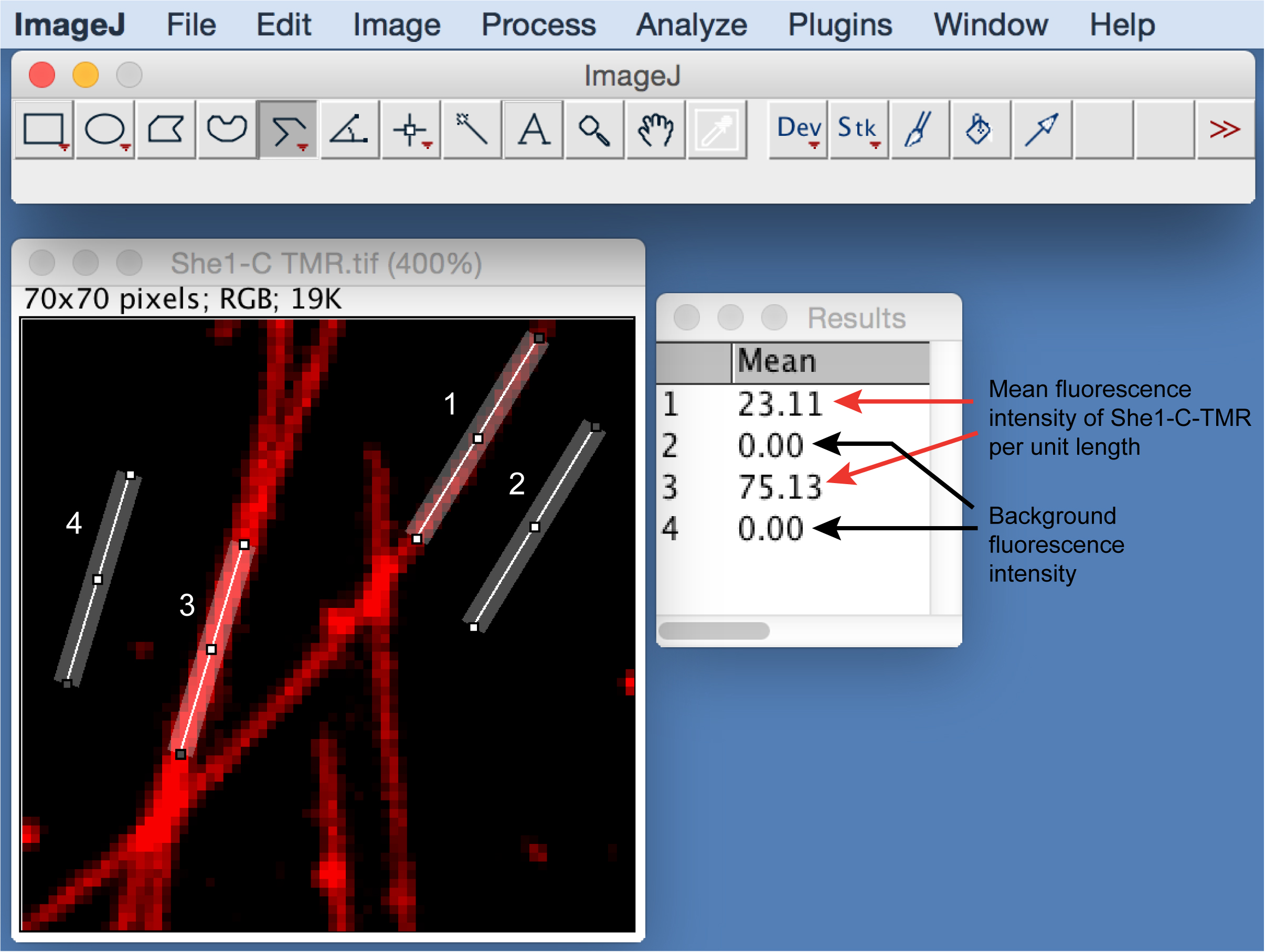
Figure 4. Example of quantifying bound MAP on microtubules. Left, sample lines drawn along individual microtubules (indicated by 1 and 3). Right, mean intensity of She1-C-TMR per unit length of the lines, as measured by ImageJ (under Analyze tab). Background intensity is measured by moving the line to an adjacent area without microtubules (indicated by 2 and 4). - Compare the values measured in the presence of MAP with that in the buffer control. Plot the distribution of the values for the experimental versus the control conditions using KaleidaGraph software. Perform a two-tailed unpaired Student's t-test to determine the statistical significance between the two distributions.
- To determine whether microtubules are crosslinked by MAP, use the segmented line tool in ImageJ to draw a line across individual microtubule structures observed in the two-color images (as illustrated in Figure 5, left panel). Use the plot profile tool in ImageJ to measure the cross-sectional fluorescence intensities along the line (Figure 5, middle panel). Perform these measurements for at least 50 different microtubule structures. Plot the fluorescence intensities as a function of the line using KaleidaGraph software (see Figure 3C in Zhu et al., 2017 for sample plots). Calculate the average peak intensity of the fluorescence profiles.
- Compare the peak intensities measured for microtubule structures observed in the presence of MAP with that in the buffer only control (see Figure 3C in Zhu et al., 2017 for examples). Perform a two-tailed unpaired Student's t-test to determine the statistical significance between the two groups of peak intensities.
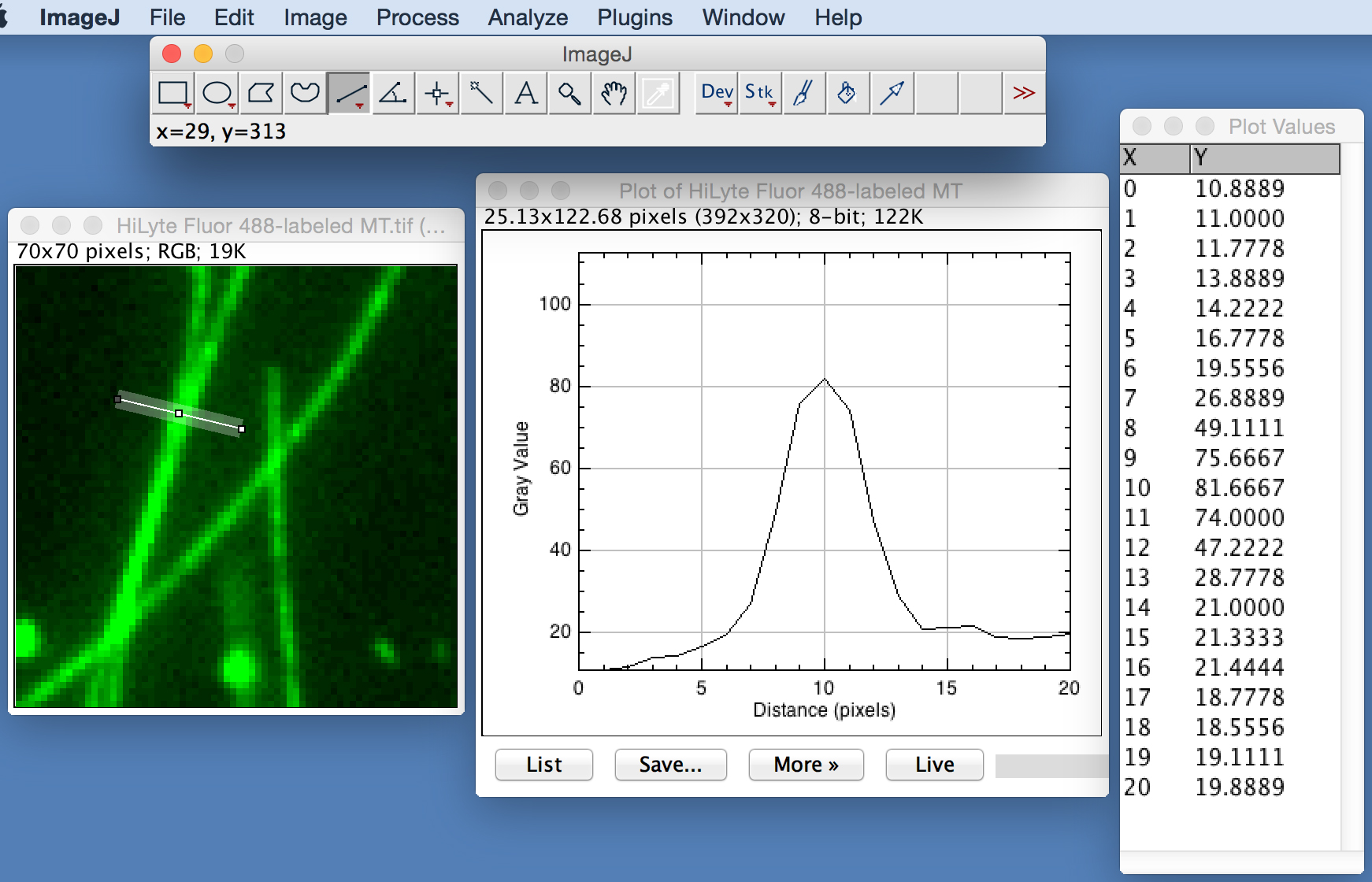
Figure 5. Example of measuring cross-sectional fluorescence intensities. Left, line across a microtubule structure. Middle, fluorescence profile along the line. Right, plot values of the profile.
Notes
- Because the imaging chamber is not sealed, the liquid in the chamber will slowly evaporate, altering sample concentration and compromising image quality. Thus, each imaging chamber should be used only for approximately 15 min of imaging at room temperature.
- Because microtubules are attached to the glass surface by anti-tubulin antibodies, capturing single focal plane images should suffice to encompass the entire thickness of the microtubule.
Recipes
- PEM Buffer (100 ml)
2.42 g of PIPES
20.33 mg of MgCl2 hexahydrate
38.04 mg of EGTA
1 ml of 100 mM GTP
Ultrapure water
Adjust the pH to 6.9 with 0.1 M NaOH - Blocking buffer (100 ml)
1 g of Pluronic F-127
100 ml of PEM buffer
Acknowledgments
This work was supported by an NIH/NIGMS grant (GM076094) to W.-L. L..
Competing interests
The authors declare no competing financial interests.
References
- Gonczy, P., Pichler, S., Kirkham, M. and Hyman, A. A. (1999). Cytoplasmic dynein is required for distinct aspects of MTOC positioning, including centrosome separation, in the one cell stage Caenorhabditis elegans embryo. J Cell Biol 147(1): 135-150.
- Goshima, G. and Scholey, J. M. (2010). Control of mitotic spindle length. Annu Rev Cell Dev Biol 26: 21-57.
- Howell, B. J., McEwen, B. F., Canman, J. C., Hoffman, D. B., Farrar, E. M., Rieder, C. L. and Salmon, E. D. (2001). Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J Cell Biol 155(7): 1159-1172.
- Khmelinskii, A., Roostalu, J., Roque, H., Antony, C. and Schiebel, E. (2009). Phosphorylation-dependent protein interactions at the spindle midzone mediate cell cycle regulation of spindle elongation. Dev Cell 17(2): 244-256.
- Markus, S. M., Kalutkiewicz, K. A. and Lee, W. L. (2012). She1-mediated inhibition of dynein motility along astral microtubules promotes polarized spindle movements. Curr Biol 22(23): 2221-2230.
- Omer, S., Greenberg, S. R. and Lee, W. L. (2018). Cortical dynein pulling mechanism is regulated by differentially targeted attachment molecule Num1. Elife 7: e36745.
- Rusan, N. M., Tulu, U. S., Fagerstrom, C. and Wadsworth, P. (2002). Reorganization of the microtubule array in prophase/prometaphase requires cytoplasmic dynein-dependent microtubule transport. J Cell Biol 158(6): 997-1003.
- Salina, D., Bodoor, K., Eckley, D. M., Schroer, T. A., Rattner, J. B. and Burke, B. (2002). Cytoplasmic dynein as a facilitator of nuclear envelope breakdown. Cell 108(1): 97-107.
- Schmidt, D. J., Rose, D. J., Saxton, W. M. and Strome, S. (2005). Functional analysis of cytoplasmic dynein heavy chain in Caenorhabditis elegans with fast-acting temperature-sensitive mutations. Mol Biol Cell 16(3): 1200-1212.
- Sharp, D. J., McDonald, K. L., Brown, H. M., Matthies, H. J., Walczak, C., Vale, R. D., Mitchison, T. J. and Scholey, J. M. (1999). The bipolar kinesin, KLP61F, cross-links microtubules within interpolar microtubule bundles of Drosophila embryonic mitotic spindles. J Cell Biol 144(1): 125-138.
- Tanenbaum, M. E., Macurek, L., Galjart, N. and Medema, R. H. (2008). Dynein, Lis1 and CLIP-170 counteract Eg5-dependent centrosome separation during bipolar spindle assembly. EMBO J 27(24): 3235-3245.
- Wadsworth, P. and Khodjakov, A. (2004). E pluribus unum: towards a universal mechanism for spindle assembly. Trends Cell Biol 14(8): 413-419.
- Wadsworth, P., Lee, W. L., Murata, T. and Baskin, T. I. (2011). Variations on theme: spindle assembly in diverse cells. Protoplasma 248(3): 439-446.
- Zhu, Y., An, X., Tomaszewski, A., Hepler, P. K. and Lee, W. L. (2017). Microtubule cross-linking activity of She1 ensures spindle stability for spindle positioning. J Cell Biol 216: 2759-2775.
- Zulkipli, I., Clark, J., Hart, M., Shrestha, R. L., Gul, P., Dang, D., Kasichiwin, T., Kujawiak, I., Sastry, N. and Draviam, V. M. (2018). Spindle rotation in human cells is reliant on a MARK2-mediated equatorial spindle-centering mechanism. J Cell Biol 217(9): 3057-3070.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Zhu, Y., Tan, W. and Lee, W. (2018). An in vitro Microscopy-based Assay for Microtubule-binding and Microtubule-crosslinking by Budding Yeast Microtubule-associated Protein. Bio-protocol 8(23): e3110. DOI: 10.21769/BioProtoc.3110.
- Zhu, Y., An, X., Tomaszewski, A., Hepler, P. K. and Lee, W. L. (2017). Microtubule cross-linking activity of She1 ensures spindle stability for spindle positioning. J Cell Biol 216: 2759-2775.
Category
Microbiology > Microbial cell biology > Cytoskeletal interaction
Biochemistry > Protein > Interaction > Protein-protein interaction
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.