- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Cluster Analysis of Endogenous HER2 and HER3 Receptors in SKBR3 Cells

Published: Vol 8, Iss 23, Dec 5, 2018 DOI: 10.21769/BioProtoc.3096 Views: 7466
Reviewed by: Xi FengTomas AparicioAkira Karasawa
Original research article
The authors used this protocol in:

May 2018
Advertisement





Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

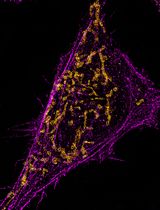
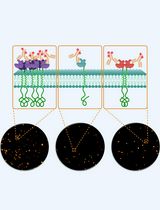
Abstract
The Human Epidermal Growth Factor Receptor (HER) family of receptor tyrosine kinases consists of four, single pass, transmembrane receptor homologs (HER1-4) that act to regulate many critical processes in normal and tumor cells. HER2 is overexpressed in many tumors, and the deregulated proliferation of cancerous cells is driven by cooperation with its preferred receptor partner, HER3. The assessment of the in-situ organization of tagged HER2 and HER3 using super-resolution microscopy reveals quantitative Single Molecule Localization Microscopy (SMLM) as an ideal bioanalytical tool to characterize receptor clusters. Clustering of receptors is an important regulatory mechanism to prime cells to respond to stimuli so, to understand these processes, it is necessary to measure parameters such as numbers of clusters, cluster radii and the number of localizations per cluster for different perturbations. Previously, Fluorescence Localization Imaging with Photobleaching (FLImP), another nanoscale, single-molecule technique, characterized the oligomerization state of HER1 [or Epidermal Growth Factor Receptors (EGFR)] in cell membranes. To achieve an unprecedented resolution (< 5 nm) for inter-molecular separations in EGFR oligomers using FLImP, very few receptors are tagged, and so this method is unsuitable for measurements of whole receptor populations in cancer cells where receptors are frequently upregulated. Here, in order to detect all receptors involved in cluster formation, we saturate endogenous HER2 and HER3 membrane receptors with ligands at a 1:1 dye to protein ratio, in the presence or absence of therapeutic drugs (lapatinib or bosutinib). This is performed in the commonly used breast cancer cell line model SKBR3 cells, where there are ~1.6 million HER2 receptors/cell and 10,000-40,000 HER3 receptors/cell. The basal state of these receptors is studied using HER2- or HER3-specific Affibodies, and likewise, the active state is probed using the natural HER3 ligand, Neuregulin-beta1 (NRGβ1). Stochastic Optical Reconstruction Microscopy (STORM), one form of SMLM, was used here to image cells, which were chemically fixed to minimize image blurring and provide data (x and y coordinates and standard deviation of the measured localizations) for cluster analysis. Further analysis can also determine proportions of receptor colocalizations. Our findings show that lapatinib-bound HER2, complexed with HER3 via a non-canonical kinase dimer structure, induces higher order oligomers. We hypothesized that nucleation of receptors creates signaling platforms that explain the counterintuitive, increase in cell proliferation upon ligand binding, in the presence of the HER2-inhibitor lapatinib.
Keywords: Epidermal growth factor receptor (EGFR)Background
Homo- and hetero-protein clustering facilitates the efficient regulation of signaling processes in cells (Sourjik and Armitage, 2010; Nussinov, 2013; Truong-Quang and Lenne, 2014; Recouvreux and Lenne, 2016). Mechanisms include, but are not limited to, the sequestering of proteins (e.g., GPI-anchored folate receptors [Mayor et al., 1994]), creation of docking sites (e.g., Syntaxin/SNARE family proteins [Sieber et al., 2007]), steric hindrance of interactions (e.g., Rictor and mSin1 in mTOR complex-2 [Chen et al., 2018]), aggregation of misfolded proteins (e.g., amyloid fibrils [Adamcik et al., 2010]), an accelerated response to pre-equilibrium signals during chemotaxis (Ventura et al., 2014), regulation of integrins at focal adhesions (Spiess et al., 2018) and formation of signaling cascades (e.g., GPCR signaling [Maurel et al., 2008; Jonas et al., 2015]). Specifically, we focus on the analysis of membrane receptor clustering/oligomerization to investigate molecular rearrangements due to drug binding. We highlight the importance of mutual cooperation resulting in an increased signaling output (Claus et al., 2018), a theme likely to be common in signaling cascades. In addition, we suggest that this protocol is generally applicable to many biological questions relating to protein clusters. Minor modifications to the sample preparation should allow localization measurements of any protein(s), either immobilized on glass or expressed in bacteria or other (e.g., mammalian) cells. Models of protein interactions and the consideration of differences between wild type proteins and equivalents comprising pertinent mutations can often lead to a more comprehensive molecular mechanism. We believe, therefore that the quantitative clustering and colocalization data delivered by this protocol advances our understanding of native and disease states even further.
The proteins of interest in this study, HER2, and HER3, are two members of the HER family of receptor tyrosine kinases (Normanno et al., 2006; Eccles, 2011). HER2 is overexpressed in many tumors, and the deregulated proliferation of cancerous cells is driven by cooperation with its preferred receptor partner, HER3 (Holbro et al., 2003). SMLM techniques (e.g., STORM for photoactivatable fluorophores or PALM for genetically-expressed tagged proteins) can image single fluorescently-tagged protein(s), like receptors, at the plasma membrane or inside the cell (Nicovich et al., 2017). STORM is a popular imaging modality thanks to a relatively simple experimental setup and procedure (Rust et al., 2006) and the availability of free, open-source software for processing, analysis and visualization of the data (Ovesný et al., 2014). We chose STORM for our application because fluorophore blinking is induced stochastically, and localization achieved one group of fluorophores at a time, the process being continued and signals accumulated until a detailed image is built. Crucially, the STORM process makes the technique compatible with saturated labeling of all endogenous HER2 protein using an Affibody ligand that is small, specific and uniformly conjugated to Alexa 488 at a ratio of 1:1 Affibody:dye (Wikman et al., 2004; Eigenbrot et al., 2010). Other HER protein affibodies are available (Friedman et al., 2007; Nordberg et al., 2007; Kronqvist et al., 2010; Gostring et al., 2012;). Cluster analysis of SMLM data can, in turn, reveal the number of molecules per cluster and the cluster size. The post analysis software we used is readily available from the research teams led by Dylan Owen (Griffié et al., 2016) and Katharina Gaus (Pageon et al., 2016) in combination with the custom written tools provided here. If required, it may be possible to use SMLM and cluster analysis to study these receptors in live cells (an extension to the analysis method is given in Griffié et al., 2018) however this has not been implemented as yet.
We also wanted to characterize protein-protein interactions within clusters, information that cannot be obtained via cluster analysis alone. Fluorescence Resonance Energy Transfer measurements using Fluorescence Lifetime Imaging Microscopy (FRET-FLIM) was also used extensively in Claus et al. to measure inhibitor-induced heterotypic interactions between HER2 and HER3 and their respective mutants (Claus et al., 2018). FRET-FLIM data complement the analysis of clusters by reporting the stability of heterodimeric complexes. However, since the length scale of FRET is 2-8 nm, the method cannot be used to report on the typically larger cluster sizes, the combination is important in providing a deeper organizational view of these receptors.
Other imaging techniques for measuring protein-protein interactions and clustering can be used in combination with cluster analysis to provide complementary information and provide a more complete picture of the underlying processes from the dynamic and structural point of view. These include two-color Single Particle Tracking (SPT), which reports the number of pairwise receptor particle interactions and the duration of these interactions (Low-Nam et al., 2011; Needham et al., 2013) allowing the distinction between multimers and dimers. However, SPT images single molecules or particles as they move on live cells and the associated blurring means that SPT often does not have enough resolution to determine the number of receptors in the “particles” imaged as they move. FLImP is an ideal method to measure precise receptor separations on static particles (Needham et al., 2013). In model cell systems the expression level of receptors can be optimized. However HER2 is highly overexpressed in the SKBR3 cell line (Eccles, 2011; Shankaran et al., 2013), and labeling would need to be kept at low levels. Nevertheless, FLImP should be able to provide information complementary to FRET on receptor-receptor interactions and the geometry of the complexes. Stimulated Emission Depletion (STED) microscopy, a distinct but complementary super-resolution imaging technique to STORM, can measure protein clusters ~50 nm in diameter. Utilizing this resolution for fixed, immuno-labeled, cell samples, Kellner et al. measured for the first time changes in the size distribution of nicotinic acetylcholine receptor (AChR) nanoclusters (Kellner et al., 2007) and Dzyubenko more recently employed this method to reveal the density of marker proteins in postsynaptic neurons (Dzyubenko et al., 2016). The direct comparison of nearest neighbor distance for active integrins derived from both techniques illustrates that STORM benefits from higher resolution than STED (Spiess et al., 2018). The latter technique, however, could extend cluster analysis to live cells and achieve higher temporal resolution. If a robust fluorophore calibration could be made, number and brightness analysis would provide stoichiometric and dynamic measurements from STED data of live cells.
Materials and Reagents
- 75 cm2 filter cap cell culture flasks (Thermo Fisher Scientific, NuncTM EasYFlaskTM, catalog number: 156499)
- 35 mm Glass Bottom Dishes, High Tolerance 1.5 Coverslip, 14 mm Glass Diameter (MatTek Corporation, catalog number: P35G-0.170-14-C)
- 5 ml individually packed sterile Serological pipettes (Corning incorporated, Costar StripetteTM, catalog number: 4487)
- 10 ml individually packed sterile Serological pipettes (Corning incorporated, Costar StripetteTM, catalog number: 4488)
- 25 ml individually packed sterile Serological pipettes (Corning incorporated, Costar StripetteTM, catalog number: 4489)
- PD10 purification columns (GE Healthcare, catalog number 17085101)
- 24-well plates (NunclonTM, catalog number: 143982)
- Aluminum foil (Terinex, catalog number: 11330)
- SK-BR-3 [SKBR3] (ATCC® HTB-30TM) cells (LGC standards) (ATCC, catalog number: HTB-30)
- Bovine Serum Albumin (BSA) (Sigma-Aldrich, catalog number: A1470) (Store at 4 °C)
- RPMI 1640 media without phenol red (Thermo Fisher Scientific, GibcoTM, catalog number: 11835063) (Store at 4 °C)
- Fetal Bovine Serum (FBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10270) (Aliquot and store at -20 °C)
- L-Glutamine (Thermo Fisher Scientific, GibcoTM, catalog number: 25030024) (Aliquot and store at -20 °C)
- Lapatinib (BioVision, catalog number: 1624) (Aliquot and store at -20 °C)
- Bosutinib (Sigma-Aldrich, catalog number: PZ0192) (Aliquot and store at -20 °C)
- Neuregulin beta-1 (NRGβ1) (Peprotech, catalog number: 100-03) (Aliquot and store at -20 °C)
- Anti-HER2, imaging agent (Affibody Incorporated, catalog number: 10.1861.01.0005) (Store powder at 4 °C, once reconstituted, aliquot and store at -20 °C)
- HER3 Affibody ligand (Plasmid was a gift from John Löfblom [Kronqvist et al., 2011], a protein expressed and purified in-house and shown to bind specifically to HER3 receptors) (Aliquot and store at -20 °C)
- Alexa FluorTM 488 NHS Ester (Thermo Fisher Scientific, InvitrogenTM, catalog number: A20100) and Maleimide (Thermo Fisher Scientific, InvitrogenTM, catalog number: A10254) (Store at -20 °C)
- Alexa FluorTM 647 NHS Ester (Thermo Fisher Scientific, InvitrogenTM, catalog number: A20006) and Maleimide (Thermo Fisher Scientific, InvitrogenTM, catalog number: A20347) (Store at -20 °C)
- Dulbecco's Phosphate-Buffered Saline (DPBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 14040091)
- Paraformaldehyde (PFA) (Electron Microscopy Sciences, catalog number: 157-4-100)
- Glutaraldehyde (Sigma-Aldrich, catalog number: G5882)
- Cysteamine hydrochloride (Sigma-Aldrich, catalog number: M6500)
- Zeiss ImmersolTM 518F Imaging Oil 23 °C (Thermo Fisher Scientific, CarlZeissTM ImmersolTM)
- Standard tool for calibration MultiSpecTM bead sample (ZEISS, catalog number: 2076-515)
- Sodium chloride (Sigma-Aldrich, catalog number: S7653)
- Potassium Chloride (Fluka, catalog number: 60128)
- Sodium phosphate dibasic (Na2HPO4, Sigma-Aldrich, catalog number: S9763)
- Potassium phosphate monobasic (KH2PO4, Sigma, catalog number: P5655)
- Tris Base (Sigma, BioUltra, catalog number: 93286)
- HEPES solution (Sigma, catalog number: H3537)
- DMSO (Invitrogen, catalog number: D12345)
- DMEM (Gibco, catalog number: 11880-028)
- DTT (Fisher, catalog number: D/P 351/43)
- Protease inhibitor cocktail (Cell Signaling Technologies, catalog number: 5871S)
- Phosphatase inhibitors (Sodium Fluoride, Sigma, catalog number: S7920 and Sodium Orthovanadate, Sigma, catalog number: S6508)
- Phosphate buffered saline (PBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 14040091 or see Recipes)
- MEA buffer (see Recipes)
Equipment
- Electronic pipette filler/dispenser (Hirschmann®, Pipetus®, model: 9907200)
- Nanodrop Spectrophotometer (Thermo Fisher Scientific, model: ND-2000C)
- Classic prestige medical Autoclave (Wolf labs, model: 210048)
- ZEISS Elyra PS.1 super-resolution microscope (ZEISS, model: ELYRA PS.1) with 1.46 NA 100x oil immersion objective (ZEISS Alpha Plan-Apochromat 100x/1.46 Oil DIC, catalog number: 420792-9800-000)
- Fume Hood
- 37 °C, 5% CO2 New BrunswickTM Incubator (Eppendorf, model: Galaxy® 170R, catalog number: CO17301002)
- SCANLAF Microbiological Safety Cabinet (Labogene, Mars Safety Class 2)
- -4 °C Fridge (LEC medical, model: LR307C, SKU number: UK444441800)
- -20 °C Freezer (LEC medical, model: ISU37C, SKU number: UK444441803)
- -80 °C Freezer (RS Biotech, model: Eclipse 100v, catalog number: 05100v-230)
- Centrifuge (Eppendorf, model: 5702)
Software
- ZEN Black 2012 SP5 FP1 (64 bit) software, version 14.0.12.201 (Zeiss)
Minimum System Requirements: Intel® Xeon X5650 6-Core 2.66 GHz, Intel® 5520 Dual chipset, 6 GB DDR3-RAM, Graphics interface PCIe x16, Graphics adapter ATI FirePro 2560 x 1600 resolution, 32-bit true color, 512 MB RAM, DirectX 8.0 or higher, Monitor 20" TFT 1600 x 1200, Hard disk 1 x 250 GB SATA2 (configured as 250 GB hard drive) and 4 x 1 TB SATA2 (configured as 2 TB RAID 10 hard drive), DVD-ROM drive, 1x free PCI Express Generation 2.0 x16 (mechanical x16, electrical x16) full height slot, Trigger-Board and Signal Distribution Box, 2 x FireWire IEEE 1394a interface, 4 x serial interface (COM1-COM4), 4 x USB interfaces, Microsoft® Windows® 7 Ultimate SP1 x64 (Multilanguage) (no special customer adapted versions of the latter 3 items), The user must be logged on as a member of the local "User" group, For the software installation procedure local administrator rights are required, The hardware requirements of modules can be higher. (taken from https://www.zeiss.com/content/dam/Microscopy/Downloads/Pdf/FAQs/zen2012-sp2_installation_guide.pdf) - MATLAB and ClusDoC package (https://github.com/PRNicovich/ClusDoC)
- Custom software (https://github.com/fbi-octopus/cluster_analysis)
The repository contains:- The ROI selection tool (in python)
- The modified cluster analysis software (in R)
- A template of LSF script for the use of computing clusters
- An example data set (input for the ROI selection tool)
- Example data from ROI ready to be clustered
- R (R Foundation for Statistical Computing, version 3.2.3 or higher)
R libraries 'splancs' and 'igraph' - Python 2.7
Modules 'shapely', 'PyQt4', 'matplotlib', 'PIL' or 'pillow' - ImageJ (https://imagej.net/Downloads)
Procedure
- Growing cells
- Seed 2.0 x 105 SKBR3 cells onto sterile BSA-coated glass-bottomed dishes and cover with 2 ml of RPMI 1640 media supplemented with 10% FBS and 2 nM L-Glutamine. BSA-coating minimizes non-specific binding of the fluorescent ligands (Zanetti-Domingues et al., 2012).
- Incubate cells with 5% CO2 in air at 37 °C for two days or until they have reached ~70% confluency.
- Drug treatment and labeling
- Conjugate HER2 and HER3 Affibody ligands to the appropriate Alexa Fluor maleimide dyes according to manufacturer’s instructions. Thanks to the unique cysteine site at the C-terminus of Affibody molecules, the dye molecule:ligand molecule can be at most 1:1, but check for incomplete labeling in a spectrophotometer. Briefly:
- Dissolve the protein at 50-100 µM in a suitable buffer at pH 7.0-7.5 (10-100 mM phosphate, Tris, HEPES) at room temperature.
- Prepare a 1-10 mM stock solution of the reactive dye in DMSO immediately before use. Protect all stock solutions from light as much as possible by wrapping containers in aluminum foil.
- Add sufficient protein-modification reagent from a stock solution to give approximately 10-20 moles of reagent for each mole of protein. Add the reagent dropwise to the protein solution as it is stirring.
- Allow the reaction to proceed for 2 h at room temperature or overnight at 4 °C.
- Separate the conjugate on a gel filtration column, such as a PD-10 purification column. Also see Yan, 2011.
The degree of labeling can be calculated using the following formula: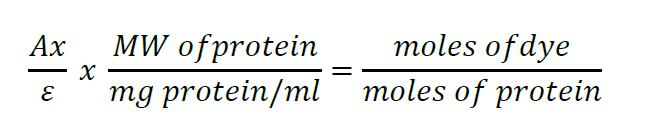
where Ax = the absorbance value of the dye at the maximum absorption wavelength. ε = molar extinction coefficient of the dye or reagent at the maximum absorption wavelength. For Alexa 488 this is 73,000 cm-1 M-1. The MW (molecular weight) of affibodies is 6,000 Da. Therefore, assuming a 1 mg/ml protein solution has A280 = 1, their molar extinction coefficients are 6,000 cm-1 M-1. - Conjugate NRGβ1 to the appropriate NHS ester Alexa Fluor dye according to manufacturer’s instructions and see above. Check dye molecule:ligand molecule ratio in a spectrophotometer and ensure that it is as close to 1:1 as possible (Protein concentration measured at 280 nm and Alexa 647 concentration measured at 647 nm. The extinction coefficient of Alexa 647, at the wavelength of maximum emission (647 nm), is 270,000 cm-1 M-1).
- Ensure that the NRG-dye conjugate is as active as the unlabeled protein by performing control Western Blot or ELISA assays for HER3 and HER4 phosphorylation. Briefly for cell culture and Western Blotting:
- Seed MCF7 cells at 0.5 x 105 cells/well in 24-well plates using DMEM supplemented with 10% FBS as growth media.
- Using FuGENE HD, transfect cells with HER3 or HER4 plasmid DNA (follow the manufacturer’s instructions).
- Twenty-four hours post-transfection, treat pre-chilled cells with ice-cold 10 nM NRG-Alexa 647 in PBS and incubate for 1 h at 4 °C.
- Lyse cells in 1x sample buffer containing 1 mM DTT, protease and phosphatase inhibitors. Sonicate and centrifuge before running on SDS-PAGE gel (using XCell apparatus from Invitrogen) alongside HiMark Prestained HMW and Novex Sharp Prestained protein standards (Invitrogen).
- Transfer to a membrane and analyze by Western blotting. Briefly, first bind anti-HER2 pY877 or anti-HER3 pY1289 from Cell Signaling Technology (follow the manufacturer’s instructions) and then the species-specific secondary antibody conjugated to HRP (Horse-radish peroxidase) from Jackson ImmunoResearch.
- Develop the blot using the Supersignal West Pico Chemiluminescent Substrate solution from Pierce. Image with a BioRad ChemiDoc MP system Imager and analyze using densitometry in ImageJ.
- After stripping the blot of antibodies, reprobe with anti-Total HER2 or anti-Total HER3, antibodies and then the species-specific secondary antibody conjugated to HRP. Develop and analyze as above.
- Change the media on the cells to serum-free RPMI 1640 media (SFM) including, if required, 14 nM Lapatinib or 41 nM Bosutinib.
- Incubate all dishes at 37 °C/5% CO2 for two hours.
- Using a serological pipette and electronic pipette filler/dispenser, remove the cell media and replace with 1 ml PBS to wash. Remove the PBS to add 1 ml of fresh PBS including the appropriate drug treatment if required.
- Chill all samples on ice at 4 °C for 10 min (to minimize receptor internalization).
- Incubate half of the samples in 1 ml of 100 nM HER2Affibody-Alexa488 and 50 nM HER3Affibody-Alexa647 diluted in ice-cold PBS and, if required, the appropriate drug (either 14 nM Lapatinib or 41 nM Bosutinib).
- Incubate the remaining dishes in 1 ml of 100 nM HER2Affibody-Alexa488 and 10 nM NRG-Alexa647 diluted in ice-cold PBS and, if required, the appropriate drug.
- Incubate all the dishes on ice for at least 1 h.
- Wash three times each with 1 ml of ice-cold PBS.
- Fix cells with 1 ml of 4% paraformaldehyde and 0.5% glutaraldehyde diluted into ice-cold PBS.
- To ensure complete fixation, incubate cells on ice at 4 °C for 30 min-1 h, then wash three times using 1 ml of PBS per wash.
- Store samples at 4 °C in PBS and prior to imaging supplement fresh PBS with 50 mM cysteamine HCl (total volume 1 ml).
- Conjugate HER2 and HER3 Affibody ligands to the appropriate Alexa Fluor maleimide dyes according to manufacturer’s instructions. Thanks to the unique cysteine site at the C-terminus of Affibody molecules, the dye molecule:ligand molecule can be at most 1:1, but check for incomplete labeling in a spectrophotometer. Briefly:
- Imaging
- Supplement objective lens (1.46 NA 100x oil immersion objective) with immersion oil and mount sample on microscope stage (Figure 1 and see
https://applications.zeiss.com/C125792900358A3F/0/8DF1FE51A0C52599C1257C1D0073F96D/$FILE/EN_41_011_061_ELYRA.pdf for microscope setup). - Locate sample using eyepiece and set up a 2 dimensional (2D), 2 color dSTORM (Endesfelder and Heilemann, 2015) experiment using the 488 nm and 641 nm excitation laser lines in separate tracks with the appropriate filters (a possible setup for this pair of dyes would be BP420-480/BP495-560/LP650). See Figure 2 (left) for parameter settings in Zen software.
- Locate a cell and position it in the center of the field of view (FOV) then refocus, ensuring that the entire cell membrane is clearly in focus (Figure 2 [right] for example image).
- Using TIRF illumination, gradually increase laser powers separately for each channel to excite the Alexa 488 and Alexa 647 fluorophores and push them towards quasi-equilibrium (Dempsey et al., 2011).
- Acquire dSTORM raw data over 10,000 frames with exposure time 20 ms, EM gain 250, alternating the lasers to image the two receptors sequentially every 300 frames.
- Use the 405 nm laser line, if necessary, at low power (< 1%) to aid fluorophore blinking at any time.
- During acquisition use excitation laser powers of ~9-10 kW/cm2 at the sample (we used 7.6-11.7 kW/cm2, average 8.9 kW/cm2 for 488 nm laser and 7.6 -13.8 kW/cm2, average 10.4 kW/cm2 for 640 nm laser). Low levels of 405 nm laser can also be used (0.001-0.005 kW/cm2).
- Repeat imaging procedure to obtain dSTORM data for at least 12 regions (25.6 µm x 25.6 µm) per treatment group with each region including at least one cell.
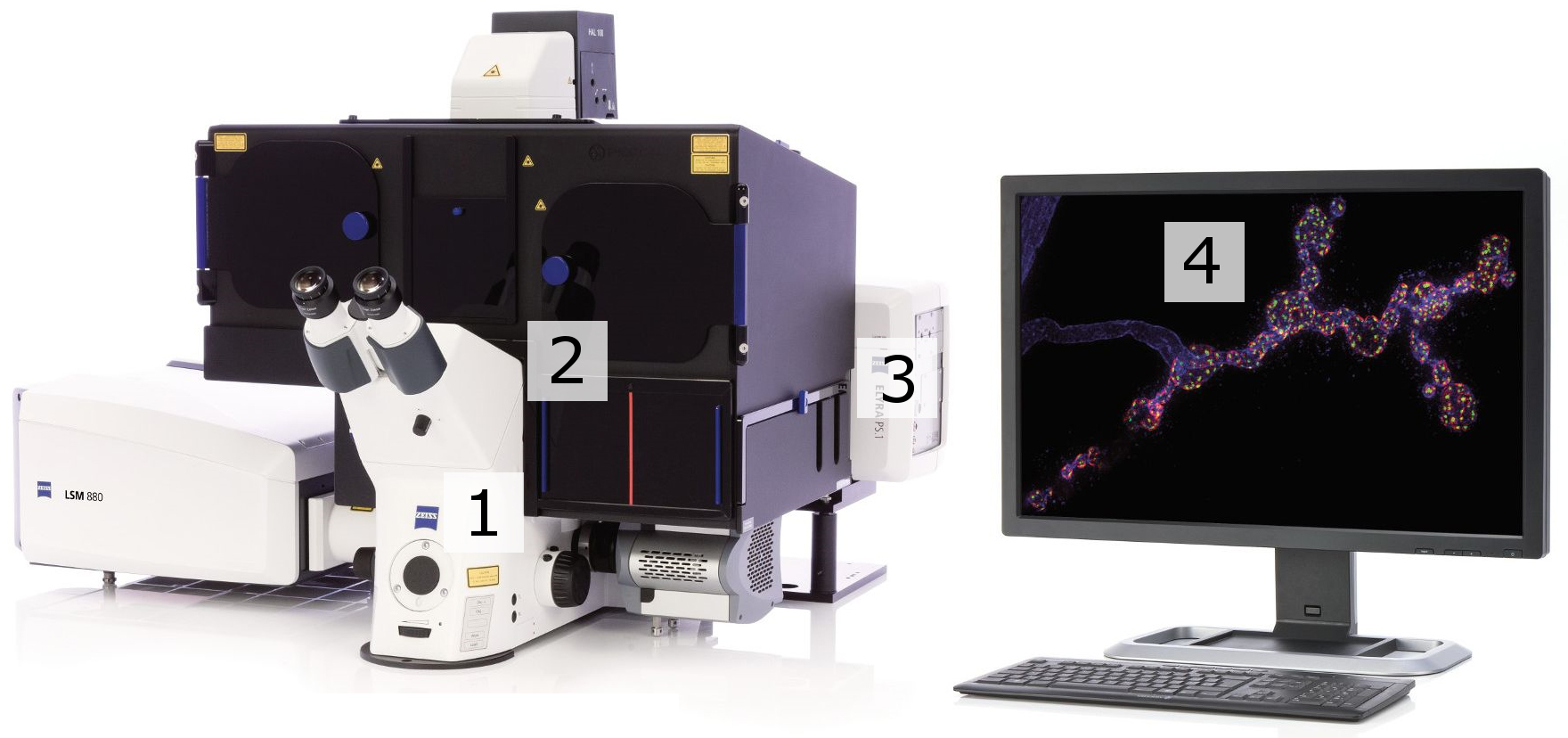
Figure 1. STORM imaging set-up. 1. Microscope: Axio Observer, Z1 (inverse stand); Incubator XL dark; Motorized Piezo XY scanning stage; Z-Piezo stage insert; Port for LSM attachment; two camera ports. 2. Objectives: Plan-APOCHROMAT 100x/1.46 Oil (DIC). 3. ELYRA Illumination and Detection: Fiber coupled solid state and diode pumped solid state lasers; 405 nm diode (50 mW); 488 nm OPSL (100 mW); 561 nm OPSL (100 mW); 642 nm diode (150 mW); Andor iXon 897 EM-CCD camera. 4. Software: ZEN (black edition), PALM module.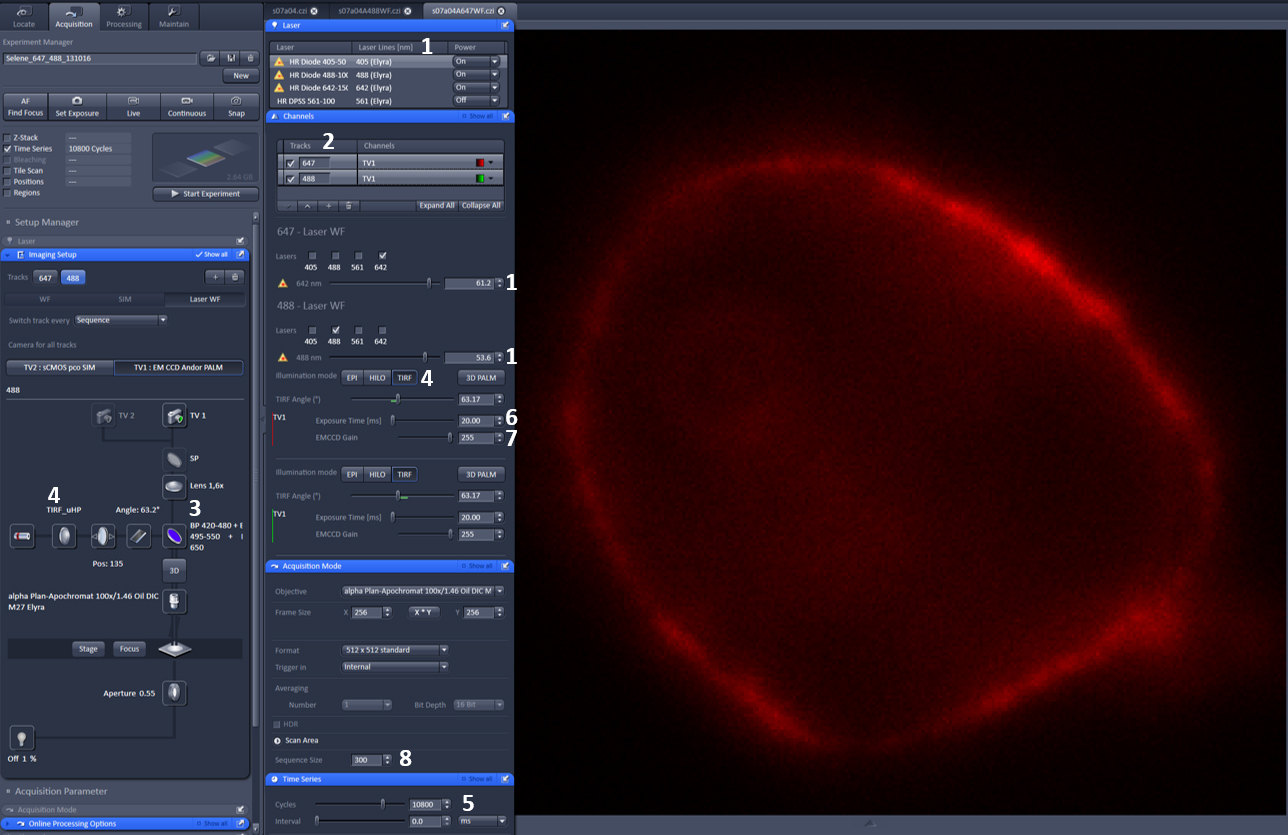
Figure 2. Zen settings for acquisition parameters (left) and Example image (right). Left: 1. Excitation lasers and power; 2. Tracks; 3. Emission filter; 4. TIRF setting and angle; 5. Number of frames; 6. Exposure time; 7. EM gain. 8. Frames per cycle. - Use the PALM function in ZEN software to process and render the images (Figure 3). Use ZEN to first define the point spread function (PSF) mask size (typically 9 pixels, max 10 pixels) and intensity to noise ratio (typically 6) then apply to localize each blinking event using a 2D Gaussian fit model.
- Take overlapping fluorophores into account using the Account for overlap setting.
- Correct for displacements of molecules in the lateral plane from drifts using feature detection and cross-correlation methods in the PAL-Drift tab.
- Implement channel alignment (Figure 4) of the reconstructed images using a standard MultiSpec bead sample.
- Co-ordinates of the localizations in the final reconstructed images (typically 30,000+ for HER2 and 5,000+ for HER3 per region, Figure 3) are passed into the Bayesian cluster identification algorithm designed by Dylan Owen et al. (Griffié et al., 2016).
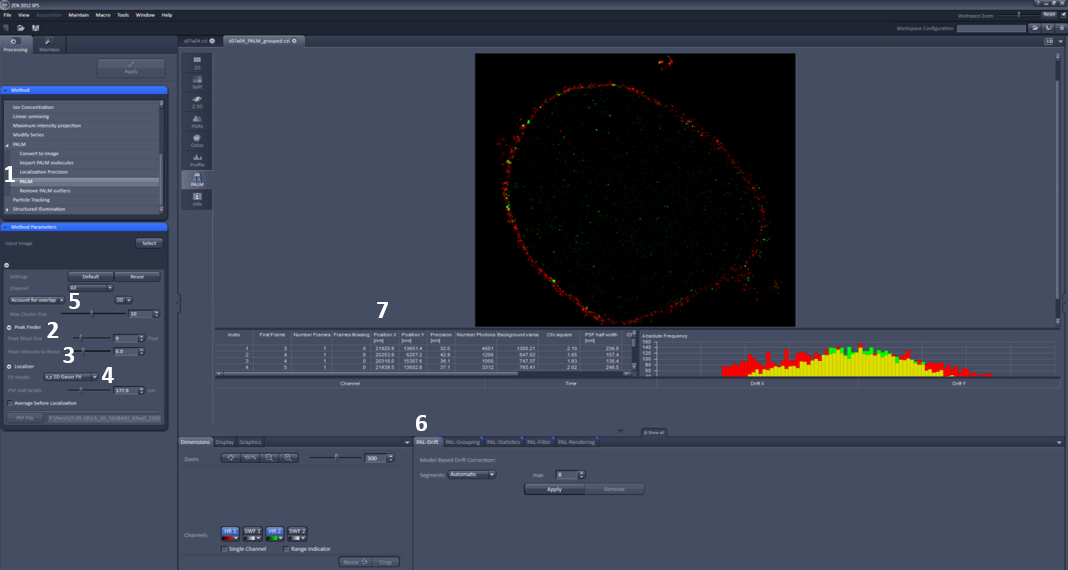
Figure 3. Zen settings for data analysis parameters. 1. PALM function. 2. PSF mask size. 3. Intensity to noise ratio. 4. Fit model. 5. Account for overlap. 6. PAL-drift tab. 7. Co-ordinates of localizations and precision.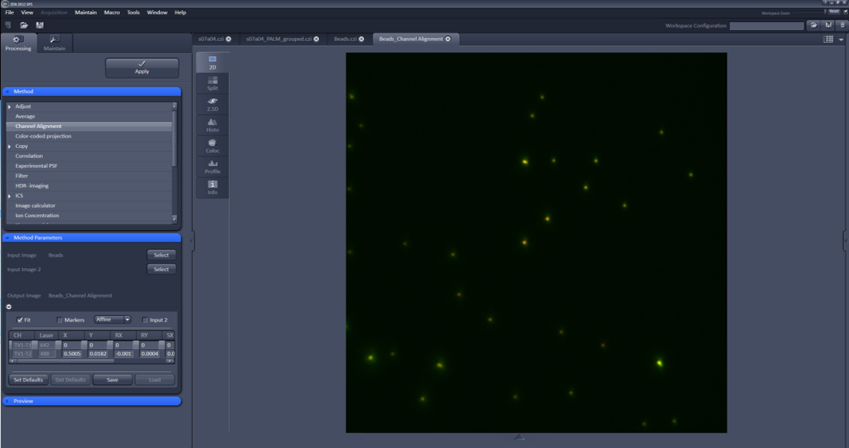
Figure 4. Channel alignment settings
- Supplement objective lens (1.46 NA 100x oil immersion objective) with immersion oil and mount sample on microscope stage (Figure 1 and see
Data analysis
- Analysis using Bayesian Cluster Algorithm
A Bayesian cluster algorithm is applied to determine the cluster radii and number of molecules in the cluster (Griffié et al., 2016). The analysis can be done under Linux or Windows OS, although the region-of-interest (ROI) selection tool has not been tested for Windows. The software requires python 2.7 and R. Both packages, including all required libraries and modules, are free and open source and available for both Linux and Windows OS. The installation depends on your specific OS. On Ubuntu 16.04, all required software can be installed using the “apt install” command, except for the R library ‘splancs’, which can be installed by running R and typing ‘install.packages(“splancs”)’, The ‘igraph’ library could be installed in this manner as well. All required python modules can alternatively be installed using python’s ‘pip’ tool. Run ‘pyrcc4 resources.qrc –o qrc_resources.py’ in the roi_selector folder before you run ‘roi_select.py’ the first time. Otherwise, the tool buttons will have text labels instead of icons (Video 1).
Once the required software is set up, the analysis consists of the following principal steps.
Table 1. Schematic of the data analysis using the Bayesian Cluster Algorithm
The data from different channels of an acquisition is stored in folders that have the same base name plus an extension “green” or “red” for the channel (such as “s07a06green” and “s07a06red” for a data set “s07a06”, see also Video 1), folder names need to end in either “green” or “red” (note this is a critical step) This is done to aid the ROI selection (note, the tool works for 1 or 2 channels). Otherwise, the conventions of the protocol (Griffié et al., 2016) are followed. The clustering algorithm expects the background and clusters to be uniformly distributed over a rectangular ROI. The analyzed images mainly show single cells. Labeled molecules of interest are in the cell membrane. The cell membrane is visible as a roughly circular shape. In order to conform to the prerequisites of the clustering algorithms, rectangular regions need to be manually or semi-automatically selected. Video 1. Preparation. What the input data looks like, what the ROI selector looks like without icons, how to compile the python resources for the ROI selector, and what the ROI selector looks like with icons.
Video 1. Preparation. What the input data looks like, what the ROI selector looks like without icons, how to compile the python resources for the ROI selector, and what the ROI selector looks like with icons.
This is accomplished with a graphical tool written in Python (provided). The tool displays the data of all channels of an acquisition, each channel in a different color. Ensure that the rectangles tightly cover the cell membrane using the most suitable angles. The semi-automatic procedure can be used to optimize a manually selected ROI (which does not need to be rectangular or of tight fit around the membrane) with a single button, -click. The whole cell membrane is covered in this way by ROIs. The data selected by these regions is rotated automatically so that the sides of the rectangles becomes parallel to the coordinate axis and the selected data is written to files, one folder for each ROI. The following workflow is used (Figure 5, Video 2):- Open the data file, key ‘ctrl-L’ or button (1).
- Select a drawing mode, button (4), if desired.
- Start drawing, key ‘d’ or button (3).
- Draw the rectangle or polygon by clicking on the image
- The rectangle will be complete after 3 points have been selected (two points for the base line and the third to determine the height), the polygon will be complete after clicking button (3) or pressing ‘d’. The first and the last point will be connected to close the polygon.
- Optimize the ROI, button (5) or key ‘s’, this will turn the highlighted ROI into a rectangle that contains the most dense regions of the highlighted ROI.
- Repeat 3-6.
- Save the rectangular ROI (it is critical to note that the polygons will not be saved), button (2) or key ‘Ctrl + S’. The first time you do this in each session, you will be asked to select the output folder. The data will be stored in sub-folders of the selected folder. The output folder also can be set from the menu: “File” → “Set base output directory”.
- Go to 1.
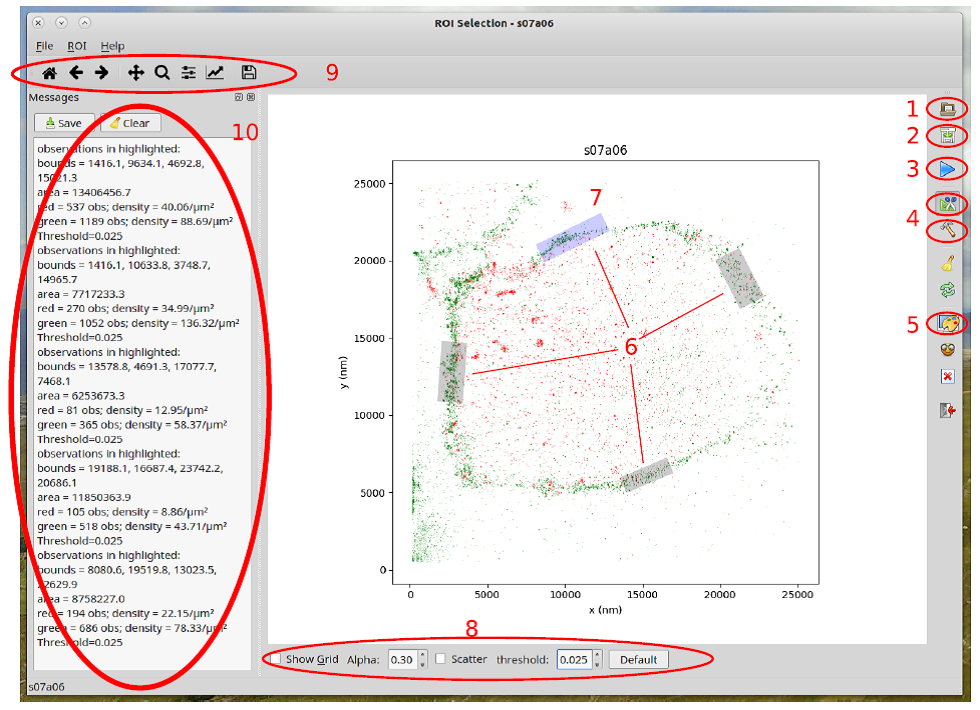
Figure 5. Features of the region of interest selection tool. Open data set (1), Save rectangular ROI (2), Start ROI drawing (3), Select ROI type: polygon or rectangle (4), automatically optimize selected ROI (5), ROI’s (6), selected ROI (7), options toolbar (8), Python’s matplotlib toolbar (9), editable message window (10). Video 2. Usage of the ROI selection tool. How to open a data set, how to activate the scatter plot display mode, how to zoom and pan, introduction to the main toolbar elements, how to use the polygon selection tool, how to use the optimize-ROI tool, how to save the selected data and the contents of the resulting output folders.
The drawing mode can be changed any time, ROI can be removed and information about the ROI (number of observations, density) can be displayed. The tool will write the required configuration-file too. The results are a series of folders such as (for Linux OS):
1) <base-output-directory>/s07a06r23415-14057_1466x3564_25red
2) <base-output-directory>/s07a06r23415-14057_1466x3564_25green
3) <base-output-directory>/s07a06r18906-21057_1101x2745_76red
4) <base-output-directory>/s07a06r18906-21057_1101x2745_76green
5) …
where each folder contains two files:
1) config.txt
2) 1/data.txt
Note: A channel may not contain any data in a particular ROI. In such a case no output is written for the channel. Note, the channels are processed independently from each other.
The files are used as input for the clustering algorithm and the algorithm was applied as described by the protocol (Griffié et al., 2016) (Video 3). The original source files (supplementary information in Griffié et al., 2016) have been modified (provided) to promote batch analysis. The analysis is executed by running:
R -f run.R --args --target=<data-folder>--config==<path-config-file>
From the folder that contains the ‘run.R’ script. Here, <data-folder>is the folder that contains data from the ROI (such as ‘<base-output-directory> /s07a06r18906-21057_1101x2745_76red’) and <path-to-config-file> is the path to the ‘config.txt’ belonging to the data (here ‘<base-output-directory> /s07a06r18906-21057_1101x2745_76red/config.txt’). Since the number of folders can be large, computing clusters may be used. We provide an example of how to run the scripts on clusters with the LSF job scheduler.
After the analysis with ‘run.R’ has completed, <data-folder>contains:
1) config.txt
2) 1/data.txt
3) 1/all_labels.txt.gz
4) 1/r_vs_thresh.txt
The files are processed with the ‘postprocessing.R’ script (Video 3):
R --slave -f postprocessing.R --args –folders=<data-folder>
where again <data-folder>is the folder that contains data from the ROI. This script adds three files to <data-folder> :
1) 1/summary.txt
2) radii.txt
3) nmols.txt
where ‘radii.txt’ contains the list of cluster sizes and ‘nmols.txt’ contains the list of molecules per cluster, which are the desired results. Video 3. Running the clustering algorithm on the command line. How to use the information from the README file, how to edit the command line, how to run the primary clustering algorithm, the result of the algorithm, how to run the post processing, result of the post processing. - Analysis using Clus DoC
- For further image analysis to compare the degree of colocalization between HER2 and HER3, with the different treatments, dSTORM images were analyzed using Clus-DoC (Cluster detection with Degree of Colocalization) program run in the MATLAB environment (Pageon et al., 2016). A schematic of this data analysis process can be found in Figure 3 of Pageon et al. (2016).
- The graphical user interface (GUI) was opened in MATLAB using the command line ‘ClusDoC’ (Figure 6), and the previously saved table of localizations from ZEISS ZEN software (in text file format) for each image was loaded as the ‘Input File’ (A).
- The destination for the data to be produced was then selected using the ‘Set Output Path’ button (B).
- Membranes of entire cells were selected by drawing around the membrane using the ‘Add ROI’ tool (C) and then double-clicking to activate.
- The ROI was saved by clicking the button ‘Export ROIs’ (D).
- A Ripley K test (E) was performed first to obtain a maximum L(r)-r value for each cell (shown on the ‘RipleyK Plots’ produced). This value was input as the L(r)-r parameter for subsequent DBSCAN and Clus-DoC tests, to filter out noise points.
- Multiple DBSCAN tests (F) were then run on each cell to determine the parameters required to obtain an average cluster area that agreed with that predicted by the Bayesian cluster identification algorithm.
- The final parameters were recorded then input into the Clus-DoC test parameters, and a Clus-DoC test (G) was run on each cell to quantify the degree of colocalization between HER2 and HER3.
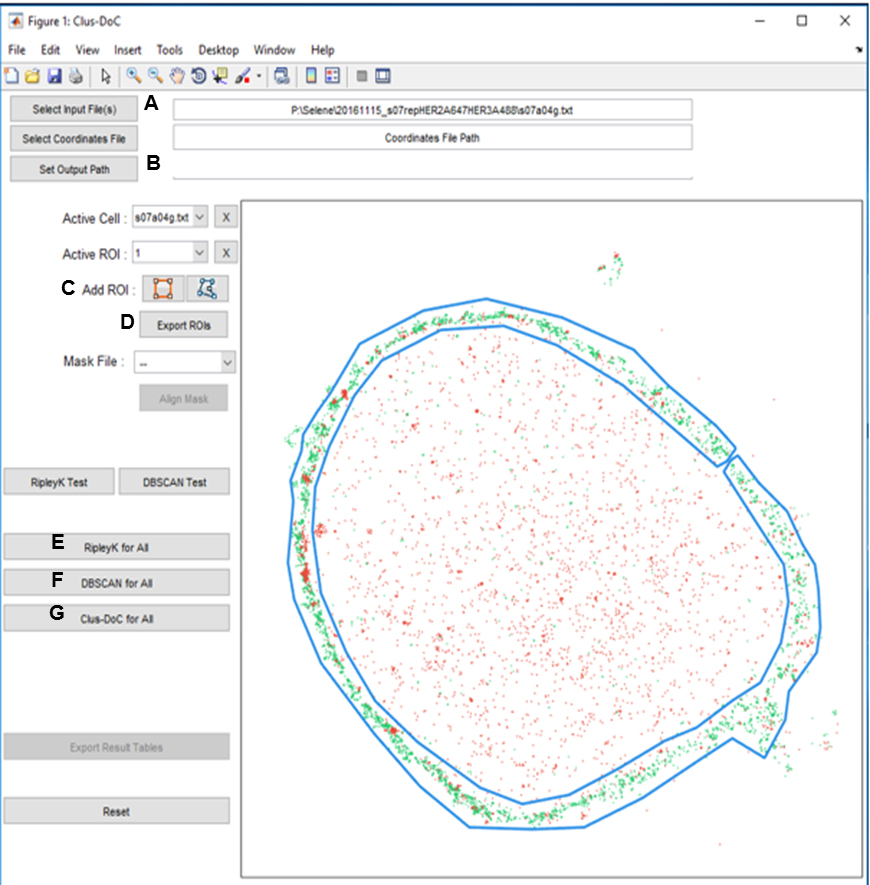
Figure 6. ClusDoC GUI with table of localizations loaded and ROI selected. Buttons of the GUI are linked to the relevant steps in the analysis procedure by labels A-G. - This procedure was repeated for every image.
- Properties such as cluster size and colocalization percentage (see Pageon et al., 2016; Table 1 for complete list) in the Excel result tables produced (in the specified output path folder) were then compared between treatment groups.
- Results can be found in Claus et al. (2018).
Recipes
- Phosphate buffered saline (PBS)
137 mM NaCl
2.7 mM KCl
10 mM Na2HPO4
1.8 mM KH2PO4
Adjust pH to 7.4 and autoclave to sterilize (121 °C, 15 min)
Store PBS at 4 °C - MEA buffer
50 mM Cysteamine hydrochloride (diluted from 100 mM stock stored at -20 °C) in PBS
Use the diluted 50 mM working solution immediately
Acknowledgments
This work has been funded by MRC grant (Ref. MR/K015591/1) from the Medical Research Council and by BBSRC grant BB/G006911/1 from the Biotechnology and Biological Sciences Research Council. This work was also supported in part by the Francis Crick Institute which receives its core funding from Cancer Research UK (FC0010130), the UK Medical Research Council (FC0010130), and the Wellcome Trust (FC0010130).
Competing interests
Authors declare no conflicts of interest or competing interests.
References
- Adamcik, J., Jung, J. M., Flakowski, J., De Los Rios, P., Dietler, G. and Mezzenga, R. (2010). Understanding amyloid aggregation by statistical analysis of atomic force microscopy images. Nat Nanotechnol 5(6): 423-428.
- Chen, X., Liu, M., Tian, Y., Li, J., Qi, Y., Zhao, D., Wu, Z., Huang, M., Wong, C. C. L., Wang, H. W., Wang, J., Yang, H. and Xu, Y. (2018). Cryo-EM structure of human mTOR complex 2. Cell Res 28(5): 518-528.
- Claus, J., Patel, G., Autore, F., Colomba, A., Weitsman, G., Soliman, T. N., Roberts, S., Zanetti-Domingues, L. C., Hirsch, M., Collu, F., George, R., Ortiz-Zapater, E., Barber, P. R., Vojnovic, B., Yarden, Y., Martin-Fernandez, M. L., Cameron, A., Fraternali, F., Ng, T. and Parker, P. J. (2018). Inhibitor-induced HER2-HER3 heterodimerisation promotes proliferation through a novel dimer interface. Elife 7: e32271.
- Dempsey, G. T., Vaughan, J. C., Chen, K. H., Bates, M. and Zhuang, X. (2011). Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods 8(12): 1027-1036.
- Dzyubenko, E., Rozenberg, A., Hermann, D. M. and Faissner, A. (2016). Colocalization of synapse marker proteins evaluated by STED-microscopy reveals patterns of neuronal synapse distribution in vitro. J Neurosci Methods 273: 149-159.
- Eccles, S. A. (2011). The epidermal growth factor receptor/Erb-B/HER family in normal and malignant breast biology. Int J Dev Biol 55(7-9): 685-696.
- Eigenbrot, C., Ultsch, M., Dubnovitsky, A., Abrahmsen, L. and Hard, T. (2010). Structural basis for high-affinity HER2 receptor binding by an engineered protein. Proc Natl Acad Sci U S A 107(34): 15039-15044.
- Endesfelder, U. and Heilemann, M. (2015). Direct stochastic optical reconstruction microscopy (dSTORM). In: Verveer, P. (Ed.) Advanced Fluorescence Microscopy. Methods Mol Biol 1251: 263-276.
- Friedman, M., Nordberg, E., Hoiden-Guthenberg, I., Brismar, H., Adams, G. P., Nilsson, F. Y., Carlsson, J. and Stahl, S. (2007). Phage display selection of Affibody molecules with specific binding to the extracellular domain of the epidermal growth factor receptor. Protein Eng Des Sel 20(4): 189-199.
- Gostring, L., Malm, M., Hoiden-Guthenberg, I., Frejd, F. Y., Stahl, S., Lofblom, J. and Gedda, L. (2012). Cellular effects of HER3-specific affibody molecules. PLoS One 7(6): e40023.
- Griffié, J., Burn, G. L., Williamson, D. J., Peters, R., Rubin-Delanchy, P., Owen, D. M. (2018). Dynamic bayesian Cluster analysis of live-cell single molecule localization microscopy datasets. Small Methods 1800008.
- Griffié, J., Shannon, M., Bromley, C. L., Boelen, L., Burn, G. L., Williamson, D. J., Heard, N. A., Cope, A. P., Owen, D. M. and Rubin-Delanchy, P. (2016). A Bayesian cluster analysis method for single-molecule localization microscopy data. Nat Protoc 11(12): 2499-2514.
- Holbro, T., Beerli, R. R., Maurer, F., Koziczak, M., Barbas, C. F., 3rd and Hynes, N. E. (2003). The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A 100(15): 8933-8938.
- Jonas, K. C., Fanelli, F., Huhtaniemi, I. T. and Hanyaloglu, A. C. (2015). Single molecule analysis of functionally asymmetric G protein-coupled receptor (GPCR) oligomers reveals diverse spatial and structural assemblies. J Biol Chem 290(7): 3875-3892.
- Kellner, R. R., Baier, C. J., Willig, K. I., Hell, S. W. and Barrantes, F. J. (2007). Nanoscale organization of nicotinic acetylcholine receptors revealed by stimulated emission depletion microscopy. Neuroscience 144(1): 135-143.
- Kronqvist, N., Malm, M., Gostring, L., Gunneriusson, E., Nilsson, M., Hoiden Guthenberg, I., Gedda, L., Frejd, F. Y., Stahl, S. and Lofblom, J. (2011). Combining phage and staphylococcal surface display for generation of ErbB3-specific Affibody molecules. Protein Eng Des Sel 24(4): 385-396.
- Kronqvist, N., Malm, M., Göstring, L., Gunneriusson, E., Nilsson, M., Guthenberg, H. I., Gedda, L., Frejd, F. Y., Ståhl, S. and Löfblom, J. (2011). Combining phage and staphylococcal surface display for generation of ErbB3-specific Affibody molecules. Protein Eng Des Sel 24: 385-396.
- Low-Nam, S. T., Lidke, K. A., Cutler, P. J., Roovers, R. C., van Bergen en Henegouwen, P. M., Wilson, B. S. and Lidke, D. S. (2011). ErbB1 dimerization is promoted by domain co-confinement and stabilized by ligand binding. Nat Struct Mol Biol 18(11): 1244-1249.
- Maurel, D., Comps-Agrar, L., Brock, C., Rives, M. L., Bourrier, E., Ayoub, M. A., Bazin, H., Tinel, N., Durroux, T., Prezeau, L., Trinquet, E. and Pin, J. P. (2008). Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: application to GPCR oligomerization. Nat Methods 5(6): 561-567.
- Mayor, S., Rothberg, K. G. and Maxfield, F. R. (1994). Sequestration of GPI-anchored proteins in caveolae triggered by cross-linking. Science 264: 1948-1951.
- Needham, S. R., Hirsch, M., Rolfe, D. J., Clarke, D. T., Zanetti-Domingues, L. C., Wareham, R. and Martin-Fernandez, M. L. (2013). Measuring EGFR separations on cells with ~10 nm resolution via fluorophore localization imaging with photobleaching. PLoS One 8(5): e62331.
- Nicovich, P. R., Owen, D. M. and Gaus, K. (2017). Turning single-molecule localization microscopy into a quantitative bioanalytical tool. Nat Protoc 12(3): 453-460.
- Nordberg, E., Friedman, M., Gostring, L., Adams, G. P., Brismar, H., Nilsson, F. Y., Stahl, S., Glimelius, B. and Carlsson, J. (2007). Cellular studies of binding, internalization and retention of a radiolabeled EGFR-binding affibody molecule. Nucl Med Biol 34(6): 609-618.
- Normanno, N., De Luca, A., Bianco, C., Strizzi, L., Mancino, M., Maiello, M. R., Carotenuto, A., De Feo, G., Caponigro, F. and Salomon, D. S. (2006). Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366(1): 2-16.
- Nussinov, R. (2013). The spatial structure of cell signaling systems. Phys Biol 10(4): 045004.
- Ovesný, M., Křížek, P., Borkovec, J., Švindrych, Z. and Hagen, G. M. (2014). ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 30(16): 2389-2390.
- Pageon, S. V., Nicovich, P. R., Mollazade, M., Tabarin, T. and Gaus, K. (2016). Clus-DoC: a combined cluster detection and colocalization analysis for single-molecule localization microscopy data. Mol Biol Cell 27(22): 3627-3636.
- Recouvreux, P. and Lenne, P. F. (2016). Molecular clustering in the cell: from weak interactions to optimized functional architectures. Curr Opin Cell Biol 38: 18-23.
- Rust, M. J., Bates, M. and Zhuang, X. (2006). Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 3(10): 793-795.
- Shankaran, H., Zhang, Y., Tan, Y. and Resat, H. (2013). Model-based analysis of HER activation in cells co-expressing EGFR, HER2 and HER3. PLoS Comput Biol 9(8): e1003201.
- Sieber, J. J., Willig, K. I., Kutzner, C., Gerding-Reimers, C., Harke, B., Donnert, G., Rammner, B., Eggeling, C., Hell, S. W., Grubmuller, H. and Lang, T. (2007). Anatomy and dynamics of a supramolecular membrane protein cluster. Science 317(5841): 1072-1076.
- Sourjik, V. and Armitage, J. P. (2010). Spatial organization in bacterial chemotaxis. EMBO J 29(16): 2724-2733.
- Spiess, M., Hernandez-Varas, P., Oddone, A., Olofsson, H., Blom, H., Waithe, D., Lock, J. G., Lakadamyali, M. and Stromblad, S. (2018). Active and inactive β1 integrins segregate into distinct nanoclusters in focal adhesions. J Cell Biol 217(6): 1929-1940.
- Truong-Quang, B. A. and Lenne, P. F. (2014). Membrane microdomains: from seeing to understanding. Front Plant Sci 5: 18.
- Ventura, A. C., Bush, A., Vasen, G., Goldin, M. A., Burkinshaw, B., Bhattacharjee, N., Folch, A., Brent, R., Chernomoretz, A. and Colman-Lerner, A. (2014). Utilization of extracellular information before ligand-receptor binding reaches equilibrium expands and shifts the input dynamic range. Proc Natl Acad Sci U S A 111(37): E3860-3869.
- Wikman, M., Steffen, A. C., Gunneriusson, E., Tolmachev, V., Adams, G. P., Carlsson, J. and Stahl, S. (2004). Selection and characterization of HER2/neu-binding affibody ligands. Protein Eng Des Sel 17(5): 455-462.
- Yan, Q. (2011). Labeling Protein with Thiol-reactive Probes. Bio-protocol Bio101: e82.
- Zanetti-Domingues, L. C., Martin-Fernandez, M. L., Needham, S. R., Rolfe, D. J. and Clarke, D. T. (2012). A systematic investigation of differential effects of cell culture substrates on the extent of artifacts in single-molecule tracking. PLoS One 7(9): e45655.
Article Information
Copyright
![]() Roberts et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Roberts et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Roberts, S. K., Hirsch, M., McStea, A., Zanetti-Domingues, L. C., Clarke, D. T., Claus, J., Parker, P. J., Wang, L. and Martin-Fernandez, M. L. (2018). Cluster Analysis of Endogenous HER2 and HER3 Receptors in SKBR3 Cells. Bio-protocol 8(23): e3096. DOI: 10.21769/BioProtoc.3096.
- Claus, J., Patel, G., Autore, F., Colomba, A., Weitsman, G., Soliman, T. N., Roberts, S., Zanetti-Domingues, L. C., Hirsch, M., Collu, F., George, R., Ortiz-Zapater, E., Barber, P. R., Vojnovic, B., Yarden, Y., Martin-Fernandez, M. L., Cameron, A., Fraternali, F., Ng, T. and Parker, P. J. (2018). Inhibitor-induced HER2-HER3 heterodimerisation promotes proliferation through a novel dimer interface. Elife 7: e32271.
Category
Cancer Biology > Proliferative signaling > Cancer therapy
Molecular Biology > Protein > Protein-protein interaction
Cell Biology > Cell imaging > Super resolution imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.