- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Separation and Visualization of Low Abundant Ubiquitylated Forms

Published: Vol 8, Iss 22, Nov 20, 2018 DOI: 10.21769/BioProtoc.3081 Views: 5538
Reviewed by: Jyotiska ChaudhuriAlvaro Eduardo GalvisMario Valentino
Original research article
The authors used this protocol in:

Jan 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







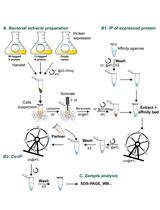
Abstract
In this protocol we describe the separation and visualization of ubiquitylated forms of the yeast mitofusin Fzo1 by Western blot. To this aim, we express HA-tagged Fzo1 in Saccharomyces cerevisiae, break the cells to extract a membrane-enriched fraction, solubilize the membranes using detergent and then specifically immunoprecipitate the tagged protein using anti-HA affinity beads. Subsequently, we separate the higher molecular weight (ubiquitylated) forms of Fzo1 via SDS-PAGE. Finally, immunoblotting and immunodecoration are used to detect the protein and its ubiquitylated forms using an HA-specific antibody. By using this protocol, it is possible to separate and visualize higher molecular weight forms of low abundant proteins such as Fzo1 and detect sharp and distinct bands above the unmodified protein by Western blot.
Keywords: Fzo1Background
Immunoprecipitation is a method to precipitate and enrich a protein from an extract by using an antibody specifically binding to it. The enrichment of a protein is of particular importance when analyzing lower abundant modified forms of it, as is usually the case for ubiquitylated forms of a protein. To be able to enrich for the ubiquitylated Fzo1 protein we tagged the protein with a Hemagglutinin (HA) epitope and use anti-HA affinity beads to precipitate this modified protein from the solubilized membrane fraction. This allows for the detection of the less abundant ubiquitylated forms of proteins as for example Fzo1 (Anton et al., 2011 and 2013; Simoes et al., 2018).
Materials and Reagents
- Centrifugation tubes (50 ml, 2 ml, 1.5 ml) (Sarstedt, catalog numbers: 62.547.254 [50 ml], 72.695.500 [2 ml], 72.690.001 [1.5 ml])
- Nitrocellulose membrane (Amersham, catalog number: 10600001)
- Filter papers (Macherey-Nagel, catalog number: 742113)
- X-Ray films (Fujifilm, catalog number: 47410 19289)
- Transparent plastic foil (Folex, catalog number: 39100440)
- Glass beads (Sartorius, catalog number: BBI-8541701)
- Yeast Saccharomyces cerevisiae BY4741 isogenic to S288c (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) (Brachmann et al., 1998)
Note: This protocol is also applicable to other yeasts strains. - D(+)-Glucose monohydrate (Roth, catalog number: 6780.2)
- 200 mM PMSF (Serva, catalog number: 32395.03) in Isopropanol (VWR, catalog number: 20842.330)
- 50x cOmpleteTM, EDTA-free Protease Inhibitor Cocktail (Roche, catalog number: 05056489001) in MilliQ water
- 5% NG310 (Anatrace, catalog number: NG310 1 MG) in TBS buffer
- 1 M DTT (MP, catalog number: 100597) in MilliQ water
- EZViewTM Red Anti-HA Affinity Gel (Sigma, catalog number: E6779-1ML)
- HA primary antibody (clone 3F10) (Roche, catalog number: 11867423001)
- Sodium azide (NaN3) (Merck, catalog number: 1.06688.0100)
- Rat secondary antibody (Invitrogen, catalog number: 31470)
- Tris (Roth, catalog number: 5429.5)
- NaCl (Roth, catalog number: 3957.2)
- Glycine (PanReac AppliChem, catalog number: 141340.0914)
- 37% HCl (VWR, catalog number: 20252.335)
- SDS (Roth, catalog number: CN30.3)
- Glycerol (Serva, catalog number: 23176.01)
- Methanol (Roth, catalog number: 8388.5)
- Bromophenol Blue (USB, catalog number: US12370)
- Rotiphorese Gel 30 (Roth, catalog number: 3029.1)
- Ponceau S (Serva, catalog number: 33429.02)
- Acetic acid (Roth, catalog number: 3738.5)
- GE Healthcare AmershamTM ECLTM Prime (Fisher Scientific, catalog number: 10308449)
- Nonfat dried milk powder (AppliChem, catalog number: A0830,1000)
- Ammonium peroxidisulfate (APS) (Merck, catalog number: 1.01201.0500)
- TEMED (Sigma, catalog number: T9281-25ML)
- Rotiphorese Gel 30 Acrylamide/Bisacrylamide stock solution (Roth, catalog number: 3029.1)
- TBS buffer (see Recipes)
- 2x Sample buffer (see Recipes)
- Ponceau S solution (see Recipes)
- Tris-Glycine buffer (see Recipes)
- Transfer buffer (see Recipes)
Equipment
- Rolling pin (e.g., a round pen or a short serological pipette)
- Shaking incubator for yeast cultures (Infors-HT, model: Multitron Standard)
- Bench-top centrifuge (Eppendorf, model: 5415R)
- Vortex (Scientific Industries, model: Vortex-Genie 2)
- Glass syringe (Hamilton, e.g., catalog number: 80565) or capillary pipette tips (VWR, catalog number: 53509-015)
- Rotator Intelli-Mixer (Elmi, model: RM-2 M)
- -20 °C freezer (Liebherr, model: LGUex 1500 MediLine)
- Photometer (Hitachi, model: Double Beam Spectrophotometer U-2900)
- Thermomixer (Eppendorf, Thermomixer comfort, model: 5355)
- Tumbling table (Biometra, model: WT12)
- Autoradiography cassette (Amersham Biosciences, HypercassetteTM Autoradiography Cassette model: RPN11642)
- Developer machine (AGFA, model: CURIX 60)
- Standard SDS-PAGE and western blot equipment
- SDS-PAGE equipment, e.g., HoeferTM SE 600 Series
- Western Blot equipment, e.g., Peqlab, PerfectBlueTM ‘Semi-Dry’-Blotter, SedecTM
Procedure
- Culture conditions
- Preculture (Day 1)
Inoculate cells in 50 ml selective minimal media supplemented with glucose (2%, w/v) and grow overnight to the exponential growth phase (less than 2 OD600 density) at 30 °C by shaking at 160 rpm. - Main culture (Day 2)
In the morning, inoculate 160 ml selective minimal media supplemented with glucose with a starting OD600 of 0.3 for 4-6 h to the exponential growth phase (less than 2 OD600 density).
- Preculture (Day 1)
- Sample preparation
- Harvest 160 OD600 of these exponentially growing cells per condition using 50 ml conical tubes by centrifuging (5 min, 4,100 x g, RT). Discard supernatant.
- Resuspend the pellets in 1 ml TBS buffer and transfer the suspension to two 2 ml round-bottom centrifugation tubes and centrifuge (1 min, 16,100 x g, 4 °C). Discard supernatant.
Note: Proceed with all following steps on ice.- Resuspend pellets in 300 µl TBS buffer each. Add 750 mg glass beads, 10 µl 0.2 M PMSF and 20 µl 50x cOmpleteTM EDTA-free protease inhibitor to each suspension.
- Disrupt the cells by vigorous vortexing using the highest setting (Vortex) (3x 30 s vortexing, 30 s on ice, alternating).
- Pool the two suspensions of corresponding conditions.
- Add 400 µl TBS buffer.
- Centrifuge mildly (2 min, 1,500 x g, 4 °C).
- Transfer the supernatant to a new 1.5 ml centrifugation tube.
- Centrifuge (15 min, 16,100 x g, 4 °C). Discard supernatant.
- Resuspend the pellet (= crude membranes) in cold 0.2% NG310 in TBS buffer.
- Solubilize the membranes by continuously rotating the tube (1 h, 10 rpm, 4 °C) using a rotator.
- During solubilization, prepare tubes.
- Input control (In): 20 µl 2x Sample buffer with freshly added 0.1 M DTT.
- Immunoprecipitation (IP): 500 µl 0.2% NG310 in TBS buffer + 23 µl Anti-HA Affinity Gel (wash the Anti-HA Affinity Gel once with TBS buffer before adding it).
- After solubilization, centrifuge (5 min, 16,100 x g, 4 °C).
- Take 20 µl of the supernatant as input control (= 4%), mix it with the 2x sample buffer in the prepared tube (In) and freeze until use at -20 °C.
- Add the remaining supernatant to the prepared IP tube and rotate the tube (overnight, 10 rpm, 4 °C) using a rotator.
- (Day 3) Centrifuge IP tube (30 s, 400 x g, 4 °C).
- Remove supernatant completely
The Anti-HA Affinity Gel consists of small gelatinous beads. As not to aspirate the beads use a syringe with a small opening or thin capillary pipette tips (refer to Equipment section for details). - Wash the Anti-HA Affinity Gel with 0.2% NG310 in TBS buffer.
- Repeat washing 3 x (Steps B16-B18).
- Add 60 µl 2x sample buffer with freshly added 0.1 M DTT.
- Thaw the input control on ice.
- Heat samples using a thermomixer (20 min, 1,400 rpm, 45 °C).
- Centrifuge (30 s, 1,500 x g, RT).
- SDS-PAGE and Immunoblotting
- Separate samples using SDS-PAGE
- Assemble gel equipment according to manufacturer’s instructions (see Equipment section for details).
- Prepare separation and stacking gel mixtures (do not add APS and TEMED yet as it will start the polymerization process).
For 2 gels: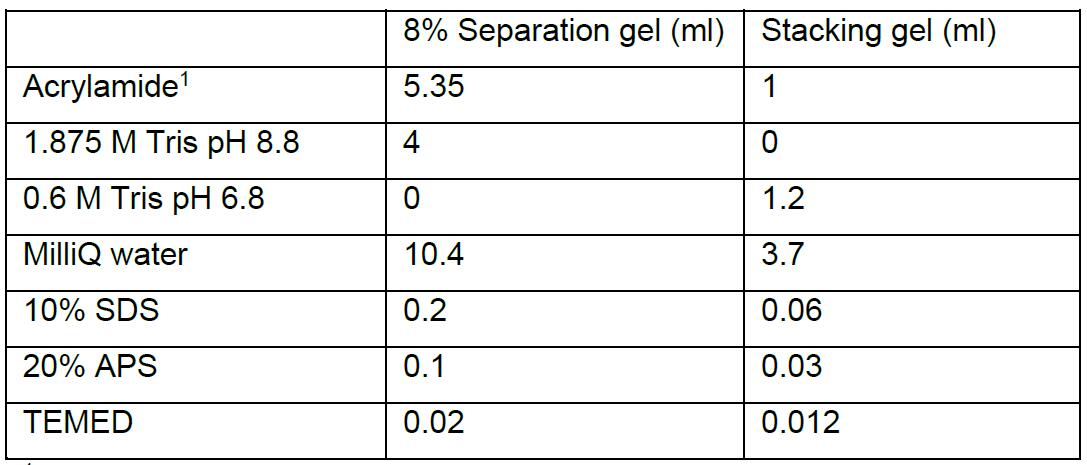
1 30% Acrylamide with 0.8% Bisacrylamide (37.5:1) - Add APS and TEMED to the separation gel mixture and mix well. For each gel, with a pipette, rapidly pour 9 ml of the separation gel mix between the two glass plates and overlay each with 1 ml of isopropanol to even out the surface. Keep the remaining gel mixture and use it to control the polymerization status. After polymerization, decant the isopropanol and wash with water carefully. Remove excess water with filter paper.
- Put in the combs, leaving 3-5 mm distance from the separation gel. Add APS and TEMED to the stacking gel mixture and mix well. Rapidly pour the stacking gel mixture on top, making sure that no bubbles appear in the stacking gel. Keep the remaining gel mixture and use it to control the polymerization status. After polymerization, remove the comb and fill the slots with 1x Tris-Glycine buffer. Load samples (e.g., 4% Input control and 100% eluate) and a pre-stained protein marker of choice (e.g., NEB, P7712).
- Fill SDS-PAGE tank with Tris-Glycine buffer to the filling line (anode). Insert gels into the tank as per manufacturer’s instructions (make sure that the bottom parts of the gels are in contact with the anode buffer). Fill cathode reservoir with enough Tris-Glycine buffer to cover the cathode.
- Run Gels at 150-200 V until the protein of interest reaches the bottom of the gel (the protein itself will not be visible on the gel, thus refer to the pre-stained protein marker to estimate when to stop the gel.)
Note: To make sure that the higher molecular weight ubiquitylation bands will be separated efficiently the unmodified protein needs to run as far as possible. - Disassemble the gels by removing them from the tanks and by taking the glass plates apart carefully with the plastic wedge coming with the SDS-PAGE electrophoresis unit. Cut off the stacking gel with the plastic wedge.
- Transfer the proteins from the acrylamide gel to a membrane by semi-dry western blotting
- Soak six filter papers, one nitrocellulose membrane and the separation gel briefly in transfer buffer.
- Assemble western blotting sandwich as described from plus to minus pole: three filter papers, one nitrocellulose membrane, gel, three filter papers. Between each layer, carefully remove bubbles by rolling over the sandwich with a rolling pin (e.g., a round pen or a short serological pipette).
- Run the transfer for 2 h for 2 mA per cm2 gel/membrane (i.e., for one 7 x 15 cm gel: 2 h, 200 mA).
- Wash the membrane with desalted water, stain the proteins with Ponceau S solution and scan the membranes (only the input control will show a staining), destain using TBS buffer, block the membranes at room temperature for 30 min in 5% milk in TBS buffer, all steps while shaking at 40 rpm using a tumbling table.
- Perform immunodecoration to detect the HA epitope (Steps a-d require shaking at 40 rpm on a tumbling table). A representative image is shown in Figure 1.
- Replace the blocking solution with the primary antibody solution: 1:1,000 anti-HA in 5% milk in TBS buffer and 0.02% NaN3 and incubate overnight at 4 °C.
- Remove the primary antibody solution (can be stored at -20 °C if desired) and wash 3 x using TBS buffer at room temperature for 10 min each.
- Replace the TBS buffer with the secondary antibody solution: 1:5,000 anti-rat in 5% milk in TBS buffer for 2 h incubation at room temperature.
- Wash 3 x for 10 min using TBS buffer at room temperature. Tilt the membrane after the last wash to remove excess liquid.
- Detect the signal.
- Place membrane in a flat container.
- Mix equal amounts of solutions A and B (e.g., 1 ml total volume) of ECL and repeatedly spread over the membrane (for 30 s).
- Tilt the membrane to remove excess liquid, place it in an autoradiography cassette and cover with a transparent plastic foil. Proceed quickly now as the signal may rapidly fade and also because big amounts of protein may burn the membranes irreparably.
Note: For big amounts of protein as here, the membrane will most likely burn at the position of the unmodified protein, therefore the development of the membrane will not be repeatable without quality loss. - In a film developing room (only red light!), place unexposed X-Ray films on the membrane for varying durations, starting with short exposures (e.g., from 1 s to 30 min). Place and remove them in a fast movement without sliding across the membrane to get a sharp signal.
- Use a developer machine to develop the films.
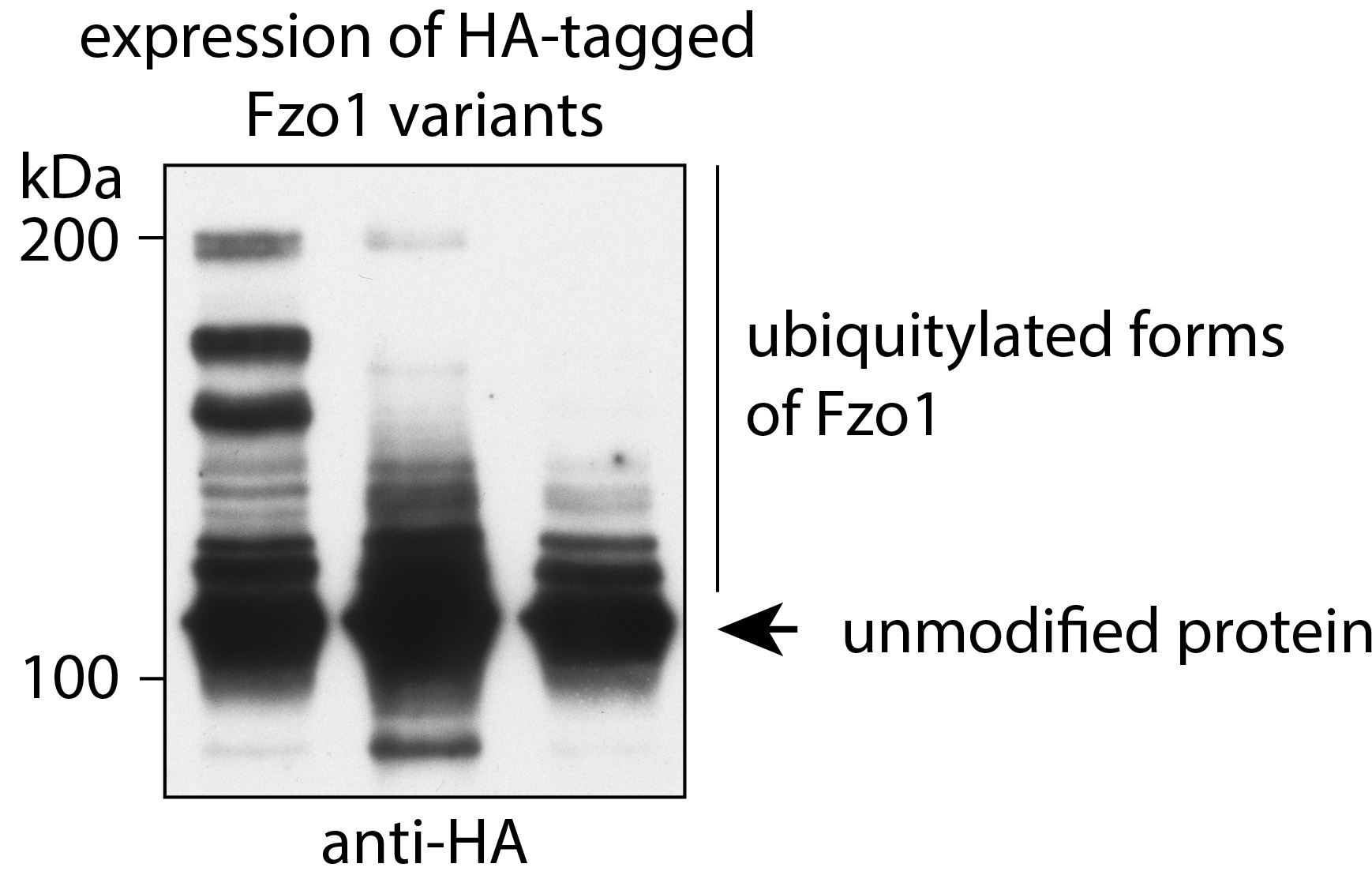
Figure 1. Visualization of low abundant ubiquitylated forms of Fzo1 by immunoprecipitation and western blot analysis
- Separate samples using SDS-PAGE
Recipes
- TBS buffer
50 mM Tris
150 mM NaCl
Adjust the pH to 7.5 using HCl 37% - 2x Sample buffer
4% SDS
20% Glycerol
135 mM Tris-HCl (pH 6.8)
0.1% Bromophenol Blue
Store at RT. Add 100 mM DTT freshly before use - Ponceau S solution
0.1% Ponceau S
5% Acetic acid - Tris-Glycine buffer
25 mM Tris
200 mM Glycine
3.5 mM SDS - Transfer buffer
200 mM Glycine
25 mM Tris
20% (v/v) Methanol
0.02% SDS
Adjust pH to 8.5 using HCl 37%
Acknowledgments
Funding: Deutsche Forschungsgemeinschaft (ES338/3-1); Universität zu Köln (German Excellence Initiative and Faculty of Mathematics and Natural Sciences); Deutsche Forschungsgemeinschaft (SFB635); Deutsche Forschungsgemeinschaft (CRC1218TPA03).
Competing interests
The authors declare they have no conflict of interest or competing interests.
References
- Anton, F., Dittmar, G., Langer, T. and Escobar-Henriques, M. (2013). Two deubiquitylases act on mitofusin and regulate mitochondrial fusion along independent pathways. Mol Cell 49(3): 487-498.
- Anton, F., Fres, J. M., Schauss, A., Pinson, B., Praefcke, G. J., Langer, T. and Escobar-Henriques, M. (2011). Ugo1 and Mdm30 act sequentially during Fzo1-mediated mitochondrial outer membrane fusion. J Cell Sci 124(Pt 7): 1126-1135.
- Brachmann, C. B., Davies, A., Cost, G. J., Caputo, E., Li, J., Hieter, P. and Boeke, J. D. (1998). Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14(2): 115-132.
- Simoes, T., Schuster, R., den Brave, F. and Escobar-Henriques, M. (2018). Cdc48 regulates a deubiquitylase cascade critical for mitochondrial fusion. Elife 7: e30015.
Article Information
Copyright
![]() Schuster et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Schuster et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Schuster, R., Simões, T., den Brave, F. and Escobar-Henriques, M. (2018). Separation and Visualization of Low Abundant Ubiquitylated Forms. Bio-protocol 8(22): e3081. DOI: 10.21769/BioProtoc.3081.
- Simoes, T., Schuster, R., den Brave, F. and Escobar-Henriques, M. (2018). Cdc48 regulates a deubiquitylase cascade critical for mitochondrial fusion. Elife 7: e30015.
Category
Biochemistry > Protein > Immunodetection > Immunoprecipitation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.