- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

RNA Immunoprecipitation Assay to Determine the Specificity of SRSF3 Binding to Nanog mRNA

Published: Vol 8, Iss 21, Nov 5, 2018 DOI: 10.21769/BioProtoc.3071 Views: 9452
Reviewed by: Dr. Amit K. TripathiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






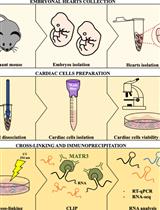
Abstract
Mammalian cells express hundreds of RNA binding proteins (RBPs) that are essential regulators of RNA metabolism. RBP activity plays a central role in the control of gene expression programs and identification of RNA-protein interactions is critical for comprehensive understanding of gene regulation in cells. In recent years, various tools and techniques to identify these RNA-protein interactions have been developed. Among those, RNA immunoprecipitation is a precise and powerful assay that can be used to establish the physical interaction of an individual RBP with its target RNAs in vivo. Here, we describe a quantitative method for determining RNA-protein interactions using RNA immunoprecipitation (RNA-IP) assay in mouse embryonic stem cells carrying ectopically expressed mutant constructs. This protocol is reliable and easily adaptable to identify the interactions of endogenous or ectopically expressed RNAs and proteins.
Keywords: RNA immunoprecipitationBackground
Embryonic stem cells express more than 500 different RBPs highlighting the potential of RNA regulation in controlling pluripotency (Kwon et al., 2013). We have established serine-arginine-rich splicing factor 3 (SRSF3) as an essential RBP to maintain pluripotency in mouse embryonic stem cells (Ratnadiwakara et al., 2018). SRSF3 binds to RNAs encoding key components of mouse pluripotency circuitry and controls their expression via multiple RNA processing mechanisms. Among these, we have specifically demonstrated that SRSF3 directly binds to the core pluripotency transcription factor Nanog mRNA to facilitate its nucleo-cytoplasmic export. Our results have shown that SRSF3 binding is essential for the efficient export of the Nanog mRNA, targeted mutation of SRSF3 binding sites in Nanog mRNA abolishing SRSF3 binding and consequently the nucleo-cytoplasmic export of Nanog.
The basic principle of RNA-IP is based on using an antibody recognizing the protein of interest to specifically pull down the RBP with the target RNA complexes. Therefore, any RNA that is physically associated with the protein can be isolated and further analyzed. We have used quantitative PCR for the downstream analysis. However, other methods such as sequencing or hybridization can also be used to identify the targeted RNAs.
Here we use cells that express an SRSF3-GFP fusion protein and used an anti-GFP antibody for the immunoprecipitation. It is important to note that an antibody against an endogenous RBP may be successfully used for RNA-IP. However, we have observed that anti-RBP antibodies can compete with RNA and thus, for instance in the case of SRSF3, cannot be used to efficiently purify SRSF3 in complex with RNA. We have used RNA immunoprecipitation (RNA-IP) to detect the physical interaction of SRSF3 not only with the endogenous Nanog mRNA, but also ectopically expressed constructs carrying mutations within SRSF3 binding sites in the Nanog mRNA. This assay allows the assessment of direct RNA-protein interactions as well as the sequence specificity of RBPs in cells.
Materials and Reagents
- 6-well cell culture plates (Sigma-Aldrich, catalog number: CLS3516)
- Serological pipettes 10 ml (Sigma-Aldrich, catalog number: CLS4488)
- 15 ml Falcon tubes (Sigma-Aldrich, catalog number: CLS430791)
- Sterile filter pipette tips 10 μl, 20 μl, 200 μl, 1,000 μl (Axygen, catalog numbers: TF10LRS, TF20LRS, TF200LRS and TF1000LRS)
- Microcentrifuge tubes (Axygen, catalog number: MCT-175-C)
- Neptune semi-skirted 96-well plates (VWR, catalog number: 89126-694)
- Optically clear adhesive seal sheets (Thermo Fisher Scientific, catalog number: AB-1170)
- Cells (here mouse embryonic stem cells)
- qPCR primers for the genes of interest
Here primers that detect both wild-type (WT) and mutant (ΔSRSF3) Nanog constructs
F: CAAGCCTCAGACAGTGGTTCA
R: ATGTCAGTGTGATGGCGAGG - Knock-Out DMEM (Thermo Fisher Scientific, catalog number: 10829018)
- ES cell grade fetal bovine serum (Sigma-Aldrich, catalog number: F9423)
- GlutaMAX (Life Technologies, catalog number: 35050-061)
- Trypsin 0.25% (Life Technologies, catalog number: 25200-056)
- Penicillin-Streptomycin (Life Technologies, catalog number: 15070-063)
- Non-Essential Amino Acids (Life Technologies, catalog number: 11140-050)
- Phosphate-buffered saline (PBS) (Life Technologies, catalog number: 14190-250)
- Complete Mini Protease Inhibitor cocktail (Roche, catalog number: 11836153001)
- RNaseOUT (Thermo Fisher Scientific, catalog number: 10777019)
- Dynabeads Protein G (see Note 1) (Invitrogen, catalog number: 10004D)
- Antibody of interest, here Anti-GFP antibody (Abcam, catalog number: AB290)
- TRI Reagent (Sigma-Aldrich, catalog number: T9424)
- Chloroform (Sigma-Aldrich, catalog number: 288306)
- Isopropanol (Sigma-Aldrich, catalog number: 278475)
- RNA grade Glycogen (Thermo Fisher Scientific, catalog number: R0551)
- Ethanol (Molecular Biology Grade)
- RNase-free water (Invitrogen, catalog number: 10977-015)
- RQ1 DNase kit (Promega, catalog number: M6101)
- SuperScript III Reverse transcription kit (Thermo Fisher Scientific, catalog number: 18080044)
- Random hexamer primer mix (Bioline, catalog number: BIIO38028)
- OligodT18 (IDT)
- SYBR Green master mix, here Luminaries HiGreen qPCR Master Mix, Low ROX (Thermo Fisher Scientific, catalog number: K0974)
- Sterile Milli-Q Water
- Ice
- Beta-mercaptoethanol (Life-technologies, catalog number: 21985-023)
- Leukemia Inhibitory Factor (LIF) (here LIF was produced by the Monash University Protein Production Unit, Australia)
Please provide the manufacture and catalog number for #item 35 if possible. - Tris-HCl
- NaCl
- Nonidet P-40
- ES culture media (see Recipes)
- NET-2 buffer (see Recipes)
Equipment
- Sterile cell culture hood (Safemate Vision 1.2 cabinet, catalog number: LDE0820)
- 37 °C cell culture incubator with 10% CO2 and 5% O2 (hypoxic conditions for embryonic stem cells) (Thermo Fisher Scientific, model: HeracellTM 150)
- Magnetic rack, MagnaRackTM (Invitrogen, catalog number: CS15000)
- Automated cell counter (NanoEnTek, catalog number: E1000)
- Vacuum pump
- Refrigerated microcentrifuge (Bio-strategy, catalog number: 75002421)
- Sonicator (Bioruptor® Plus sonication device, Diagenode, catalog number: B01020001)
- Tube revolver (Thermo Fisher Scientific, model: 88881002)
- qPCR machine (7500 Real-Time PCR System) (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4351105)
- Milli-Q water purification system (Milli-Q Direct Water Purification System, MERK)
- Fluorescence Microscope (Nikon Eclipse TS100 Inverted Routine Microscope)
- Vortexer
- Freezer
Software
- SDSv2.4 (Thermo Fisher Scientific, www.thermofisher.com/au/en/home/technical-resources/software-downloads/applied-biosystems-7900ht-fast-real-timespcr-system.html)
- GraphPad Prism 7 (GraphPad Sowtware, Inc, www.graphpad.com)
- Microsoft Excel Version 15.41 (Microsoft)
- Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/)
Procedure
- Cell culture
- Maintain cells using standard cell culture procedures for the cell line of interest. We used mouse embryonic stem (ES) cells (JM8.A) expressing BAC-SRSF3-GFP or NLS-GFP grown on irradiated mouse embryonic fibroblast (MEF) feeder cells. A detailed description of the generation and culture properties of the ES cell lines used here can be found in Ratnadiwakara et al. (2018) and the media composition can be found under Recipes.
- Transfect the cells with WT-Nanog or ΔSRSF3-Nanog vectors as described in Ratnadiwakara et al. (2018) and 24 h later collect the cells using 0.1% trypsin. WT-Nanog construct carries the wildtype version of Nanog mRNA including the 3’UTR whereas in the ΔSRSF3-Nanog construct synonymous mutations have been introduced within the SRSF3 binding sites.
- Deplete the MEF feeder cells by seeding the cells on non-gelatine coated plates and incubating for 45 min. This step can be omitted when using feeder-free cell lines.
- Collect the feeder-free embryonic stem cells in suspension and centrifuge 470 x g for 3 min at 20 °C.
- Count the cells manually or by using an automated cell counter. We prefer automated counter for consistency and accuracy. Use 5 x 106 cells per immunoprecipitation (see also Note 1).
- Magnetic bead preparation (Figure 1–step 1)
- Add 50 μl of Protein G Dynabeads to a fresh microcentrifuge tube corresponding to each of the experimental samples.
- Wash beads twice each with 900 μl of NET-2 buffer using a magnetic rack.
- Re-suspend the beads in 100 μl NET-2 buffer with 2-10 μg of antibody of your interest per reaction. We used 10 μg rabbit ChIP grade anti-GFP antibody per 50 μl beads. It is critical to include a negative control in the experiment. Here we used NLS-GFP expressing cells with anti-GFP coupled beads as a negative control. If an antibody against an endogenous RPB instead of an epitope-tag is used, beads coupled to IgG (antibody isotype control) should be prepared as a negative control reaction.
Note: The amount of the antibody needs to be optimized before the first use. The selection of protein G or A beads may depend on the source species of the antibody (Refer to: https://www.neb.com/tools-and-resources/selection-charts/affinity-of-protein-ag-for-igg-types-from-different-species). - Rotate the tubes for 60 min at room temperature to allow for antibody coupling to the beads.
Note: The cell extract can be prepared during this time. - Place the tubes on the magnetic rack and wash two times each with 900 μl NET-2 buffer to remove any unbound antibody and leave the beads in the last wash. Remove the buffer only when ready to proceed to Step D1.
- Preparation of the cell extract (Figure 1–step 2)
- Wash the cell pellet by re-suspending it in 1ml of PBS and centrifuging for 3 min at 470 x g at 20 °C.
- Re-suspend the washed cell pellet in 1 ml of ice-cold NET-2 buffer supplemented with 1x protease inhibitor and 1:1,000 RNaseOUT.
Note: From here onwards perform all steps on ice.- Sonicate the cells using Bioruptor® for 5 cycles of 20 s on/30 s off at low intensity at 4 °C to disrupt the cell and nuclear membranes and to fragment chromatin.
- Spin for 10 min at 15,000 x g at 4 °C to remove cell debris.
- Transfer the supernatant to a fresh microfuge tube.
- Save 50 μl (5%) of the lysate as input sample and store on ice until the IP is complete.
- Immunoprecipitation (Figure 1–step 3)
- Add the remaining lysate to the antibody-coupled beads prepared in Step B5 and rotate at 4 °C for 2 h on a tube rotator to facilitate the binding of the antibody to RBP-RNA complexes (here SRSF3-GFP-RNA and NLS-GFP negative control).
- Place the tubes on the magnetic rack and carefully remove and discard the supernatant using a vacuum pump.
Note: When performing the IP for the first time, the supernatant can be collected and used for Western blot analysis together with the input and bead eluate to determine how well the antibody depletes the cell extract of the protein of interest, and to confirm the specificity of the antibody. - Wash the beads five times by carefully re-suspending in 1 ml of ice-cold NET-2 buffer during each wash. Collect the beads using a magnetic rack between washes.
Notes:- To improve washing efficiency, the beads can be rotated with the wash buffer for 5 min at 4 °C for the final wash.
- Leave behind a small amount of the buffer solution during the early washing steps to prevent the loss of beads. Use the recommended amount of the buffer through subsequent washes (10x bead volume or more).
- RNA extraction (Figure 1–step 4)
- Add 500 μl of TRI Reagent to the tubes with beads and inputs.
- Add 100 μl of chloroform and shake vigorously for 15 s. Allow the tubes to stand for 10 min at room temperature.
- Centrifuge the mixture at 12,000 x g for 15 min at 4 °C.
- Transfer the aqueous layer into a fresh tube and add 250 μl of 2-propanol and 1 μl of Glycogen.
Note: Addition of glycogen helps the maximum recovery of the RNA and visualization of the RNA pellet. - Mix carefully and allow to stand at room temperature for 10 min to precipitate the RNA.
- Collect the precipitated RNA by centrifugation at 12,000 x g for 10 min at 4 °C.
- Remove the supernatant and wash the RNA pellet by adding 500 μl of 75 % ethanol, vortex and centrifuge at 7,500 x g for 5 min at 4 °C.
- Briefly air-dry the RNA pellet for 5-10 min.
Note: Avoid over-drying the pellet as this will greatly decrease its solubility. - Re-suspend RNA in 10 μl of RNase-free water and proceed to cDNA Synthesis.
Note: Use the complete 10 μl of RNA as a template for cDNA synthesis for each sample allowing the calculation of the enrichment over input (IP/input).

Figure 1. Summary of the workflow of RNA immunoprecipitation in mouse embryonic stem cells using protein G Dynabeads and rabbit anti-GFP antibody - Quantitative PCR
- Perform DNase I treatment on all samples to remove any genomic DNA contamination that can affect the downstream analysis. We used Promega RQ1 DNase kit according to the manufacturer’s protocol.
Note: Adjust the volumes to total of 13 μl at the end of the DNaseI treatment allowing to proceed directly to cDNA synthesis in a single tube. - Carry out the reverse transcription of the input and IP samples using a reverse transcription kit. We used SuperScript III Reverse transcriptase according to the manufacturer's protocol with 1:1 of random hexamers and oligodT18 for reverse priming.
Note: A mixture of random hexamers and oligodT18 can improve the sensitivity of the cDNA synthesis. - Dilute the cDNA 1:10 with nuclease-free water for quantitative PCR.
- Perform quantitative PCR with primers specific for the gene of interest. Use the optimized manufacturer’s protocol for specific SYBR Green master mixes. We used 5 µl of Luminaries 2x HiGreen qPCR Master Mix per reaction, optimized concentration of each forward and reverse primers (typically 0.15-0.30 µM end concentration) and 2 µl of diluted cDNA per reaction. Perform two or more technical replicates for each sample (see Note 2).
- Perform DNase I treatment on all samples to remove any genomic DNA contamination that can affect the downstream analysis. We used Promega RQ1 DNase kit according to the manufacturer’s protocol.
Data analysis
- Upon the completion of the PCR, run use the machine specific software to analyze the data. We used SDS software (see Note 3).
- Export Ct values into an Excel spreadsheet and calculate the average of three replicates for each reaction.
- Normalize the Ct average of individual IP samples to the Ct value of their respective input samples to obtain ∆Ct value.
∆Ct = (Average Ct of IP - Average Ct of input) - Normalize the ∆Ct of each sample to ∆Ct of the no antibody control to obtain ∆∆Ct.
∆∆Ct = (∆Ct of IP - ∆Ct of no antibody control) - Calculate the ratio IP/input for each IP (Figure 2). Here we show data for n = 2 but for statistical analysis, n ≥ 3 is required.
IP/Input = 2(-∆∆Ct)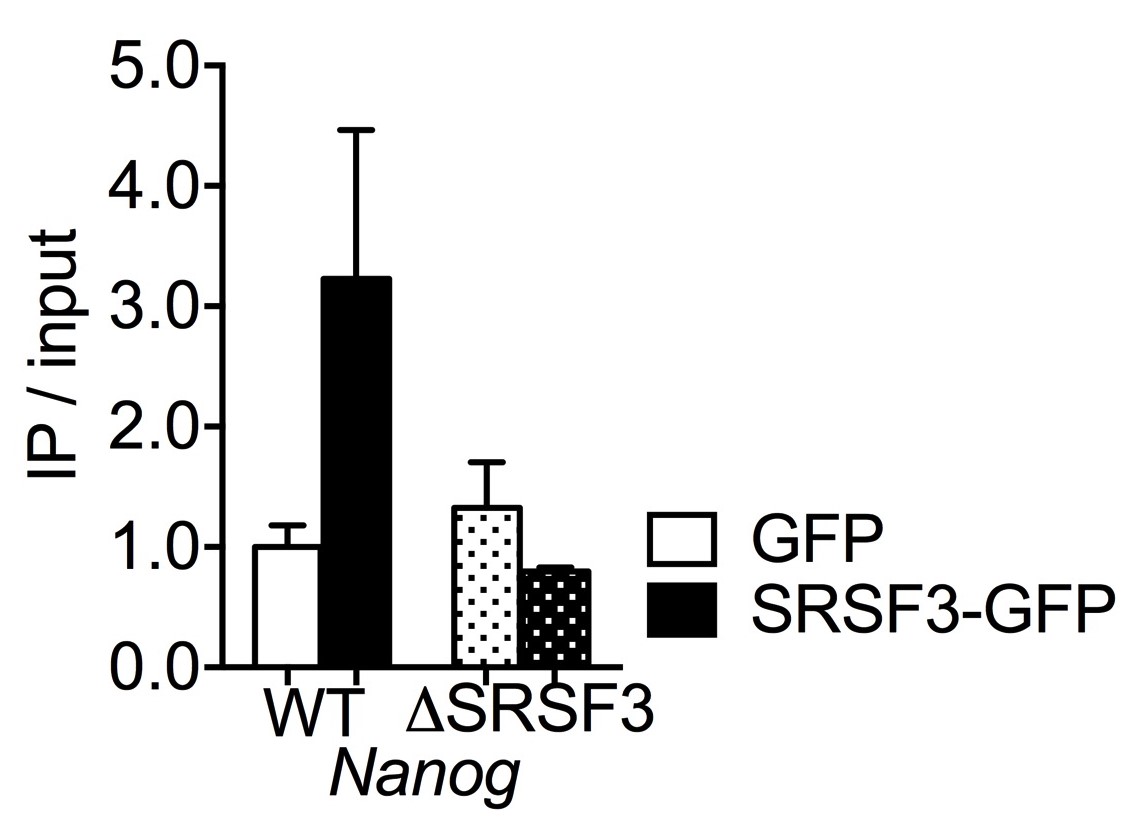
Figure 2. Representative data of RNA immunoprecipitation (IP) of WT- and ∆SRSF3- Nanog mRNA in SRSF3-GFP or GFP-only expressing mouse embryonic stem cells as in (Ratnadiwakara et al., 2018). The Nanog mRNA enrichment is presented as IP/input and normalized to GFP-only control cells expressing WT-Nanog (mean ± SEM, n = 2). The graph was generated in GraphPad Prism 7 (Ratnadiwakara et al., 2018).
Notes
- Ultraviolet (UV) cross-linking can be used at this stage to stabilize the RNA-protein interaction before the immunoprecipitation.
- The primer pairs used for quantitative PCR should be pre-optimised for optimal amplification by using cDNA serial dilutions (plot-log10[cDNA concentration] vs. Ct to calculate primer efficiency). We used Primer3 software to design primers with the following primer picking settings:
- Primer Tm–60 °C
- Product size range–100-200 nucleotides
- Primer size–21 nucleotides
- The dissociation curve should produce a single peak for each reaction indicating the amplification of a single specific product. The MIQE Guidelines in Bustin et al. (2009) provide further information on publication of quantitative real-time PCR experiments.
Recipes
- ES culture media
1x Knock-Out DMEM
15% ESC grade fetal bovine serum
2 mM GlutaMAX
1 mM Non-essential amino acids
1x Penicillin-streptomycin
Just before use, add Leukemia Inhibitory Factor (LIF) and beta-mercaptoethanol to the final concentration of 1,000 U/ml and 1 μM, respectively - NET-2 buffer
50 mM Tris-HCl (pH 7.5)
150 mM NaCl
0.05% Nonidet P-40
Dissolve in sterile Milli-Q water and store and use at 4 °C
Acknowledgments
MLA was supported by National Health and Medical Research Council (NHMRC) GNT1043092 and GNT1138870, Aatos and Jane Erkko Foundation and Monash Biomedicine Discovery Fellowship.
The protocol was adapted from a previously published study (Anko et al., 2010).
Competing interests
The authors have no conflicts of interest or competing interests.
References
- Anko, M. L., Morales, L., Henry, I., Beyer, A. and Neugebauer, K. M. (2010). Global analysis reveals SRp20- and SRp75-specific mRNPs in cycling and neural cells. Nat Struct Mol Biol 17(8): 962-970.
- Bustin, S. A., Benes, V., Garson, J. A., Hellemans, J., Huggett, J., Kubista, M., Mueller, R., Nolan, T., Pfaffl, M. W., Shipley, G. L., Vandesompele, J. and Wittwer, C. T. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55(4): 611-622.
- Kwon, S. C., Yi, H., Eichelbaum, K., Fohr, S., Fischer, B., You, K. T., Castello, A., Krijgsveld, J., Hentze, M. W. and Kim, V. N. (2013). The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol 20(9): 1122-1130.
- Ratnadiwakara, M., Archer, S. K., Dent, C. I., Ruiz De Los Mozos, I., Beilharz, T. H., Knaupp, A. S., Nefzger, C. M., Polo, J. M. and Anko, M. L. (2018). SRSF3 promotes pluripotency through Nanog mRNA export and coordination of the pluripotency gene expression program. Elife 7: e37419.
Article Information
Copyright
![]() Ratnadiwakara and Änkö. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Ratnadiwakara and Änkö. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Ratnadiwakara, M. and Änkö, M. (2018). RNA Immunoprecipitation Assay to Determine the Specificity of SRSF3 Binding to Nanog mRNA. Bio-protocol 8(21): e3071. DOI: 10.21769/BioProtoc.3071.
- Ratnadiwakara, M., Archer, S. K., Dent, C. I., Ruiz De Los Mozos, I., Beilharz, T. H., Knaupp, A. S., Nefzger, C. M., Polo, J. M. and Anko, M. L. (2018). SRSF3 promotes pluripotency through Nanog mRNA export and coordination of the pluripotency gene expression program. Elife 7: e37419.
Category
Molecular Biology > RNA > RNA-protein interaction
Stem Cell > Pluripotent stem cell > Cell pluripotency
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.