- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Nuclear/Cytoplasmic Fractionation of Proteins from Caenorhabditis elegans

Published: Vol 8, Iss 20, Oct 20, 2018 DOI: 10.21769/BioProtoc.3053 Views: 10971
Reviewed by: Matthias RieckherAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
C. elegans is widely used to investigate biological processes related to health and disease. To study protein localization, fluorescently-tagged proteins can be used in vivo or immunohistochemistry can be performed in whole worms. Here, we describe a technique to localize a protein of interest at a subcellular level in C. elegans lysates, which can give insight into the location, function and/or toxicity of proteins.
Keywords: C. elegansBackground
Subcellular fractionation has been used in different model organisms to identify and study protein function in nuclei, membranes and cytoplasm. For example, aggregation-prone proteins may be more toxic when they are localized in the nucleus or in the cytosol (Kontopoulos et al., 2006; Barmada et al., 2010). Here we provide a protocol (adapted from Chen et al., 2000 and La Rocca et al., 2007) to localize specific proteins in the nuclear and cytoplasmic fractions of C. elegans.
Materials and Reagents
- 94 mm plates (Greiner Bio One International, catalog number: 633185 )
- 15 ml conical tube (SARSTEDT, catalog number: 62.554.502 )
- 1.5 ml tubes (Greiner Bio One International, catalog number: 616201 )
- Pellet pestle (Sigma-Aldrich, catalog number: Z359947-100EA )
- Glass slides (Fisher Scientific, catalog number: 12164682 )
- Nitrocellulose (Bio-Rad Laboratories, catalog number: 1620112 )
- C. elegans strain
- Escherichia coli OP50 strain
- Deionized water (dH2O)
- Cholesterol (Fisher Scientific, catalog number: 10263660 )
- Ethanol absolute (Merck, catalog number: 1009831000 )
- Magnesium sulphate (MgSO4) (Fisher Scientific, catalog number: 10264630 )
- Calcium dichloride (CaCl2) (Fisher Scientific, catalog number: 10171800 )
- di-Potassium hydrogen phosphate (K2HPO4) (Merck, catalog number: 105101 )
- Potassium dihydrogen phosphate (KH2PO4) (Merck, catalog number: 104873 )
- Casein digest, Difco (BD, catalog number: 211610 )
- Select agar (Thermo Fisher Scientific, InvitrogenTM, catalog number: 30391049 )
- Disodium hydrogen phosphate (Na2HPO4) (Acros Organics, catalog number: 424380010 )
- Sodium chloride (NaCl) (Merck, catalog number: 106404 )
- DL-Dithiothreitol (DTT) (Sigma-Aldrich, catalog number: D0632 )
- HEPES (Sigma-Aldrich, catalog number: H4034 )
- Potassium hydroxide (KOH) (Fisher, catalog number: 10705921 )
- Potassium chloride (KCl) (Fisher Scientific, catalog number: 10010310 )
- Magnesium dichloride (MgCl2) (Fisher Scientific, catalog number: 10518060 )
- Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, catalog number: E6758 )
- Sucrose (Fisher Scientific, catalog number: 10386100 )
- Tween 20 (Sigma-Aldrich, catalog number: P1379-100ML )
- Protease inhibitors (cOmplete) (Roche, catalog number: 11697498001 )
- Benzonase nuclease (Merck, catalog number: 70746 )
- PierceTM BCA Protein Assay Kit (Thermo Fisher Scientific, catalog number: 23225 )
- Liquid nitrogen
- Ammonium persulfate (APS) (Thermo Fisher Scientific, catalog number: A/6120/60 )
- N,N,N′,N′-Tetramethylethylenediamine (TEMED) (Fisher Scientific, catalog number: 10142863 )
- 40% Acrylamide/Bis solution (29:1) (Bio-Rad Laboratories, catalog number: 1610146 )
- Sodium dodecyl sulfate (SDS) (Merck, Calbiochem, catalog number: 428015 )
- Tris Base (Roche Diagnostics, catalog number: 11814273001 )
- Hydrochloric acid 37% (Acros Organics, catalog number: 124630010 )
- Glycine (Fisher Scientific, catalog number: 10070150 )
- Methanol (Merck, catalog number: 106009 )
- Bromophenol blue (Acros Organics, catalog number: 403160100 )
- Glycerol (Sigma-Aldrich, catalog number: G5516 )
- β-Mercaptoethanol (Merck, catalog number: 444203 )
- Milk powder (Campina)
- PageRulerTM Plus Prestained Protein Ladder (Thermo Fisher Scientific, catalog number: 26619 )
- Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare, catalog number: RPN2236 )
- Antibodies (see Table 1)
Table 1. List of primary and secondary antibodies
Primary antibody Host Company/Catalog number Dilution primary antibody Secondary antibody α-LMN-1 Rabbit Novus Biologicals,
catalog number:
385300021:1,000 Anti-rabbit (1:10,000)
(Bio-Rad Laboratories, catalog number: 1706515 )α-tubulin Mouse Sigma-Aldrich,
catalog number:
T60741:10,000 Anti-Mouse (1:10,000)
(Bio-Rad Laboratories, catalog number: 1706516 )α-GFP
(Living Colors® A.v. Monoclonal Antibody (JL-8)Mouse Takara Bio,
catalog number:
6323811:10,000 Anti-Mouse (1:10,000)
(Bio-Rad Laboratories, catalog number: 1706516 ) - M9 buffer (see Recipes)
- Phosphate buffered saline (PBS) (see Recipes)
- PBS-T (see Recipes)
- Blocking solution (see Recipes)
- Nematode growth medium (NGM) plates (see Recipes)
- Phosphate buffer (see Recipes)
- Hypotonic buffer (see Recipes)
- Hypertonic buffer (see Recipes)
- 25x protease inhibitors (see Recipes)
- 1 M DTT (see Recipes)
- 12% acrylamide gel (see Recipes)
- Running buffer (see Recipes)
- Transfer buffer (see Recipes)
- SDS lysis buffer (see Recipes)
Equipment
- Single channel pipettes (Gilson, models: P2G , P20G , P200G , P1000G )
- Orbital shaker (Thermo Fisher Scientific, model: MaxQTM 2000)
- 20 °C incubator (Winecooler, LIEBHERR, model: WK 4126 )
- Stereo microscope (Leica, model: MZ7.5 )
- Pellet pestle (motor) (Sigma-Aldrich, catalog number: Z359971 )
- Tabletop centrifuge, cooled (Eppendorf, model: 5424 R )
- Centrifuge (Thermo Fisher Scientific, model: SL 40R )
- Autoclave (VWR, model: VAPOUR-Line Lite )
- -80 °C freezer (Sanyo, model: VIP plus )
- Mini PROTEAN 3 system (Bio-Rad Laboratories, catalog number: 1658001edu )
- WB Imager (GE Healthcare, model: ImageQuant LAS 4000 mini, catalog number: 28955813 )
Software
- ImageJ (Open source: https://imagej.nih.gov/ij/)
- Microsoft Excel (Microsoft Corporation, Redmond, USA)
Procedure
- Collection of worms
- Prepare 10-15 plates of bleach synchronized worms, with approximately 800-1,000 worms per 9 cm NGM plate in order to get relatively large nuclear fractions.
Note: We performed subcellular fractionation experiments with as few as 5 x 9 cm NGM plates per condition. However, working with 10 x 9 cm NGM plates will yield a more workable nuclear fraction, as the pellet will be more easily visible during the washing steps. - Incubate worms at 20 °C for approximately 72 h, until day 1 of adulthood (D1).
Note: Subcellular fractionation can be performed at different ages of worms. Here, we describe a protocol for worms at the first day of adulthood (D1). However, the protocol is also feasible for L4-staged animals and older worms. One should keep in mind that it is easier to break the cuticle of larvae with a pellet pestle than older worms. Thus, the number of strokes should be adjusted accordingly to the age of the worms (7-12 times). - Collect D1-staged worms with ~4 ml M9 per plate in a 15 ml conical tube and wash until the supernatant is clear (3-5 times).
- Wash the worm pellet twice with 1 ml cold hypotonic buffer.
- Prepare 2-5 ml ‘complete hypotonic buffer’ per sample by adding 1 M DTT (final concentration: 1 mM DTT) and 25x protease inhibitors (final concentration: 2x protease inhibitors) to a few ml of standard hypotonic buffer.
Note: It is suggested to calculate the volume of ‘complete hypotonic buffer’ that is required for the experiment. Pipette the required volume of hypotonic buffer in a tube and add DTT and protease inhibitors; compounds that should both be stored at -20 °C. - Remove all hypotonic buffer and add ‘complete hypotonic buffer’, as prepared in Step A5 (~500 μl). Transfer the worm suspension into a 1.5 ml tube.
Note: Estimate the volume of the worm pellet and add the same volume of ‘complete hypotonic buffer’. - Avoid freezing the worm pellet at this point, as this may damage the nuclei and cause leakage of nuclear proteins in the cytosol.
- Prepare 10-15 plates of bleach synchronized worms, with approximately 800-1,000 worms per 9 cm NGM plate in order to get relatively large nuclear fractions.
- Fractionation
- Homogenize worms with a pellet pestle for 1 min and let the worms rest for 1 min, repeat this 5 to 10 times. Keep the worms on ice during the whole process. Pipette 5 µl of the worm suspension onto a glass slide after 4-5 times grinding the pellet with the pestle to evaluate the degree of worm lysis. Homogenization is complete when 70%-80% of the worms are disrupted and only a few worm remnants are visible (see Figure 1).

Figure 1. Example of worm homogenization. A. Incomplete homogenization. B. Complete homogenization. - Pellet the worm bodies and debris by spinning at 500 x g, 4 °C for 5 min.
- Transfer the supernatant to a new 1.5 ml tube and spin again at 500 x g, 4 °C for 5 min to make sure you get rid of all the worm debris.
- Transfer the supernatant to a new 1.5 ml tube. Save 25-30 µl of this fraction in a new 1.5 ml tube (‘input fraction’) and keep on ice. Write down the total volume of supernatant. Discard the pellet.
- Pellet the nuclei at 4,000 x g, 4 °C for 5 min.
- Transfer the supernatant to a new 1.5 ml tube and centrifuge at maximal speed (17,000 x g), 4 °C for 30 min. Transfer the supernatant to a new 1.5 ml tube; this will contain the ‘cytoplasmic fraction’.
- Wash the pellet from Step B5 with 500 µl ‘complete hypotonic buffer’ (as prepared in Step A5) and centrifuge the samples at 4,000 x g, 4 °C for 5 min.
- Discard the supernatant and add 500 µl of fresh ‘complete hypotonic buffer’ (as prepared in Step A5) to resuspend the nuclear pellet and pipette the suspension into a new 1.5 ml tube. Centrifuge the samples at 4,000 x g, 4 °C for 5 min.
Note: It is important to change tubes after each wash, as tubulin and fatty substances tend to stick to the tube walls, which may contaminate the nuclear fraction. - Prepare ‘complete hypertonic buffer’ by adding 1 M DTT (final concentration: 1 mM DTT) and 25x protease inhibitors (final concentration: 2x protease inhibitor) to the hypertonic buffer.
- Remove the supernatant and dissolve the pellet in a small volume of ‘complete hypertonic buffer’ (as prepared in Step B9) (half of or less the volume that was noted in Step B4). Write down the volume that was used to dissolve the nuclear fraction. Transfer the suspension to a new 1.5 ml tube; this will contain the ‘nuclear fraction’.
- OPTIONAL: For better protein extraction add 25 U/µl of Benzonase nuclease to the ‘nuclear fraction’ and incubate the sample at 4 °C for 45 min on an orbital shaker.
- Determine at this point the protein concentration of the ‘input’ (~20 µg/µl), ‘cytosolic fraction’ (~18 µg/µl) and ‘nuclear fraction’ (~5 µg/µl) with the PierceTM BCA Protein Assay Kit.
- Safe stopping point: Flash freeze samples in liquid nitrogen and store at -80 °C until further use (see Figure 2 for a schematic overview of the fractionation steps).
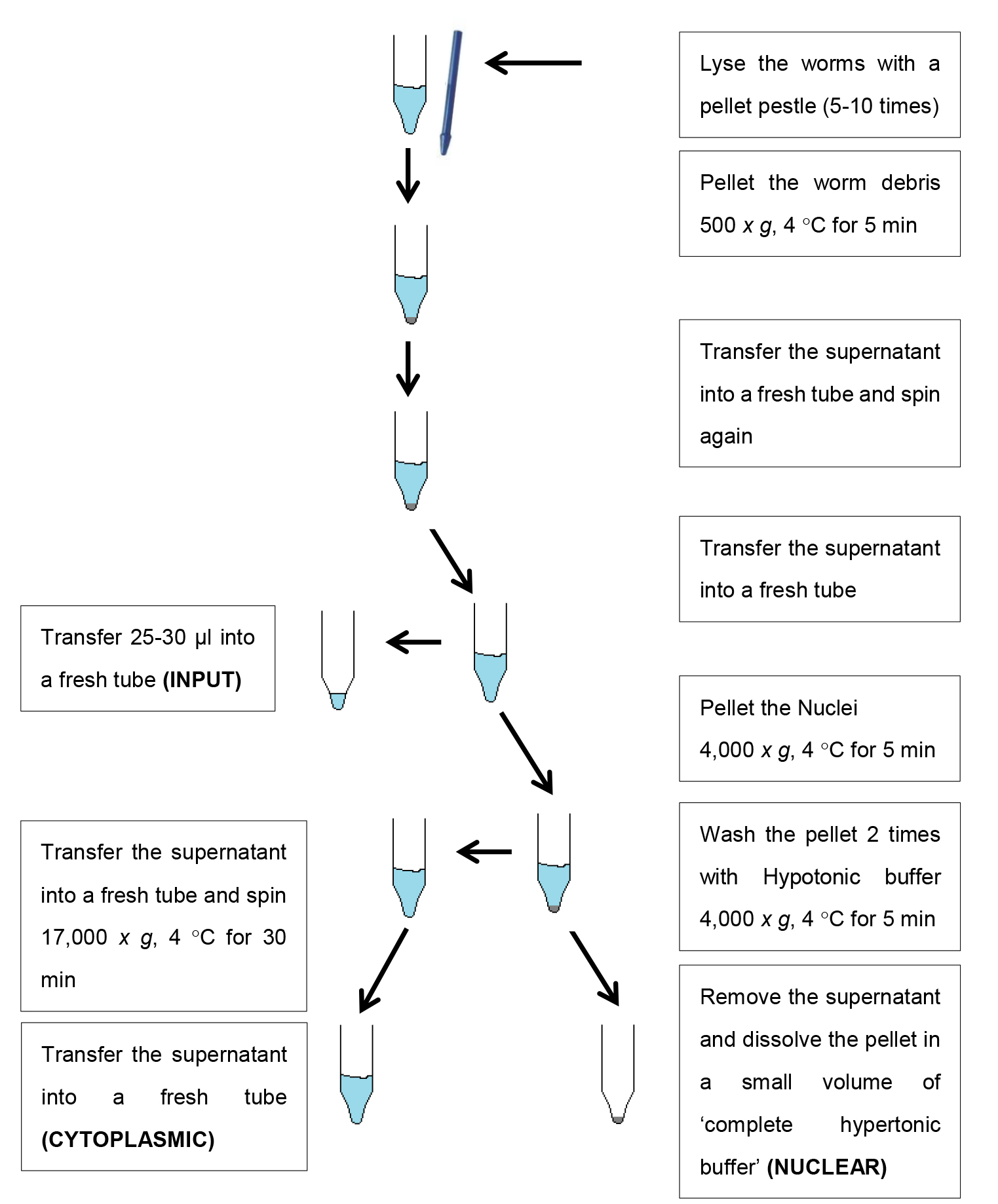
Figure 2. Schematic overview of the fractionation protocol
- Homogenize worms with a pellet pestle for 1 min and let the worms rest for 1 min, repeat this 5 to 10 times. Keep the worms on ice during the whole process. Pipette 5 µl of the worm suspension onto a glass slide after 4-5 times grinding the pellet with the pestle to evaluate the degree of worm lysis. Homogenization is complete when 70%-80% of the worms are disrupted and only a few worm remnants are visible (see Figure 1).
- Western blotting
- Thaw the samples containing the ‘input’, ‘cytosolic fraction’ and ‘nuclear fraction’. Pipette a volume of the samples (either the same amount of protein for all fractions or the same volume) into new 1.5 ml tubes.
Note: When equal volumes are loaded, one can actually compare the amount of specific protein between the different fractions within a sample. When equal protein amounts are loaded, one can look for an enrichment of a protein within specific fractions between conditions or worm strains. Importantly, always write down the volume of the total input fraction and the volume in which the nuclear fraction is dissolved. Enriching the nuclear fraction, i.e., dissolving the pellet in less volume, increases the concentration of proteins, which may be useful during immunoblotting. However, in this situation the signal cannot be compared directly to that of the input, as this fraction is not enriched in the same way. Therefore, corrections based on the described volumes should be performed afterward. - Add ¼ volume of SDS lysis buffer (5x).
- Boil the samples at 95 °C for 10 min.
- Prepare a 12% SDS polyacrylamide gel.
- Load the samples and the protein marker onto the 12% SDS polyacrylamide gel. Fill the tank with running buffer and start run at 100 V until the samples have run through the stacking gel and then continue at 120-140 V until the dye front reaches the bottom of the gel.
- Blot the gel to a nitrocellulose membrane with a wet transfer in the transfer buffer at 95 V for 55-60 min at 4 °C.
- Block the nitrocellulose membrane for 1 h in blocking solution.
OPTIONAL: Cut the nitrocellulose membrane just below the 70 kDa marker (LMN-1 normally runs around 70 kDa and tubulin around 55 kDa). But keep in mind the size of your protein of interest. - Incubate the membrane with the appropriate antibodies for 1 h at room temperature or overnight at 4 °C (see Table 1). LMN-1 is used as a nuclear marker (~70 kDa) and tubulin (~55 kDa) as a cytosolic marker.
- Wash the membranes 3 x 10 min with 1x PBS-T.
- Incubate the membranes with the appropriate HRP-conjugated secondary antibodies for 1 h at room temperature (see Table 1).
- Wash the membranes 3 x 10 min with 1x PBS-T.
- Add the ECL to the membranes and develop the blots with the Las 4000 mini.
OPTIONAL: When not cutting the membranes, one can strip the membranes and re-probe with a different antibody of interest.
- Thaw the samples containing the ‘input’, ‘cytosolic fraction’ and ‘nuclear fraction’. Pipette a volume of the samples (either the same amount of protein for all fractions or the same volume) into new 1.5 ml tubes.
Data analysis
Figure 3 shows an example of an immunoblot. At least three independent experiments should be performed and an example of data analysis can be found in Figures 5 and S5C of Sin et al. (2017).
The fraction levels were quantified by densitometry using ImageJ. A detailed tutorial is available at https://imagej.nih.gov/ij/docs/menus/analyze.html (under “Gels Submenu”) and at http://lukemiller.org/index.php/2010/11/analyzing-gels-and-western-blots-with-image-j/.
To calculate the cytosol/input ratio, the signal of interest can be normalized against the signal of the cytosolic marker (α-tubulin). The nuclear fraction is a concentrated sample, and therefore one should use the input volume and the volume in which the nuclear fraction was resuspended to calculate the “real” value.
Figure 3. Subcellular fractionation of wild type (N2) worms and worms expressing 40 polyglutamine repeats fused to YFP (Q40-YFP, AM141). A representative immunoblot was probed with α-GFP antibody to detect polyglutamine. LMN-1 (nuclear marker) and α-tubulin (cytosolic marker) were used as controls. More details of the experiment can be found in Sin et al. (2017).
Recipes
- M9 buffer (1 L)

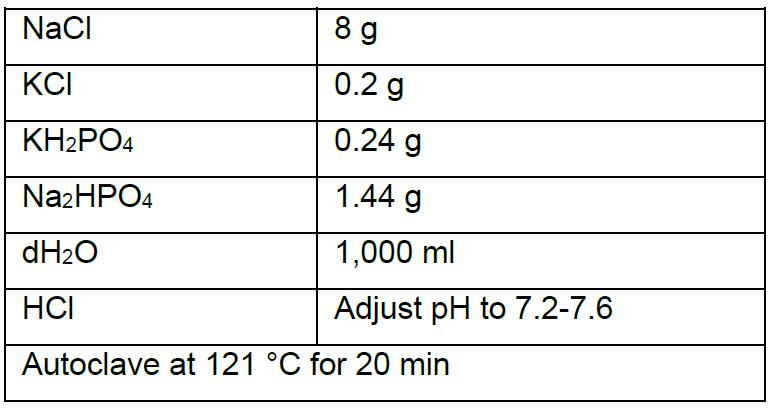
- PBS (1 L)
- PBS-T
PBS containing 0.01% Tween 20 - Blocking solution
5% Milk in PBS-T - NGM plates (1 L)

After cooling add the following buffers:
- Phosphate buffer 1 M pH 6.0 (1 L)

- Hypotonic buffer
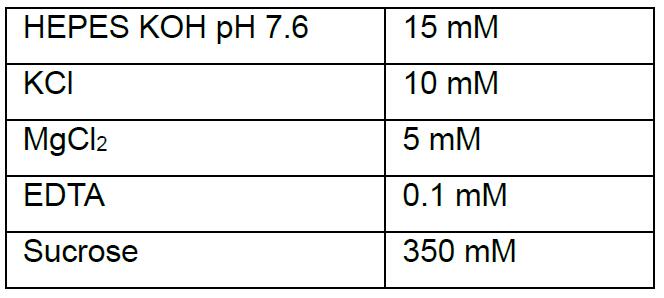
- Hypertonic buffer
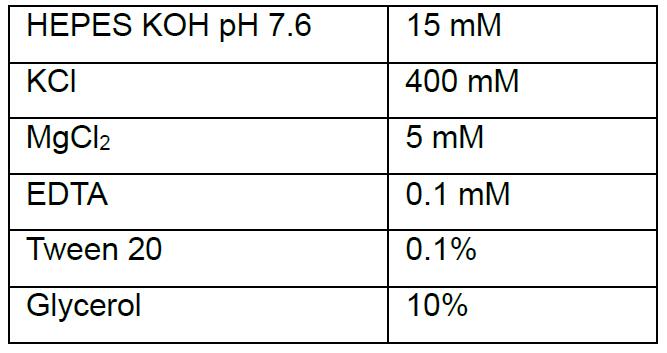
- 25x protease inhibitors
Dissolve 1 tablet Complete in 2 ml dH2O (store at -20 °C) - 1 M DTT
Dissolve 0.154 g DTT in 1 ml dH2O (store at -20 °C) - 12% acrylamide gel (1.0 mm cassette)
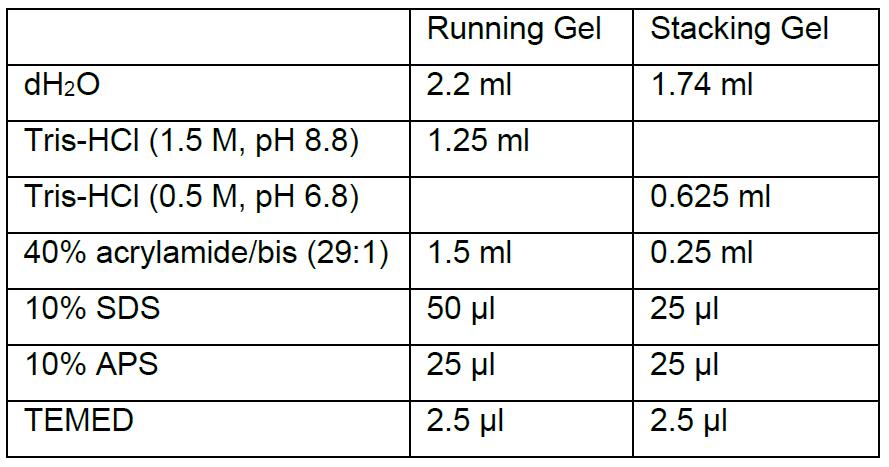
- Running/transfer buffer
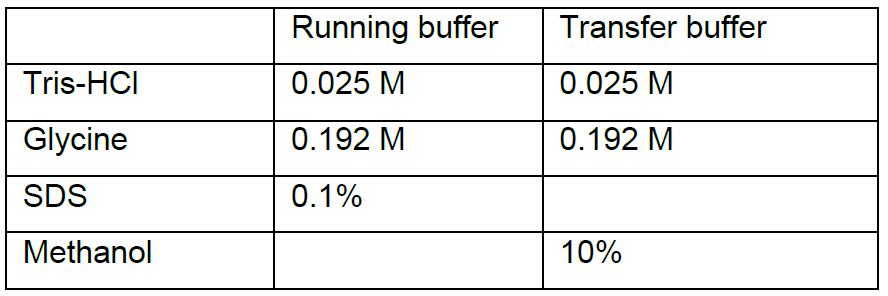
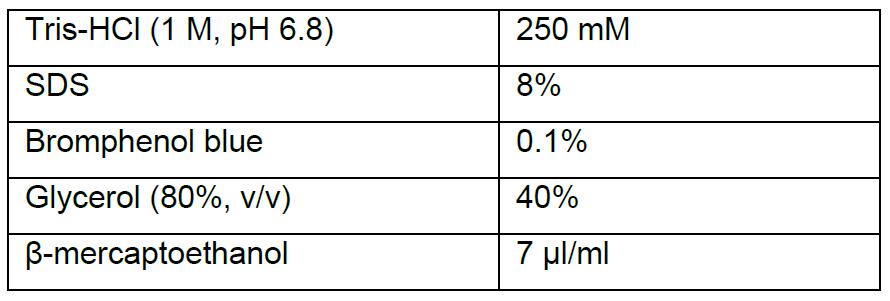
- SDS lysis buffer(5x)
Acknowledgments
We thank the Caenorhabditis Genetics Centre (funded by the National Institutes of Health National Centre for Research Resources) and the Mitani laboratory for the C. elegans strains (funded by the Japan National BioResource Project). This project was funded by a European Research Council (ERC) starting grant (to E.A.A.N.), the Fundação para a Ciência e Tecnologia fellowship (SFRH/BPD/51009/2010) (to O.S.), the Alumni chapter Gooische Groningers facilitated by the Ubbo Emmius Fonds (to E.A.A.N) and a Groningen University Institute for Drug Exploration (GUIDE) fellowship (O.S.). This protocol is adapted from Chen et al. (2000) and La Rocca et al. (2007).
Competing interests
The authors declare no conflicts of interest or competing interests.
References
- Barmada, S. J., Skibinski, G., Korb, E., Rao, E. J., Wu, J. Y. and Finkbeiner, S. (2010). Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci 30(2): 639-649.
- Chen, F., Hersh, B. M., Conradt, B., Zhou, Z., Riemer, D., Gruenbaum, Y. and Horvitz, H. R. (2000). Translocation of C. elegans CED-4 to nuclear membranes during programmed cell death. Science 287(5457): 1485-1489.
- Kontopoulos, E., Parvin, J. D. and Feany, M. B. (2006). Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet 15(20): 3012-3023.
- La Rocca, G., Burgio, G. and Corona, D. F. (2007). A protein nuclear extract from D. melanogaster larval tissues. Fly (Austin) 1(6): 343-345.
- Sin, O., de Jong, T., Mata-Cabana, A., Kudron, M., Zaini, M. A., Aprile, F. A., Seinstra, R. I., Stroo, E., Prins, R. W., Martineau, C. N., Wang, H. H., Hogewerf, W., Steinhof, A., Wanker, E. E., Vendruscolo, M., Calkhoven, C. F., Reinke, V., Guryev, V. and Nollen, E. A. (2017). Identification of an RNA polymerase III regulator linked to disease-associated protein aggregation. Mol Cell 65(6): 1096-1108.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Mata-Cabana, A., Sin, O., Seinstra, R. I. and Nollen, E. A. A. (2018). Nuclear/Cytoplasmic Fractionation of Proteins from Caenorhabditis elegans. Bio-protocol 8(20): e3053. DOI: 10.21769/BioProtoc.3053.
Category
Biochemistry > Protein > Quantification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.