- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Generation of Gene Knockout and Gene Replacement with Complete Removal of Full-length Endogenous Transcript Using CRISPR-Trap


(*contributed equally to this work) Published: Vol 8, Iss 20, Oct 20, 2018 DOI: 10.21769/BioProtoc.3052 Views: 9604
Reviewed by: Renate WeizbauerIndranil MalikMolly Leung
Original research article
The authors used this protocol in:

Jan 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
This protocol describes the application of the CRISPR-Trap from designing of the gene targeting strategy to validation of successfully edited clones that was validated on various human cell lines, among them human induced pluripotent stem cells (hiPSCs). The advantage of CRISPR-Trap over conventional approaches is the complete removal of any endogenous full-length transcript from the target gene. CRISPR-Trap is applicable for any target gene with no or little coding sequence in its first exon. Several human cell lines and different genes have so far been edited successfully with CRISPR-Trap.
Keywords: CRISPRBackground
The advent of CRISPR/Cas9 technology facilitated the genomic targeting for the generation of gene knockouts and gene editing. The conventional method to perform a knockout relies on the introduction of a frameshift leading to premature termination codons (PTCs), truncating the open reading frame (ORF) and subsequent degradation of the transcript of the targeted gene by nonsense-mediated mRNA decay (NMD). A possible pitfall of this approach is full-length transcripts which may escape NMD and give rise to C-terminal truncated proteins harboring residual or even dominant negative functions. This protocol presents the CRISPR-Trap, a method we recently established (Reber et al., 2018), which upon successful editing will prevent the expression of any full-length transcript from the target gene locus (Figure 1). Simply put, this approach targets the first intron of the gene of interest with CRISPR/Cas9. Using homology-directed repair (HDR) a customizable cassette flanked by a strong 3’-splice site and a strong polyadenylation signal is introduced in the first intron, thereby generating an artificial second and effectively last exon. Since transcription is terminated by the introduced polyadenylation signal, only the first endogenous exon and the inserted cassette is transcribed. The customizable cassette can be used to introduce a selection marker, thereby enabling easy selection for at least heterozygous edited clones. If a gene replacement is wanted, the customizable cassette can be used to introduce the replacement gene, followed by an internal ribosomal entry site (IRES) and a selection marker (Reber et al., 2016 and 2018).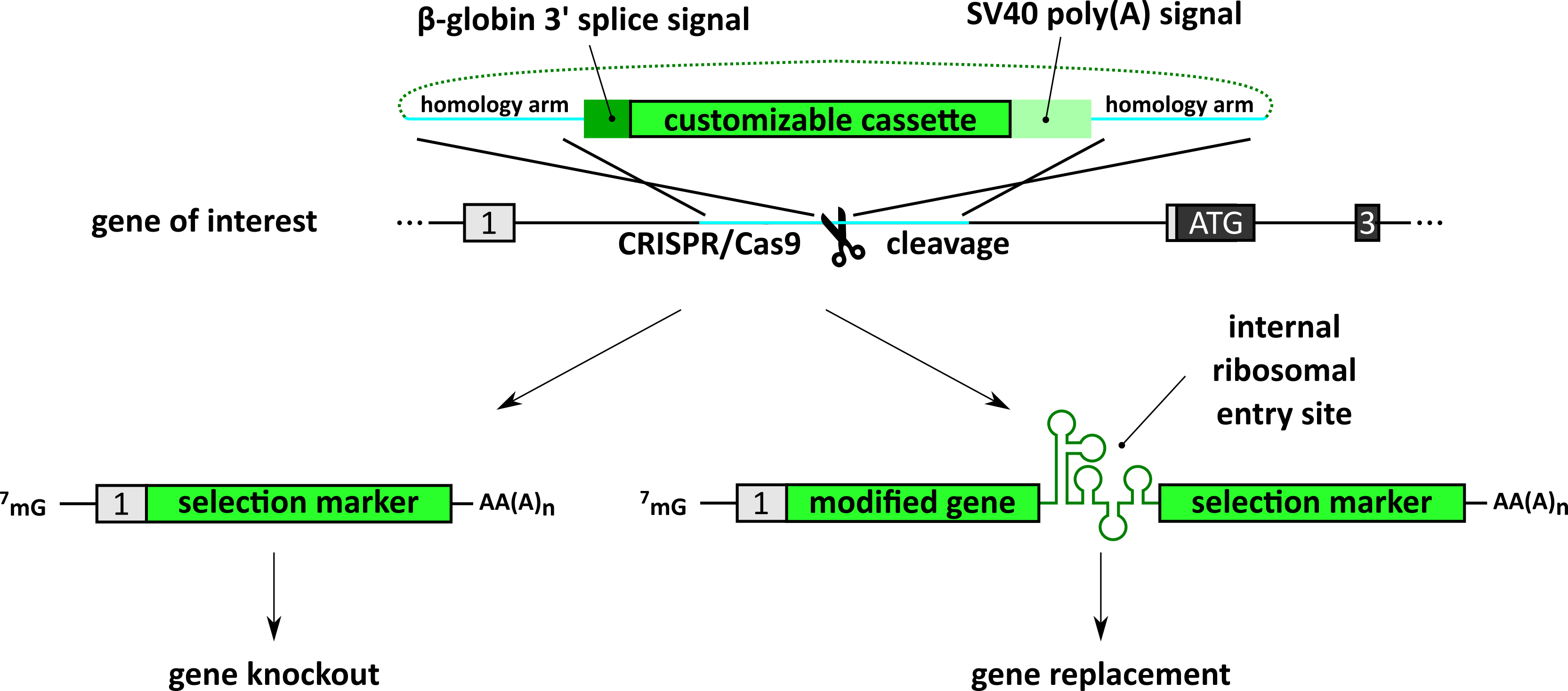
Figure 1. Schematic of the application of the CRISPR-Trap. The first intron of the target gene is cleaved using the CRISPR/Cas9 system and template DNA for homology-directed repair is provided. The template DNA contains a strong 3’ prime splice signal (dark green), a customizable cassette (light green) and a strong polyadenylation signal (turquoise). The customizable cassette can be utilized to either knockout (left) or replace the target gene (right). The cassette contains a selection marker that will be under the control of the endogenous promoter of the target gene upon successful editing. For gene replacements, an IRES is introduced in between the replacement gene and the selection marker. Figure adapted from Reber et al. (2018).
Materials and Reagents
- Pipette tips
- 10 µl sapphire bulk non-sterile pipette tips (Greiner Bio One International, catalog number: 771250 )
- 200 µl polypropylene universal pipette tips with graduation (Greiner Bio One International, catalog number: 739282 )
- 1,250 µl sapphire bulk non-sterile pipette tips (Greiner Bio One International, catalog number: 750250 )
- TPP® tissue culture plates
- TPP cell scraper 20 cm (MIDSCI, catalog number: TP 99010 )
- Cloning cylinder sterile, 3/16’’ ID x 5/16’’ H (SP Scienceware - Bel-Art Products - H-B Instrument, catalog number: 37847-0100 )
- Cell strainer (EASYstrainer, 70 µm) (Greiner Bio One International, catalog number: 542070 )
- Bel-ArtTM SP SciencewareTM BelpenTM black markers (SP Scienceware - Bel-Art Products - H-B Instrument, catalog number: F13374-0000 )
- Plasmid of choice, serving as homology directed repair donor template (donor plasmid)
- Plasmid of choice for Cas9 and sgRNA expression, e.g., pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene, catalog number: 42230 )
- Transformation competent E. coli strain of choice for cloning and plasmid amplification (e.g., XL10 Gold) (Agilent, catalog number: 200314 )
- Transfection reagent of choice
- Cell detachment solutions
- hiPSCs
- Other human cell lines: Trypsin-EDTA (0.05%), phenol red (Thermo Fisher Scientific, catalog number: 25300054 )
- hiPSCs
- To grow hiPSCs as single cells: Y-27632, RHO/ROCK pathway inhibitor (STEMCELL Technologies, catalog number: 72302 )
- ZeocinTM (InvivoGen, catalog number: ant-zn )
- Puromycin dihydrochloride (Santa Cruz Biotechnology, catalog number: sc-108071A )
- PBS (Ca2+, Mg2+ free) (Thermo Fisher Scientific, GibcoTM, catalog number: 10010023 )
- TRIZOL (Sigma-Aldrich, catalog number: T9424 )
- Dow Corning® High-Vacuum silicone Grease (Sigma-Aldrich, catalog number: Z273554 )
- TOPOTM TA CloningTM Kit, Dual Promoter (Thermo Fisher Scientific, catalog number: 450640 )
- Chloroform for analysis EMSURE® ACS, ISO, Reag. Ph Eur (Merck, catalog number: 102445 )
- Absolute EtOH (Sigma-Aldrich, catalog number: 59176 )
- Trisodium citrate (Sigma-Aldrich, catalog number: W302600 )
- 2-propanol for analysis EMSURE® ACS, ISO, Reag. Ph Eur (Merck, catalog number: 109634 )
- RNase free glycogen (Thermo Fisher Scientific, catalog number: R0551 )
- DNA extraction kit (Quick-DNATM miniprep Kit) (ZYMO RESEARCH, catalog number: D3025 )
- NaOH (Sigma-Aldrich, catalog number: 306576 )
- Maximo Taq DNA Polymerase 2X-preMix/PCR Master Mix (GeneON, catalog number: S113 )
- KAPA Taq ReadyMix PCR Kit (KAPA Biosystems, catalog number: KK1006)
Equipment
- Pipettes
- Mammalian culture equipment
- Heat block (VWR, catalog number: 444-0938 )
- Thermocycler (VWR, catalog number: 732-2551 )
- NanoDrop (Thermo Fisher Scientific, model: NanoDropTM 2000 , catalog number: ND-2000)
- Cooling Centrifuge (Eppendorf, model: 5424 R , catalog number: 5404000010)
- Wide-field microscope
- Vortex (Scientific Industries, catalog number: SI-0266 )
Software
- Clone manager (http://www.scied.com/pr_cmpro.htm) or other cloning software
Procedure
Notes:
- One has to be aware that the first exon of the target gene will still be expressed following the CRISPR-Trap strategy. Therefore, the ideal target gene has no or only few amino acids coded in its first exon.
- To allow direct selection for genome edited cells, the gene has to be expressed in the cells in which the editing is performed.
- As every target behaves differently depending on the specific gene and cell line one might have to adjust various parameters.
- Design of sgRNAs and donor plasmids
sgRNA Design
Identify a target site within the first intron of your target gene and generate a suitable expression plasmid (hereafter called pCas9/sgRNA)- Most CRISPR/Cas9 expression plasmids use a CMV promoter for the expression of the Cas9 endonuclease. In our hands, this promoter resulted in low Cas9 expression levels in human stem cells. While, this still resulted in successful generation of CRISPR-trap knockouts (Reber et al., 2018) replacement of the CMV promoter with an EF1α promoter could be considered, for specific cell lines in which the CMV promoter displays low activity.
- For the intronic sgRNA target site:
- Make sure to leave a spacer from the 3’ end of the first exon not to disrupt intronic splicing enhancers, which are typically located within the first 100 nucleotides downstream of the 5’ splice site (Aznarez et al., 2008).
- Introduce the cassette upstream of the intronic branch point to ensure proper splicing to the first exon of the target gene. Branch points are typically located < 50 nucleotides upstream of the 3’ splice site, with 90% of all human branch points located 37 nucleotides upstream of the 3’ splice site (Mercer et al., 2015). However, in certain human genes, the branch point can be located further upstream. Hence, we recommend the use of a splice site predictor such as the Human Splicing Finder available at http://www.umd.be/HSF3/ to identify the ideal target region.
- If possible, find two or more adjacent sgRNA sequences that can trigger HDR with the same donor plasmid, thereby enhancing the success rate.
- Option 1: For many genes, pre-designed and validated sgRNAs in suitable expression plasmids as well as custom sgRNA design are commercially available (e.g., CRISPR/Cas Nuclease RNA-guided Genome Editing from Sigma-Aldrich available at https://www.sigmaaldrich.com/technical-documents/articles/biology/crispr-cas9-genome-editing.html#purified or from Addgene available at https://www.addgene.org/crispr/)
- Option 2: Use a dedicated online-tool (we typically used the CRISPR Design Tool by the Feng Zhang group available at http://crispr.mit.edu/) to identify CRISPR/Cas9 target sequences within the first intron of the gene of interest. Subsequently, clone the guide sequence into a suitable CRISPR/Cas9 expression plasmid (e.g., pX330-U6-Chimeric_BB-CBh-hSpCas9, Addgene Plasmid # 42230 ). Follow the Zhang Lab General Cloning protocol available at https://www.addgene.org/crispr/zhang/.
Donor plasmid design
Generate donor plasmid containing the customizable cassette either by cloning or by gene synthesis into a plasmid of choice. This donor plasmid is subsequently called pHDR-Don. The donor plasmids used in (Reber et al., 2018) for knockouts and gene replacements (pHDR-FUS KO-Zeo: generation of FUS knockouts & pHDR-TDP43 repl-Puro: gene replacement of TDP-43 with DD-TDP43) are available as Genbank files. Furthermore, pHDR-Don example constructs for gene knockouts are provided for genes with and without coding sequence in the first exon: pHDR-Zeo-Don for genes with a non-coding first exon, pHDR-Zeo-Don_f1 for genes where the splice site after the first exon is located directly after a codon, pHDR-Zeo-Don_f2 for genes where the splice site is located after 1 extra nucleotide following the last codon, and pHDR-Zeo-Don_f3 for genes where the splice site is located after 2 extra nucleotides following the last codon. Furthermore, an example construct for gene replacement with a non-coding first exon is provided (pHDR-IRES-Puro-Don).- We used Zeocin and puromycin as selection markers, however, depending on the targeted cell line alternative resistance markers such as blasticidin could be beneficial.
In case that the endogenous gene contains some coding sequence in its first exon, make sure that the resistance marker/gene replacement will be spliced in-frame. - Cas9 cuts 3 nucleotides upstream of the PAM motif. Homology arms 5’ and 3’ of the customizable cassette should therefore be identical to the sequences upstream (5’) and downstream (3’) of the Cas9 cleavage site. To allow efficient homology-directed repair mediated by pHDR-Don, we typically use homology arms with a length of 700-800 nucleotides (per homology arm) (see Figure 2).
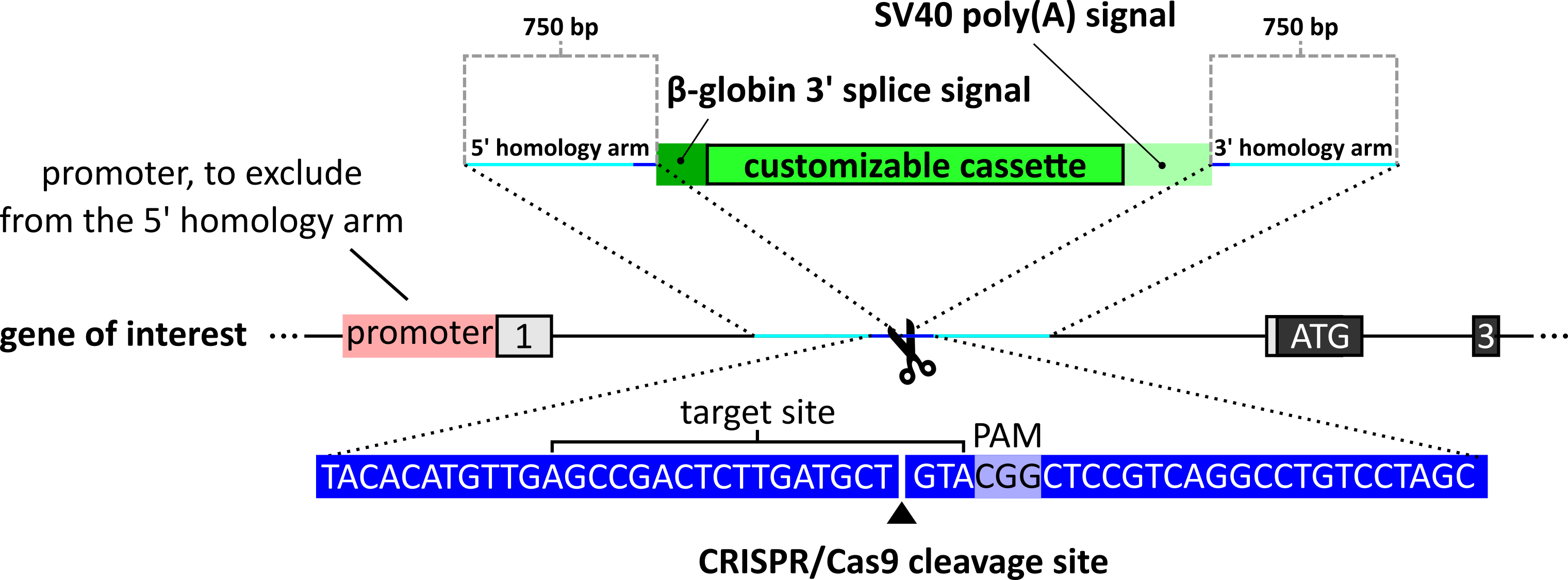
Figure 2. Schematic of the pHDR-Don design. Cas9 cuts 3 nucleotide positions upstream of the PAM motif. The homology arms should be identical to the sequences upstream (5’ homology) and downstream (3’ homology) of the cleavage site. Each homology arm should be approximately 750 base pairs long to ensure efficient HDR. In case of short introns, the 750 base pairs of the 5’ homology might contain the first exon and parts of the promoter region of the gene of interest. In that case, make sure not to include promotor sequences in your 5’ homology arm to prevent expression of the cassette directly from the plasmid. - The upstream (5’) homology arm has to be shortened, if it would contain the promoter sequence of the targeted gene. Otherwise, the cassette will be expressed directly from the pHDR-Don plasmid.
- Transfect cells
- Grow cells in conditions suitable for the respective cell line (growth medium, growth surface, growth conditions). These conditions can be maintained during the whole protocol. Seed cells the day before transfection in all wells of a 6-well plate. In order to seed the cells follow these steps:
- Aspirate medium from the plate containing the cells.
- Wash the cells once by adding a sufficient amount of PBS to cover the surface of the plate by gently swirling. Subsequently aspirate the PBS.
- Add the appropriate reagent to detach the cells (e.g., Trypsin, Accutase) and wait until the cells detach.
- Resuspend the cells in appropriate medium and count the cells (e.g., by using a hemocytometer).
- Transfer the desired amount of cells into the wells. The optimal amount of cells per well will vary by the cell line and the transfection reagent used. Follow the transfection reagent manufacturer’s protocol for an optimal transfection efficiency using a brightfield microscope to estimate density.
- Use the appropriate transfection reagent for your cell line. In our hands, the most efficient transfection reagents were DogTor for HeLa cells, Mirus TransIT®-LT1 for HT1080, and human induced pluripotent stem cells (hiPSCs) and LipofectamineTM 3000 for SH-SY5Y neuroblastoma cells. Transfect each well with pCas9/sgRNA and pHDR-Don. Table 1 gives empirical values for successful edited cell lines in our hands (values each for the transfection of one well of a 6-well plate).
Table 1. Empirical values for amounts of transfected plasmids. Empirical values for transfected pCas9/sgRNA and pHDR-Don plasmids in successful editings of various cell lines.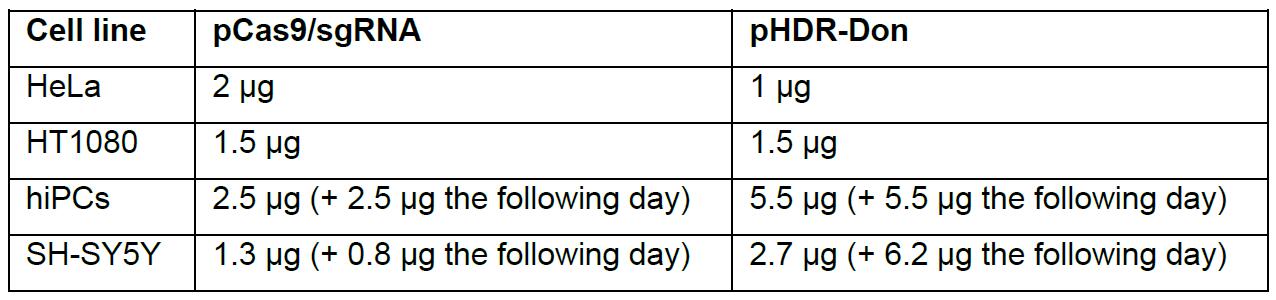
Note: When sgRNAs for two adjacent targets were cloned, transfect 3 wells with one sgRNA and the other 3 wells with the other sgRNA. The 6-wells can also be used to vary either the total amount of transfected plasmids or the ratio between pCas9/sgRNA and pHDR-Don. A good starting point is to try molarity ratios of pCas9/sgRNA:pHDR-Don of 1:6 to 2:1. If 6-wells are not sufficient for all variation, more wells can be transfected. Since CRISPR-Trap relies on homology-directed repair, small molecules increasing the likelihood of these events such as L755507 (Yu et al., 2015; Li et al., 2017) can be added at this stage. - Two to four days after transfection, detach cells using the appropriate cell detachment solution and pool the cells from all wells in one 15 cm dish. Let cells attach, and then start with selection using the selection marker introduced with the cassette. Table 2 gives empirical values for successful edited cell lines in our hands.
Table 2. Empirical values for antibiotic selection. Empirical values for antibiotic concentrations and duration of selection in successful editings of various cell lines.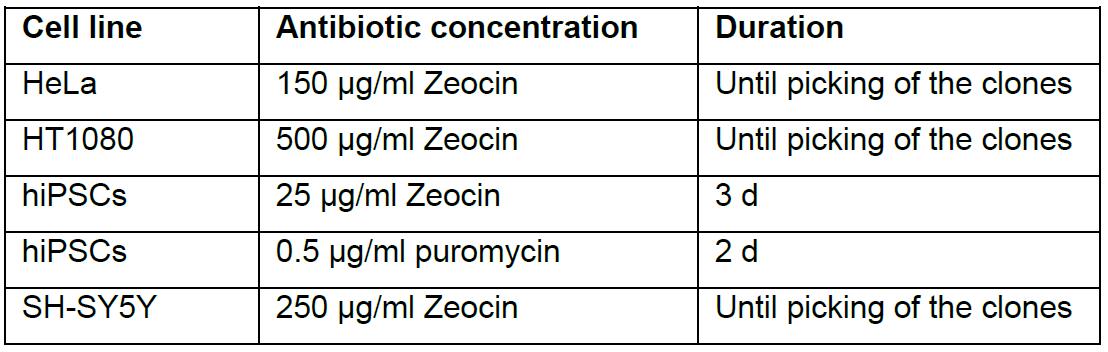
- Grow cells in conditions suitable for the respective cell line (growth medium, growth surface, growth conditions). These conditions can be maintained during the whole protocol. Seed cells the day before transfection in all wells of a 6-well plate. In order to seed the cells follow these steps:
- Pick selected clones
It is best to pick colonies while looking through a microscope, as this gives better control and reduces the risk of mixing multiple colonies. Here, we describe a method that allows manual picking of single colonies when picking under a microscope under sterile conditions is not possible. If a microscope for manual picking under sterile conditions is not available, follow this protocol. Otherwise, pick colonies directly under the microscope under sterile conditions and continue from Step C10.- Upon finishing the selection process, allow single cells to grow into colonies. It is important however to not let the colonies grow into to close proximity of one another, to ensure that each colony picked grew from one single cell. If the colonies grow to close to each other, split the cells to a new plate (use a cell strainer to ensure new colonies emerge from single cells).
Note: As rule of thumb, pick colonies consisting of at least 10 cells and 1 cm of space from the edge to the edge of the next colonies. - Count and note the number of colonies which are good to pick (i.e., consisting of at least 10 cells and 1 cm of space from the edge to the edge of the next colonies). Use a pen to mark all colonies which are good to pick by circling them on the underside of the 15 cm plate (see Figure 3).
Note: If two colonies are too close to each other, forego to pick them to avoid picking of mixed colonies. - Apply vacuum grease on a fresh sterile dish and use a sterile cell scraper to generate a grease film of approximately 5 mm thickness (Figures 3A and 3B).
- Prepare cloning cylinders in the number assessed in Step C2. Put cloning cylinders with their narrow side onto the grease. Try to avoid any horizontal movement of the cloning cylinders once they are sticking to the grease (Figure 3C).
- Sterilize 20 min under UV light.
- Aspirate medium from the 15 cm plate containing cells and wash once with PBS by adding 10 ml of PBS, distributing the PBS among the plate by gently swirling and subsequently aspirating the PBS.
- Remove cloning cylinders one by one from the grease film and put them on the colonies with the greased side facing the plate. If you are picking without concurrent usage of a microscope, use the marks from Step C2 as a guide to where to put the cloning cylinders (Figure 3D).
Note: Apply a little pressure for a watertight sealing.
Figure 3. Preparation of vacuum grease and cloning cylinders for manual clone picking. A. Apply grease on sterile dish. B. Generate an approximately 5 mm thick grease film using a sterile cell scraper. C. Put cloning cylinders with their narrow side onto the grease. D. Put the cloning cylinders with the greased side first onto the colonies. - Add 20 μl of the appropriate reagent to detach the cells (e.g., Trypsin, Accutase) into each cloning cylinder and wait until the cells detach.
- As soon as the cells detach, add 80 μl of appropriate medium into each cloning cylinder and directly transfer all liquid into a separate well of a 24-well plate for each colony. Subsequently top up medium in each well to 500 μl.
- Let the colonies grow over the next couple of days in the 24-well plate and transfer the cells into a well of a 6-well plate as soon as they reach confluence.
Note: It is also possible to harvest clones directly from 24-wells following the same protocol from Procedure D.
- Upon finishing the selection process, allow single cells to grow into colonies. It is important however to not let the colonies grow into to close proximity of one another, to ensure that each colony picked grew from one single cell. If the colonies grow to close to each other, split the cells to a new plate (use a cell strainer to ensure new colonies emerge from single cells).
- Validation of successful edited clones
- As soon as the clones reach confluence on the 6-well plates (or 24-well plates), detach the cells and split them into two parts. Freeze half the cells to continue with in case of a successful genome editing, while extracting the DNA and RNA from the other half to assess the genotype. Hereto we re-suspend the cells in TRIZOL to isolate gDNA as well as RNA.
Note: A DNA extraction kit can be used as an alternative to the extraction using TRIZOL. The advantage of the TRIZOL extraction, however, is the possibility to simultaneously extract RNA. Using RT-qPCR allows for a quick check if any RNA from the target gene downstream from the introduced gene trap is expressed. Low traces of such RNA can indicate a mixed population of cells (picked clone originates not from a single cell).
Furthermore, samples in TRIZOL can be stored for long-term at -20 °C allowing batch analysis of a high number of clones simultaneously. - Isolation of gDNA from TRIZOL (protocol modified from the manufacturer's protocol)
- Add 0.5 ml TRIZOL per well (6-well or 24-well) and pipette up and down in order to lyse the cells.
- Transfer the cell lysate into a 1.5 ml Eppendorf tube.
- Add 0.1 ml chloroform, close the lid tightly and shake vigorously for 15 sec.
- Incubate for 2-15 min at RT.
- Centrifuge for 15 min at 12,000 x g at 2-8 °C.
- Remove upper phase (contains RNA, store at -20 °C).
- Add 0.3 ml 100% EtOH and 1.5 μl glycogen, mix by inversion.
Note: Adding the glycogen is essential if you work with a low amount of cells–without it, you won’t see a pellet in the later steps. - Incubate for 2-3 min at RT.
- Centrifuge for 5 min at 2,000 x g at 2-8 °C.
- Remove supernatant.
- Wash DNA solution twice:
- Add 1 ml 0.1 M trisodium citrate, 10% EtOH.
- Incubate (with occasional mixing, inversion) for at least 30 min at RT.
- Centrifuge for 5 min at 2,000 x g at 2-8 °C.
- Remove supernatant.
Note: The DNA pellet is typically not attaching strongly to the tube, be careful not to aspirate it along with the supernatant; better leave a small amount of supernatant.
- Resuspend pellet in 1 ml 75% EtOH.
- Incubate for 10-20 min at RT.
- Centrifuge for 5 min at 2,000 x g at 2-8 °C.
- Air-dry pellet by opening the lid of the Eppendorf and placing it with the opening facing down on a clean tissue at room temperature for 15 min.
- Dissolve the pellet in 50-100 μl 8 mM NaOH with repeated pipetting.
- Use a Spectrophotometer (e.g., NanoDrop) to assess DNA concentration.
Note: DNA isolated from a low amount of cells following this protocol usually results in a high ethanol/phenol peak measuring DNA concentration using a Spectrophotometer (e.g., NanoDrop). These contaminants however did not interfere with downstream PCR reactions in our hands (using Maximo Taq DNA Polymerase 2x-preMix [ S113 , GeneON] or KAPA Taq ReadyMix [KK1006, KAPA Biosystems]). - Store at -20 °C.
- Isolation of RNA from TRIZOL (Protocol modified from the manufacturer's protocol)
- Continue with upper phase from Step D2f from DNA isolation.
- Add 600 μl isopropanol + 1.5 μl glycogen, invert to mix multiple times.
- Spin for 30 min 16,000 x g 4 °C.
- Wash pellet twice in 70% EtOH.
- Vortex.
- 15 min at 7,500 x g, 4 °C.
- Remove supernatant.
- Air-dry pellet by opening the lid of the Eppendorf and placing it with the opening facing down on a clean tissue at room temperature for 10 min.
- Resuspend in 100 μl RNase free water.
- Use a Spectrophotometer (e.g., NanoDrop) to assess RNA concentration.
- Store at -80 °C.
- Use PCR to amplify the purified DNA. Use PCR product for Sanger sequencing
Note: If possible, use primers annealing outside the homology arms of the donor plasmid to avoid amplifying possible traces of pHDR-Don plasmids. - If the reads are ambiguous/not clean (e.g., overlap of multiple signals in the sequencing chromatogram after a certain point in the sequence), use TOPO-TA cloning to assess if you have a heterozygous clone or a mixed population. In case of a heterozygous clone, the TOPO-TA cloning will yield E.coli colonies containing the allelic sequences in a 1:1 ratio.
- As soon as the clones reach confluence on the 6-well plates (or 24-well plates), detach the cells and split them into two parts. Freeze half the cells to continue with in case of a successful genome editing, while extracting the DNA and RNA from the other half to assess the genotype. Hereto we re-suspend the cells in TRIZOL to isolate gDNA as well as RNA.
Notes
Homozygous clones should then finally be analyzed by Western Blotting to demonstrate the absence of the targeted endogenous protein. An example for cell harvesting, extract preparation and immunoblotting is described in the original CRISPR-trap publication (Reber et al., 2018). In our hands, CRISPR-trap yielded no homozygous clones when targeting essential genes (lethal knockout). In this case, a gene replacement with a degron-tagged cDNA (see pHDR-TDP43 repl-Puro as example) should be considered to allow an inducible depletion of the protein of interest.
The success rate of CRISPR-trap will depend on cell line and target gene. It is therefore difficult to predict the number of clones that need to be sequenced before a homozygous clone is detected. In our hands, the success rate for homozygous clones ranged from 10 % to 90 % of the analyzed clones.
Acknowledgments
This work was made possible by the support of the NOMIS Foundation, the National Centre of Competence in Research (NCCR) RNA & Disease funded by the Swiss National Science Foundation, and the support of the UK Dementia Research Institute. The protocol is based on the publications (Reber et al., 2016 and 2018) in The EMBO Journal and Molecular Biology of the Cell.
Competing interests
The authors declare that they have no competing or conflicting interests.
References
- Aznarez, I., Barash, Y., Shai, O., He, D., Zielenski, J., Tsui, L. C., Parkinson, J., Frey, B. J., Rommens, J. M. and Blencowe, B. J. (2008). A systematic analysis of intronic sequences downstream of 5' splice sites reveals a widespread role for U-rich motifs and TIA1/TIAL1 proteins in alternative splicing regulation. Genome Res 18(8): 1247-1258.
- Li, G., Zhang, X., Zhong, C., Mo, J., Quan, R., Yang, J., Liu, D., Li, Z., Yang, H. and Wu, Z. (2017). Small molecules enhance CRISPR/Cas9-mediated homology-directed genome editing in primary cells. Sci Rep 7(1): 8943.
- Mercer, T. R., Clark, M. B., Andersen, S. B., Brunck, M. E., Haerty, W., Crawford, J., Taft, R. J., Nielsen, L. K., Dinger, M. E. and Mattick, J. S. (2015). Genome-wide discovery of human splicing branchpoints. Genome Res 25(2): 290-303.
- Reber, S., Stettler, J., Filosa, G., Colombo, M., Jutzi, D., Lenzken, S. C., Schweingruber, C., Bruggmann, R., Bachi, A., Barabino, S. M., Muhlemann, O. and Ruepp, M. D. (2016). Minor intron splicing is regulated by FUS and affected by ALS-associated FUS mutants. EMBO J 35(14): 1504-1521.
- Reber, S., Mechtersheimer, J., Nasif, S., Benitez, J. A., Colombo, M., Domanski, M., Jutzi, D., Hedlund, E. and Ruepp, M. D. (2018). CRISPR-Trap: a clean approach for the generation of gene knockouts and gene replacements in human cells. Mol Biol Cell 29(2): 75-83.
- Yu, C., Liu, Y., Ma, T., Liu, K., Xu, S., Zhang, Y., Liu, H., La Russa, M., Xie, M., Ding, S. and Qi, L. S. (2015). Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 16(2): 142-147.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Mechtersheimer, J., Reber, S. and Ruepp, M. (2018). Generation of Gene Knockout and Gene Replacement with Complete Removal of Full-length Endogenous Transcript Using CRISPR-Trap. Bio-protocol 8(20): e3052. DOI: 10.21769/BioProtoc.3052.
Category
Molecular Biology > DNA > Mutagenesis
Molecular Biology > DNA > Gene expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.