- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Filter Retardation Assay for Detecting and Quantifying Polyglutamine Aggregates Using Caenorhabditis elegans Lysates

Published: Vol 8, Iss 19, Oct 5, 2018 DOI: 10.21769/BioProtoc.3042 Views: 9335
Reviewed by: Saumik BasuManoj B. MenonAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Protein aggregation is a hallmark of several neurodegenerative diseases and is associated with impaired protein homeostasis. This imbalance is caused by the loss of the protein’s native conformation, which ultimately results in its aggregation or abnormal localization within the cell. Using a C. elegans model of polyglutamine diseases, we describe in detail the filter retardation assay, a method that captures protein aggregates in a cellulose acetate membrane and allows its detection and quantification by immunoblotting.
Keywords: Protein aggregationBackground
One pathological feature of neurodegenerative diseases like Parkinson’s, Alzheimer’s and polyglutamine diseases is the presence of protein aggregates in distinct areas of the brain (reviewed in Soto, 2003; Stroo et al., 2017). In the case of polyglutamine diseases, abnormal expansion of glutamine (CAG) repeats in the coding sequence disturbs the native folding of the protein. As a result, the misfolded protein exposes regions of its amino acid sequence, which makes it prone to aggregate with other proteins, forming large, insoluble aggregates that can hamper normal cellular function (reviewed in Kuiper et al., 2017).
Several methods have been developed for the detection of insoluble protein aggregates including, for instance, dye binding assays (e.g., Thioflavin T, Congo red, NIAD-4) and electron microscopy. Filter retardation assay is a quick and sensitive method that detects and quantifies protein aggregates formed in vivo and in vitro, including polyglutamine (Scherzinger et al., 1997; Wanker et al., 1999), alpha-synuclein (Recasens et al., 2018), and amyloid-beta aggregates (Bieschke et al., 2009). In this assay, sodium dodecyl sulfate (SDS)-resistant protein aggregates are filtered and retained in a cellulose acetate membrane, while monomeric intermediate species are not captured. The protein aggregates retained in the membrane are subsequently detected by antibodies, which allows for their quantification.
This protocol describes a method to detect and quantify SDS-resistant polyglutamine aggregates in the nematode Caenorhabditis elegans and can be applied to investigate the aggregation of aggregation-prone proteins in vivo and in vitro.
Materials and Reagents
- 200 µl pipette tips (Greiner Bio-one, catalog number: 741065 )
- 1000 µl pipette tips (Greiner Bio-one, catalog number: 741045 )
- 15 ml conical tubes (SARSTEDT, catalog number: 62.554.502 )
- 2 ml Screw cap tubes (SARSTEDT, catalog number: 72.693.105 )
- 0.5 mm Glass beads (Carl Roth, catalog number: N030.1 )
- 11.3 x 7.7 cm Bio-Dot/Bio-Dot SF Filter Paper (Bio-Rad Laboratories, catalog number: 1620161 )
- Cellulose acetate membranes, 0.22 micron, Sphaero (Sterlitech, catalog number: CA023001 )
- 1.5 ml microfuge tube (Greiner Bio One International, catalog number: 616201 )
- Rubber seal
- Plastic wrap
- 50 ml conical tube (SARSTEDT, catalog number: 62.547.004 )
- Worms
- PierceTM BCA Protein Assay Kit (Thermo Fisher Scientific, catalog number: 23227 )
- Complete protease inhibitors (Roche Diagnostics, catalog number: 11697498001 )
- ECLTM Prime Western Blotting Detection (GE Healthcare, Amersham, catalog number: RPN2232 )
- Liquid nitrogen
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 )
- DL-Dithiothreitol (DTT) (Sigma-Aldrich, catalog number: D0632 )
- 1 M Tris-HCl
- 5 M NaCl
- 10% SDS
- Milli-Q water
- Non-fat dry milk powder (Campina)
- Antibodies against green fluorescent protein (GFP)/yellow fluorescent protein (YFP) and tubulin (Table 1)
Table 1. List of primary and secondary antibodiesPrimary antibody Host Company/catalog number Dilution primary antibody Secondary antibody α-GFP Mouse TaKaRa Bio,Clontech Laboratories ( 632381 )1:5,000 α-tubulin Mouse Sigma-Aldrich ( T6074 ) 1:5,000 Anti-mouse (1:10,000)
Bio-Rad ( 1706516 ) - Sodium phosphate dibasic heptahydrate (Na2HPO4•7H2O) (Acros Organics, catalog number: 424380010 )
- Sodium chloride (NaCl) (Merck, catalog number: 1064041000 )
- Potassium chloride (KCl) (Fisher Chemicals, catalog number: 10010310 )
- 10x Phosphate buffered saline (PBS) (see Recipes)
- 1x PBS-Triton 0.1% (see Recipes)
- 1x PBS-Triton 0.05% (see Recipes)
- Filter trap assay (FTA) sample buffer (see Recipes)
- FTA wash buffer (see Recipes)
- 1 M dithiothreitol (DTT, see Recipes)
- Blocking solution (see Recipes)
Equipment
- P20 Pipetman (Gilson, catalog number: F123600 )
- P200 Pipetman (Gilson, catalog number: F123601 )
- P1000 Pipetman (Gilson, catalog number: F123602 )
- 30-300 μl multi-channel pipette (Eppendorf, catalog number: 3125000052 )
- 10 ml Serological glass pipettes (SARSTEDT, catalog number: 86.1254.001 )
- Centrifuge (Thermo Fisher Scientific, model: SL 40R , rotor: TX-750 )
- Bead beater (MP Biomedicals, model: FastPrep-24 )
- 48-well Bio-Dot Microfiltration System (Bio-Rad Laboratories, catalog number: 1703938 )
- Vacuum pump
- 20 °C incubator (LIEBHERR, model: WK 4126 )
- Vortex (Fisher scientific, model: ZX Wizard vortex, catalog number: 11746744 )
- Fuji Super RX-N 13x18 film (Fujifilm, catalog number: RX1318 )
- Autoclave
Software
- ImageJ (Open source: https://imagej.nih.gov/ij/)
- Microsoft Excel (Microsoft Corporation, Redmond, USA)
Procedure
- Sample preparation
- Prepare 10-15 plates with approximately 800-1,000 synchronized L4 worms per 9 cm NGM plate.
- Wash worms off from plates with 8-10 ml 1x PBS using a 10 ml serological glass pipette and collect worms into a 15 ml conical tube.
- Centrifuge worms at 1,000 x g for 2 min at room temperature. Discard supernatant without disturbing the worm pellet.
- Repeat Step A2 (washing with 1x PBS) a total of three times or until supernatant is clear. Remove supernatant and leave 1.5-2.0 ml (or just enough volume to cover the worm pellet).
- Pre-wet a P1000 blue tip by pipetting PBS with 0.05% Triton X-100 “up-and-down”. This helps to decrease worms sticking to the wall of the plastic tip.
- Use this P1000 blue tip and transfer worms to one 2 ml screw cap tube and briefly centrifuge at 1,000 x g for 2 min to sediment the worms. Discard supernatant.
- Wash worms once with cold 500 μl FTA wash buffer.
- Add cold FTA sample buffer to the worm pellet in a 1:1 ratio (e.g., 10-15 plates yield about 250 μl of worm pellet).
- Add 0.5 mm glass beads in a 2:1 ratio by volume.
- Bead beat samples for 7 cycles of 20 sec at 4 m/sec with 5 min rest in between cycles. Keep the samples on ice whenever possible.
Note: This protocol is performed using a bead beater at room temperature. Therefore, it is important to keep the samples on ice whenever possible (e.g., in between the 5 min rest cycles). - Transfer supernatant to a 1.5 ml microfuge tube.
Safe stopping point: Flash freeze samples in liquid nitrogen and store at -80 °C until further use. - Measure protein concentration by using the PierceTM BCA Protein Assay Kit.
Note: For 10-15 plates of synchronized L4 worms, we find that the protein yield varies between 2-5 mg/ml. This variability will depend on factors such as the efficiency of the worm synchronization by bleaching (therefore the number of worms per plate to start with), how well the worms were washed off of the plates and transferred to microfuge tubes, etc. - Transfer 100 μg of total protein to a 1.5 ml microfuge tube and add cold FTA sample buffer to a final volume of 400 μl (final concentration 100 μg/400 μl). For loading control, take 1-5 μg of sample to run separately by a 10 or 12% SDS–polyacrylamide gel electrophoresis (SDS-PAGE).
- Add 20 μl 1 M DTT.
- Boil samples at 98 °C for 5 min.
- Prepare two dilutions for each sample in new 1.5 ml microfuge tubes:

- Setting up the Bio-Dot microfiltration system
- Soak two 11.3 x 7.7 cm Bio-Dot/Bio-Dot SF filter papers and one cellulose acetate membrane (cut to similar size) in about 25 ml of FTA wash buffer.
- Place these on the base of the Bio-Dot Microfiltration System, with the cellulose acetate membrane on top of the filter papers.
- Roll out any air bubbles using a glass pipette.
- Place the rubber seal and lid onto the Bio-Dot Microfiltration System, insert and tighten all four screws diagonally so the pressure is evenly spread (Figure 1).

Figure 1. Bio-Dot Microfiltration System with rubber seal and diagonal tightening of the screws - Apply vacuum and tighten the screws further. Turn off the vacuum.
- Using a multi-channel pipette, wash each well of the Bio-Dot Microfiltration System with 100 μl FTA wash buffer and apply full vacuum until the wells are empty.
- Repeat this step a total of three times.
- While the vacuum is turned off, turn the T-shaped connector 180° and place a P1000 blue tip on it to apply low vacuum (Figure 2).

Figure 2. Schematic representation of how to set up the Bio-Dot Microfiltration System to low vacuum
- Load 100 μl of the undiluted, 5-fold and 25-fold diluted samples to adjacent wells while placing the pipette tip to the side of the well to avoid air bubbles.
- Apply low vacuum until the wells are empty (should take about 2 to 3 min).
- Wash the wells three times each with 100 μl FTA wash buffer.
- Dismantle the apparatus and clean it properly with demineralized water.
- Immunodetection of protein aggregates on cellulose acetate membrane
- Apply 15 ml of blocking solution (5% milk in 1x PBS-Triton 0.1%) per membrane and incubate at room temperature for 30 min.
- Remove blocking solution, then apply 15 ml of primary antibody diluted in blocking solution (see Table 1 for dilution). Incubate at 4 °C overnight.
- Wash the membranes three times for 5 min in PBS-Triton 0.1%.
- Apply 15 ml of secondary antibody diluted in blocking solution (see Table 1) and incubate at room temperature for 1 h.
- Wash the membranes three times for 5 min in PBS-Triton 0.1%.
- Place the membranes in a plastic wrap and apply 1 ml of ECLTM Prime Western Blotting Detection Reagent per membrane. Incubate for 1 min.
- Remove excess liquid and air bubbles and develop.
Data analysis
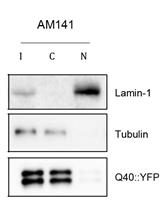
In Sin et al. (2017), a forward genetic screen was performed on worms that express 40 glutamine (Q) repeats fused to YFP (here referred as Q40 worms) to search for mutants with reduced number of polyglutamine aggregates (Sin et al., 2017) (Figure 3A). The study identified a regulator of RNA polymerase III transcription–moag-2/lir-3–as an aggregation-promoting factor. The inactivation of this gene (by point mutation or deletion of the allele) reduced polyglutamine aggregation (Figure 3).
At least three independent experiments should be performed and alternative options for equal protein loading controls can be used (e.g., actin). If using a fluorescent reporter gene to monitor protein aggregation (e.g., GFP, mCherry), one should quantify its expression levels to exclude the possibility that the effects observed are due to changes in the expression level of the reporter gene.
Immunoblots were quantified by densitometry using ImageJ. An example of data analysis can be found in Figure 1F and S1E of Sin et al. (2017). To quantify the relative amount of SDS-insoluble protein, a ratio (fold change) was calculated by dividing the values of Q40; moag-2/lir-3 mutants (undiluted sample of the point-mutation allele pk2183 and deletion allele tm813 shown on Figure 3B, top) by their corresponding wild-types (wt, previously normalized to α-tubulin as a loading control). In this study, Q40-YFP and α-tubulin expression were included as controls (Figure 3B, bottom). A more detailed tutorial is also available at https://imagej.nih.gov/ij/docs/menus/analyze.html (under “Gels Submenu”) and at http://lukemiller.org/index.php/2010/11/analyzing-gels-and-western-blots-with-image-j/.
Figure 3. Filter retardation assay with 5- and 25-fold dilutions of crude protein lysate from Q40 worms and Q40; moag-2/lir-3 mutant worms (point-mutation allele pk2183 and deletion allele tm813). A. Representative image of a Q40 worm (strain AM141) and a Q40; moag-2/lir-3 (pk2183) worm (Sin et al., 2017). Scale bar = 75 µm. B. On top, a representative immunoblot which was probed with α-GFP antibody to detect polyglutamine aggregates. On the bottom, an input control is shown by standard Western immunoblot analysis of worm lysates probed with α-GFP (control for YFP expression) and α-tubulin (control for equal protein loading) antibodies. C. Quantification of filter retardation assays by densitometry using ImageJ. The average of three biological replicates is represented. Data are represented as mean ± SEM and significance was calculated using a one-tailed unpaired Student’s t-test. *P < 0.05. More details of the experiment can be found in Sin et al. (2017).
Recipes
- 10x PBS (1 L)
- Mix 25.6 g Na2HPO4•7H2O, 80 g NaCl, 2 g KCl, 2 g KH2PO4
- Bring to 1 L with distilled water
- Autoclave for 20 min at 121 °C
- 10x PBS is kept at room temperature
- Dilute to 1x with distilled water, and adjust pH to 7.0 for working solution
- The 1x PBS can be kept at room temperature
- 1x PBS with 0.1% Triton X-100 (10 ml)
Add 1 ml of Triton X-100 to 10 ml of 1x PBS and mix - 1x PBS with 0.05% Triton X-100 (10 ml)
Add 500 μl of Triton X-100 to 10 ml of 1x PBS and mix - Filter Trap Assay (FTA) sample buffer (50 ml, make fresh prior to use)
- Add all of the components:
500 μl of 1 M Tris-HCl pH 8.0 (final concentration: 10 mM)
1.5 ml of 5 M NaCl (final concentration: 150 mM)
10 ml of 10% SDS (final concentration: 2%)
Add Complete protease inhibitors (1 tablet per 10 ml) - Fill up to 50 ml with Milli-Q water
- Add all of the components:
- Filter Trap Assay (FTA) wash buffer (50 ml, make fresh prior to use)
- Add all of the components:
500 μl of 1 M Tris-HCl pH 8.0 (final concentration: 10 mM)
1.5 ml of 5 M NaCl (final concentration: 150 mM)
500 μl of 10% SDS (final concentration: 0.1%)
Add Complete protease inhibitors (1 tablet per 10 ml) - Fill up to 50 ml with Milli-Q water
- Add all of the components:
- 1 M dithiothreitol (DTT) (10 ml)
- Dissolve 1.5 g of DTT (MW = 154.25) in 8 ml of deionized or distilled water
- Adjust volume to 10 ml and make 1 ml aliquots
- Store at -20 °C (for long-term)
- Blocking solution (5% milk in 1x PBS-Triton 0.1%) (50 ml, make fresh prior to use)
- Weigh 2.5 g of non-fat dry milk powder into a 50 ml conical tube and add up to 50 ml of PBS-Triton 0.1%
- Vortex to mix thoroughly
Acknowledgments
We thank the Caenorhabditis Genetics Centre (funded by the National Institutes of Health National Centre for Research Resources) and the Mitani laboratory for the C. elegans strains (funded by the Japan National BioResource Project). This project was funded by a European Research Council (ERC) starting grant (to E.A.A.N.), the Fundação para a Ciência e Tecnologia fellowship (SFRH/BPD/51009/2010) (to O.S.), the Alumni chapter Gooische Groningers facilitated by the Ubbo Emmius Fonds (to E.A.A.N) and a Groningen University Institute for Drug Exploration (GUIDE) fellowship (O.S.). This protocol was adapted from procedures published in Scherzinger et al. (1997) and Wanker et al. (1999).
Competing interests
The authors declare no conflicts of interest or competing interests.
References
- Bieschke, J., Cohen, E., Murray, A., Dillin, A. and Kelly, J. W. (2009). A kinetic assessment of the C. elegans amyloid disaggregation activity enables uncoupling of disassembly and proteolysis. Protein Sci 18(11): 2231-2241.
- Kuiper, E. F., de Mattos, E. P., Jardim, L. B., Kampinga, H. H. and Bergink, S. (2017). Chaperones in polyglutamine aggregation: Beyond the Q-Stretch. Front Neurosci 11: 145.
- Recasens, A., Carballo-Carbajal, I., Parent, A., Bove, J., Gelpi, E., Tolosa, E. and Vila, M. (2018). Lack of pathogenic potential of peripheral α-synuclein aggregates from Parkinson's disease patients. Acta Neuropathol Commun 6(1): 8.
- Scherzinger, E., Lurz, R., Turmaine, M., Mangiarini, L., Hollenbach, B., Hasenbank, R., Bates, G. P., Davies, S. W., Lehrach, H. and Wanker, E. E. (1997). Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90(3): 549-558.
- Sin, O., de Jong, T., Mata-Cabana, A., Kudron, M., Zaini, M. A., Aprile, F. A., Seinstra, R. I., Stroo, E., Prins, R. W., Martineau, C. N., Wang, H. H., Hogewerf, W., Steinhof, A., Wanker, E. E., Vendruscolo, M., Calkhoven, C. F., Reinke, V., Guryev, V. and Nollen, E. A. (2017). Identification of an RNA polymerase III regulator linked to disease-associated protein aggregation. Mol Cell 65(6): 1096-1108 e1096.
- Soto, C. (2003). Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 4(1): 49-60.
- Stroo, E., Koopman, M., Nollen, E. A. and Mata-Cabana, A. (2017). Cellular regulation of amyloid formation in aging and disease. Front Neurosci 11: 64.
- Wanker, E. E., Scherzinger, E., Heiser, V., Sittler, A., Eickhoff, H. and Lehrach, H. (1999). Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods Enzymol 309: 375-386.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sin, O., Mata-Cabana, A., Seinstra, R. I. and Nollen, E. A. A. (2018). Filter Retardation Assay for Detecting and Quantifying Polyglutamine Aggregates Using Caenorhabditis elegans Lysates. Bio-protocol 8(19): e3042. DOI: 10.21769/BioProtoc.3042.
Category
Biochemistry > Protein > Quantification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.