- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR/Cas9-mediated ssDNA Recombineering in Corynebacterium glutamicum



(*contributed equally to this work) Published: Vol 8, Iss 19, Oct 5, 2018 DOI: 10.21769/BioProtoc.3038 Views: 8405
Reviewed by: David CisnerosJuan Facundo Rodriguez AyalaPeter E Burby
Original research article
The authors used this protocol in:

Nov 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Corynebacterium glutamicum is a versatile workhorse for industrial bioproduction of many kinds of chemicals and fuels, notably amino acids. Development of advanced genetic engineering tools is urgently demanded for systems metabolic engineering of C. glutamicum. Recently unveiled clustered regularly interspaced short palindromic repeats (CRISPR) and their CRISPR-associated proteins (Cas) are now revolutionizing genome editing. The CRISPR/Cas9 system from Streptococcus pyogenes that utilizes NGG as protospacer adjacent motif (PAM) and has good targeting specificity can be developed into a powerful tool for efficient and precise genome editing of C. glutamicum. In this protocol, we described the general procedure for CRISPR/Cas9-mediated ssDNA recombineering in C. glutamicum. Small modifications can be introduced into the C. glutamicum chromosome with a high editing efficiency up to 90%.
Keywords: CRISPR/Cas9Background
The Gram-positive soil bacterium Corynebacterium glutamicum is a versatile workhorse for industrial bioproduction of amino acids, biofuels, and polymer building blocks (Becker et al., 2016). At the early stage of engineering of C. glutamicum, random mutagenesis combined with positive selection by phenotypic resistance to amino acid analogs was the most commonly used strategy (Vertes et al., 2005). Genetic manipulations in C. glutamicum were initiated in 1984 and became a key enabling strategy for strain improvement (Ozaki et al., 1984). A routinely used method for gene disruption and insertion in C. glutamicum is based on integration of a suicide vector into its chromosome, followed by a second recombination event to remove the plasmid backbone and a counter-selection step using a conditionally lethal marker. Nevertheless, due to the frequent spontaneous inactivation of the counter-selectable marker sacB, up to 45% of colonies obtained in the screening process were false-positive, making this multi-step procedure time-consuming and inefficient (Schafer et al., 1994). To engineer C. glutamicum more efficiently, simple but versatile genome editing tools are still in urgent demand.
Recently, clustered regularly interspaced short palindromic repeats (CRISPR) and their CRISPR-associated proteins (Cas) have been explored as a leading-edge tool for bacterial genome editing (Choi et al., 2016). The CRISPR/Cas9 system accepts NGG protospacer adjacent motif (PAM) and has good targeting specificity (Jiang et al., 2013). Therefore, the CRISPR/Cas9 system is expected to possess abundant editing targets in GC-rich C. glutamicum. We successfully developed a CRISPR/Cas9 toolbox for efficient and comprehensive engineering of C. glutamicum strains (Liu et al., 2017; Wang et al., 2018). By using the tailor-made CRISPR/Cas9 system, efficient deletion and insertion of large DNA fragments using plasmid-borne editing templates were achieved. By combining CRISPR/Cas9 and ssDNA recombineering, small modifications were introduced into the genome with efficiencies up to 90% (Liu et al., 2017). Besides, targeted based editing without donor DNA was also realized using Cas9 and cytidine deaminase fusions (Wang et al., 2018). The toolbox developed is simple and versatile, which is expected to overcome the major limitations of existing genome editing tools of C. glutamicum and advance the genetic manipulation of this industrial workhorse. Herein, we described the detailed protocol for CRISPR/Cas9-mediated ssDNA recombineering in C. glutamicum.
Materials and Reagents
- 0.2 ml PCR tubes (Biosharp, catalog number: BS-02-P )
- 1.5 ml Eppendorf tubes (Biosharp, catalog number: BS-15-M )
- Pipette tips (Biosharp, catalog numbers: BS-10-T , BS-200-T , BS-1000-T )
- Petri dishes (NEST Biotechnology, catalog numbers: 752001 )
- Competent Escherichia coli DH5α cells (CWBIO, catalog number: CW0808 )
- pgRNA5 plasmid (Liu et al., 2017)
- pCas9 plasmid (Liu et al., 2017)
- Agarose (Biowest, catalog number: 111860 )
- GoldView I Nuclear Staining Dyes (10,000x) (Solarbio, catalog number: G8140 )
- AxyPrepTM DNA Gel Extraction Kit (Corning, Axygen®, catalog number: AP-GX-250 )
- Tryptone (Oxoid, catalog number: LP0042 )
- Yeast extract (Oxoid, catalog number: LP0021 )
- Kanamycin (Solarbio, catalog number: K8020 )
- Chloramphenicol (Fisher Scientific, catalog number: BP904-100 )
- Q5® High-Fidelity DNA polymerase (New England Biolabs, catalog number: M0491L )
- AxyPrepTM Plasmid Miniprep Kit (Corning, Axygen®, catalog number: AP-MN-P-250 )
- Vazyme ClonExpress® II One Step Cloning Kit (Vazyme Biotech, catalog number: C112-01/02 )
- 2x EasyTaq PCR SuperMix (+dye) (Beijing TransGen Biotech, catalog number: AS111-11 )
- Tris base (Solarbio, catalog number: T8060 )
- Glacial acetic acid (Sinopharm Chemical Reagent, catalog number: 10000218 )
- EDTA (Solarbio, catalog number: E8030 )
- NaCl (Sinopharm Chemical Reagent, catalog number: 10019328 )
- Agar (Solarbio, catalog number: A8190 )
- (NH4)2SO4 (Solarbio, catalog number: A8821 )
- K2HPO4 (Solarbio, catalog number: D9880 )
- NaH2PO4 (Solarbio, catalog number: S5830 )
- MgSO4•7H2O (Sinopharm Chemical Reagent, catalog number: 10013018 )
- Brain Heart Infusion (BHI) (BD, catalog number: 237500 )
- Tween 80 (Solarbio, catalog number: T8360 )
- DL-threonine (DL-Thr) (Tokyo Chemical Industry, catalog number: T3105 )
- Glycine (Gly) (Solarbio, catalog number: G8200 )
- Glucose (Beijing Dingguo Changsheng Biotechnology, catalog number: DS063 )
- Trisodium citrate (Solarbio, catalog number: S8220 )
- Sorbitol (Solarbio, catalog number: S8090 )
- Isonicotinic acid hydrazide (INH) (Sigma-Aldrich, catalog number: I3377 )
- IPTG (Solarbio, catalog number: I8070 )
- Sodium propionate (Sigma-Aldrich, catalog number: P1880 )
- Oligo for ssDNA recombineering:
AGGTAAGGCGCACACGAGCGACCTTACGAAGAGCAGAGTTAGGCTTGCGAGGGGTGGTGGTGTACACACGGGTGCATACGCCACGACGCT (synthesized and page-purified in Genewiz, Suzhou, China) - Primers for gRNA plasmid construction (synthesized and page-purified in Genewiz, Suzhou, China):
gRNA-1: AGAGCAGAGTTAGGCTTCTTGTTTTAGAGCTAGAAATAGCAAG
gRNA-2: TGCTCTTTCATTGATGGCTGG
gRNA-3: CAGCCATCAATGAAAGAGCAAC
gRNA-4: AAGAAGCCTAACTCTGCTCTTGAATTACACTGTACCTGTTGCGTC - 50x TAE electrophoresis buffer (see Recipes)
- 1x TAE electrophoresis buffer (see Recipes)
- LB medium (see Recipes)
- LB solid medium (see Recipes)
- BHI medium (see Recipes)
- NCM medium (see Recipes)
- BHIS medium (see Recipes)
- LBHIS solid medium (see Recipes)
- 25 mg/ml kanamycin stock (see Recipes)
- 5 mg/ml chloramphenicol stock (see Recipes)
- 0.1 M IPTG stock (see Recipes)
- 500 g/L sodium propionate stock (see Recipes)
Equipment
- Pipettes (Eppendorf, catalog number: 022575442 )
- Thermal cycler (Thermo Fisher Scientific, Thermo ScientificTM, model: ArktikTM Thermal Cycler )
- Water bath (Beijing Ever Bright Medical Treatment Instrument, model: DZKW-S-4 )
- NanoDrop (Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM 2000 )
- Milli-Q H2O dispenser (Thermo Fisher Scientific, Thermo ScientificTM, model: BarnsteadTM GenPureTM Pro , catalog number: 50131950)
- Centrifuge (Eppendorf, model: 5084 R , catalog number: 5805000696)
- Electroporation cuvette (Bio-Rad Laboratories, catalog number: 1652082 )
- -20 °C freezer (Panasonic, catalog number: MDF-U548D-C )
- pH meter (Fisher Scientific, model: Fisher ScientificTM accumetTM AB150, catalog number: 13-636-AB150 )
- DNA electrophoresis apparatus (Beijing LIUYI Biotechnology, catalog numbers: DYY-6C and DYCP-32B )
- Electroporator (Eppendorf, model: Electroporator 2510 )
- Autoclave (SANYO, catalog number: MLS-3750 )
Software
- Primer Premier 5 (Premier Biosoft International)
- sgRNAcas9 (Xie et al., 2014)
Procedure
- Design mutation strategy and prepare ssDNA editing templates
- Select the target position where the mutation will be introduced. Upload the sequence of the target region (23 mer upstream and downstream the target position, respectively, totally 47 mer) and the genome sequence of C. glutamicum to sgRNAcas9 software (Xie et al., 2014) to design gRNAs and evaluate the potential off-target sites.
- Choose a proper gRNA with no potential off-target sites and make sure that the target mutation position locates in its seed region (green letters in Figure 1) or the “GG” region of the PAM sequence (Figure 1).
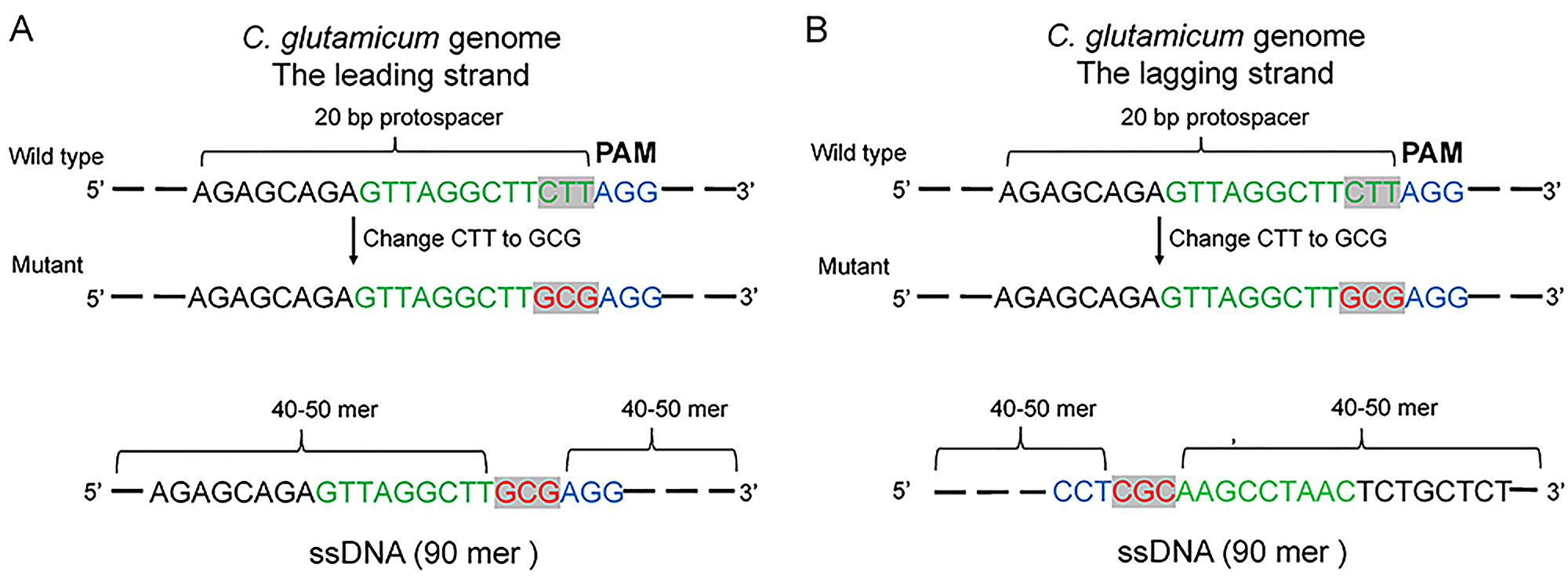
Figure 1. Design of protospacer, PAM site and ssDNA. A. Introducing mutations (red letters) with a protospacer and a PAM sequence (5’-NGG-3’, blue letters) in the leading strand of C. glutamicum genome. B. Introducing mutations (red letters) with a protospacer and a PAM sequence (5’-NGG-3’, blue letters) in the lagging strand of C. glutamicum genome. The target mutation position should locate in the seed region (green letters) or the “GG” region of the PAM sequence. The ssDNA (90 mer) editing templates should be complementary to the lagging strand. - Design a 90 mer ssDNA using the 40-50 mer upstream and downstream the target position, respectively. Make sure that the 90 mer ssDNA is complementary to the lagging strand (Figure 1).
Note: A previous study demonstrated that ssDNA that is complementary to the lagging strand results in a higher recombination efficiency (Binder et al., 2013). According to the genome sequence of C. glutamicum ATCC 13032 published by Kalinowski et al. (2003), the bi-directional replication of the genome starts at the proposed oriC sequence near the dnaA gene (cg0001) and ends near to the calculated replication terminus at around 1.6 Mb. Therefore, regarding to genes located in 0-1.6 Mb, just copy the genome sequence to get the lagging strand sequence. For genes located in 1.6-3.3 Mb, the reverse complementary sequence of the genome sequence is the lagging strand sequence. - Synthesize PAGE-purified ssDNA (10 μg is used for each transformation).
- Construct plasmid pgRNA-target
To construct plasmid pgRNA-target that is required for expression of specific gRNA, replace the original 20 bp spacer sequence of pgRNA5 with the target spacer sequence designed previously (Figure 2). Use Vazyme ClonExpress® II One Step Cloning Kit to construct the plasmid (this cloning technology is based on homologous recombination and overlapped sequences of 15-20 bp on both 5’- and 3’-end of PCR fragments are needed). Add the 20 bp spacer sequence in the primer and use the 20 bp spacer sequence as the overlap. Detailed procedure is as follows: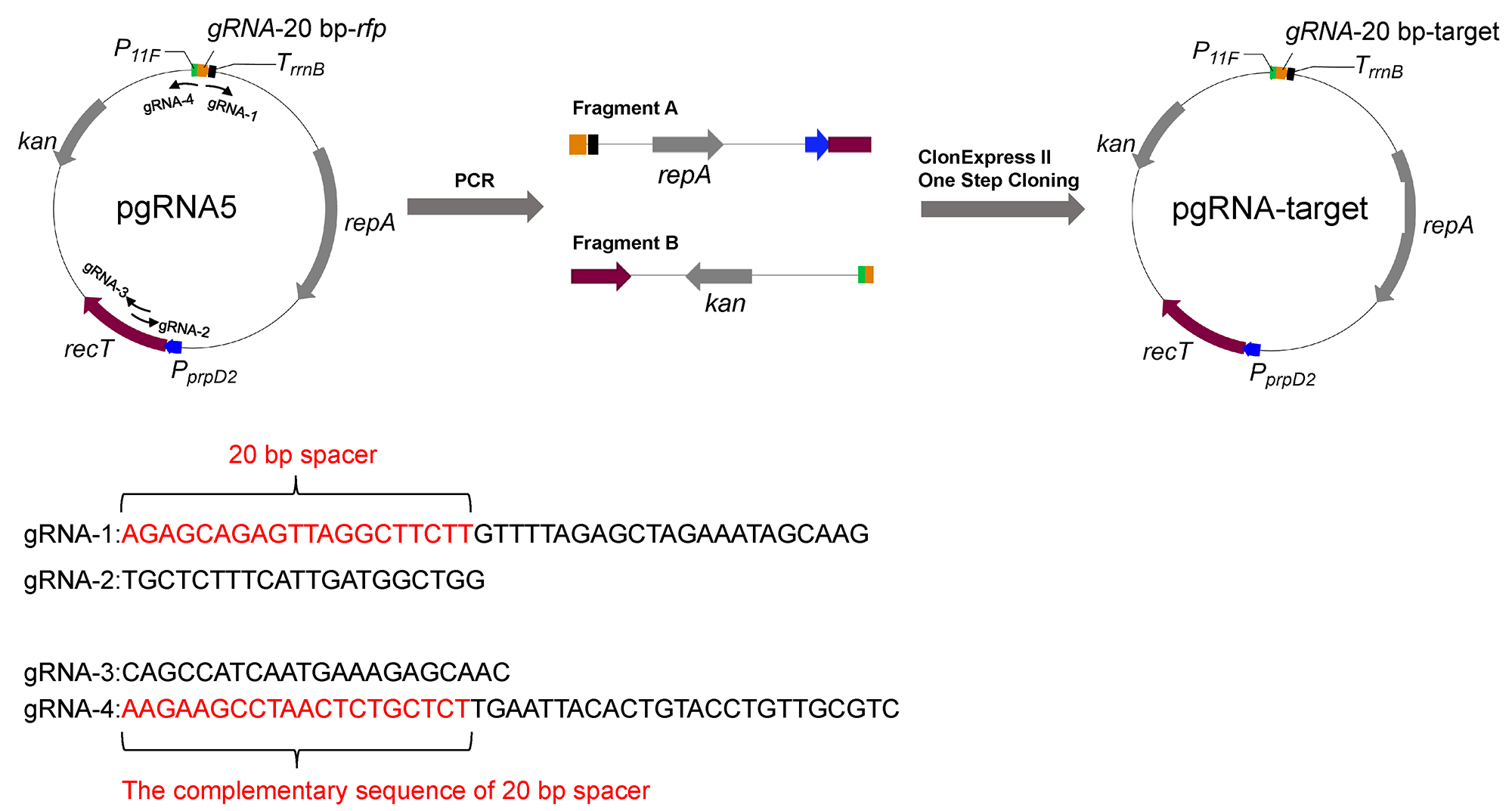
Figure 2. Construction of plasmid pgRNA-target- Design primers for construction of plasmid pgRNA-target.
Primer gRNA-1: 20 bp spacer sequence + GTTTTAGAGCTAGAAATAGCAAG
Primer gRNA-2: TGCTCTTTCATTGATGGCTGG (permanent sequence)
Primer gRNA-3: CAGCCATCAATGAAAGAGCAAC (permanent sequence)
Primer gRNA-4: reverse complement of 20 bp spacer sequence + TGAATTACACTGTACCTGTTGCGTC - Amplify Fragment A (3421 bp) and Fragment B (2812 bp) via PCR using pgRNA5 as a template and primer pairs gRNA-1/gRNA-2 and gRNA-3 and gRNA-4, respectively.
- PCR program using Q5® High-Fidelity DNA polymerase:

- Purify PCR products by 1% agarose gel electrophoresis in 1x TAE electrophoresis buffer followed by gel extraction using AxyPrepTM DNA Gel Extraction Kit (Corning, Axygen®). Store the purified PCR product at -20 °C until used.
Note: GoldView I Nuclear Staining Dyes (10,000x) are used for DNA staining. According to the manufacturer’s instructions, GoldView I should be protected from light and stored at room temperature. For 100 ml agarose gel solution, 10 μl GoldView I Nuclear Staining Dyes (10,000x) is used.
- PCR program using Q5® High-Fidelity DNA polymerase:
- Construct pgRNA-target plasmid
- Use Vazyme ClonExpress® II One Step Cloning Kit to construct pgRNA-target plasmid. The recombination reaction mixture (20 μl) contains 60-120 ng Fragment A, 60-120 ng Fragment B, 4 μl 5x CE II Buffer, 2 μl Exnase II. Incubate the reaction mixture at 37 °C in a thermocycler for 30 min and immediately place it on ice.
- Transform 100 μl chemically competent E. coli strain DH5α with 10 μl of the recombination products (make sure the transformation efficiency of competent cells is > 107 cfu/μg). Plate the transformation mix on LB solid medium containing kanamycin (25 μg/ml). Incubate the plate at 37 °C overnight.
- Verify the transformants via PCR using an upstream primer specific to the 20 bp spacer sequences and a downstream primer gRNA-5 (AGTCTTTCGACTGAGCCTTTCG) that locates on the plasmid backbone. The size of the expected fragment generated by PCR is approximately 150 bp.
- Cultivate the correct colonies and extract plasmids using AxyPrepTM Plasmid Miniprep Kit. Store the purified pgRNA-target plasmid at -20 °C until used.
- Design primers for construction of plasmid pgRNA-target.
- CRISPR/Cas9-mediated ssDNA recombineering in C. glutamicum
The procedure for CRISPR/Cas9-mediated ssDNA recombineering is shown in Figure 3.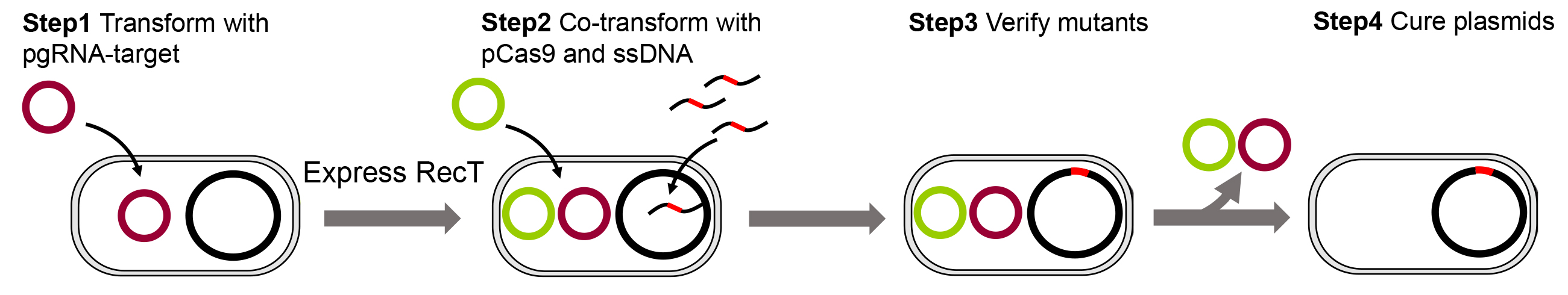
Figure 3. The procedure for CRISPR/Cas9-mediated ssDNA recombineering- Transform C. glutamicum with pgRNA-target plasmid
Prepare competent cells for C. glutamicum and transform plasmids according to the protocols described previously (Ruan et al., 2015).- Activate C. glutamicum strain by cultivation on LBHIS solid medium at 30 °C for 24 h.
- Inoculate a pre-culture of 20 ml BHI medium (in 100 ml shake flask) from a single colony and incubate at 30 °C with vigorous shaking (220 rpm) overnight.
- Dilute the overnight culture to an OD600nm of 0.3 in NCM medium (100 ml) containing 0.46% INH, 0.11% Tween 80, 1.11% DL-Thr and 3.59 % Gly, and incubate at 30 °C with vigorous shaking (220 rpm) until the OD600nm value reaches 0.9-1.0 (ca. 5-6 h).
- Chill the culture on ice for 15 min and harvest cells by centrifugation at 4 °C, 4,000 x g for 10 min.
- Discard the supernatant and resuspend the pellet in 50 ml ice-cold, sterile 10% glycerol. Centrifuge the resuspended cells at 4 °C, 4,000 x g for 10 min. Repeat this wash step four times.
- Discard as much supernatant as possible. Immediately resuspend the cell pellet in 0.4 ml ice-cold, sterile 10% glycerol, and divide the resuspended cells into 100 μl aliquots.
- Add 0.5-1 μg pgRNA-target plasmid (up to 5 μl) to the competent cells and transfer the mixture to a 2 mm electroporation cuvette (Bio-Rad Laboratories). After electroporation at 2.5 kV, 5 msec, add 1 ml BHIS medium to the cuvette immediately, transfer the mixture into a 1.5 ml tube, and incubate the tube at 46 °C for 6 min. Then incubate the tube at 30 °C with vigorous shaking (220 rpm) for 2 h.
- Spread 50-100 μl of the mixture on LBHIS solid medium containing 25 μg/ml kanamycin. Incubate the plates at 30 °C for 24-36 h. The transformants are C. glutamicum (pgRNA-target) strains.
- Transform C. glutamicum (pgRNA-target) with pCas9 plasmid and ssDNA
- Prepare competent cells for C. glutamicum (pgRNA-target). Follow the procedures described above except that add 1 g/L sodium propionate into NCM medium to induce RecT recombinases expression.
- Transform C. glutamicum (pgRNA-target) competent cells with 1-2 μg pCas9 and 6-15 μg synthetic ssDNA (total volume up to 10 μl). After electroporation at 2.5 kV for 5 msec, add 1 ml BHIS medium to the cuvette immediately, transfer the mixture into a 1.5 ml tube, and incubate the tube at 46 °C for 6 min. Then incubate the tube at 30 °C with vigorous shaking (220 rpm) for 5-6 h.
- Collect the cells by centrifugation at 5,000 x g for 1 min. Remove the supernatant and resuspend the cells using 100 μl BHIS medium. Spread all the cells on LBHIS solid medium containing 25 μg/ml kanamycin, 5 μg/ml chloramphenicol and 0.01 mM IPTG. Incubate the plates at 30 °C for 48-72 h until colonies appear.
- Screen edited mutants
Design a primer pair that locates at approximately 200 bp upstream and downstream of the mutation site, respectively (Figure 4). Amplify the target fragment by using colony PCR and the above-mentioned primer pair. Sequence the PCR product by Sanger sequencing to verify the mutation was introduced and no other unexpected mutations were introduced.
Note: According to our experience, the editing efficiency is over 80%. Therefore, screening 5 colonies can ensure the acquisition of edited mutants.
For colony PCR, pick a speck of colony using a 10 μl pipette tip and mix it with the PCR reaction mixture to introduce DNA template.
Colony PCR program using 2x EasyTaq PCR SuperMix (+dye):
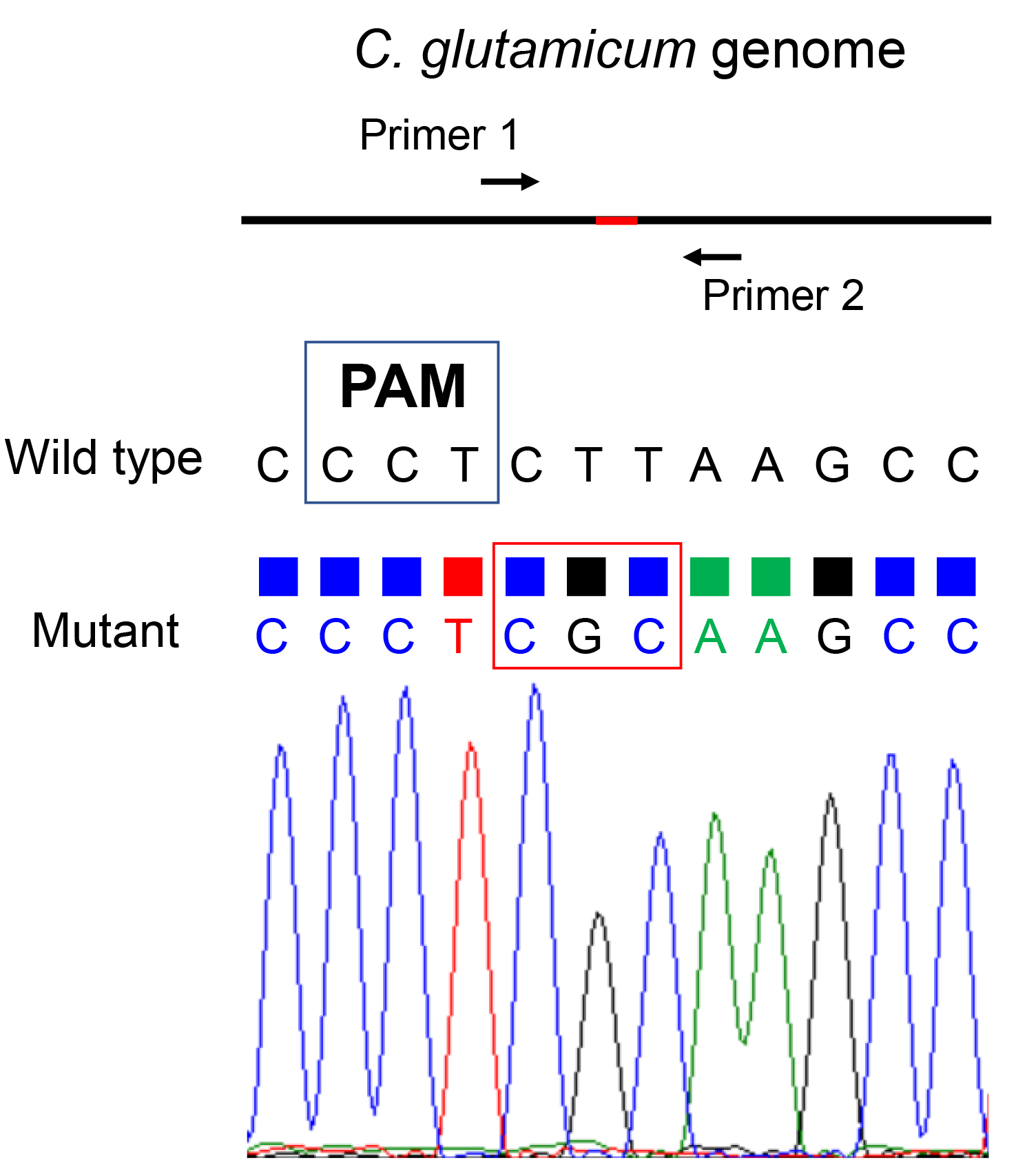
Figure 4. Verification of the mutants by Sanger sequencing
- Transform C. glutamicum with pgRNA-target plasmid
- Plasmid curing
To remove pCas9 and pgRNA-target plasmids from the edited cells, inoculate the edited colony into 100 ml of BHI medium without antibiotics and cultivate it at 30 °C, 220 rpm for 12 h. Transfer 100 μl of the culture into 100 ml fresh BHI medium without antibiotics and cultivate it at 30 °C, 220 rpm for 12 h. Repeat this step for another time. Dilute the culture and spread 100 μl on LBHIS solid medium without antibiotics. Pick 10 to 20 isolates and inoculate LBHIS solid media with and without 25 μg/ml kanamycin (or 5 μg/ml chloramphenicol). Cultivate the plates at 30 °C overnight. The isolates that do not grow on LBHIS solid medium with 25 μg/ml kanamycin and LBHIS solid medium with 5 μg/ml chloramphenicol have been cured of the editing plasmid.
Note: According to our experience, the efficiency observed for curing strains of the plasmids is approximately 25%.
Data analysis
To verify the genome editing events, amplify the target region via colony PCR and subsequently sequence the PCR product. Align the reference sequence (wild-type) with the sequenced results to analyze whether the target mutations were introduced via CRISPR/Cas9-mediated ssDNA recombineering. The editing efficiency = No. of correct mutants/No. of colonies tested.
Note: After the CRISPR/Cas9-mediated ssDNA recombineering, some abnormally large colonies may appear on the plates (Figure 5). According to our experience, these abnormally large colonies are very likely false positives, which lose the cas9 expression plasmid during the editing process.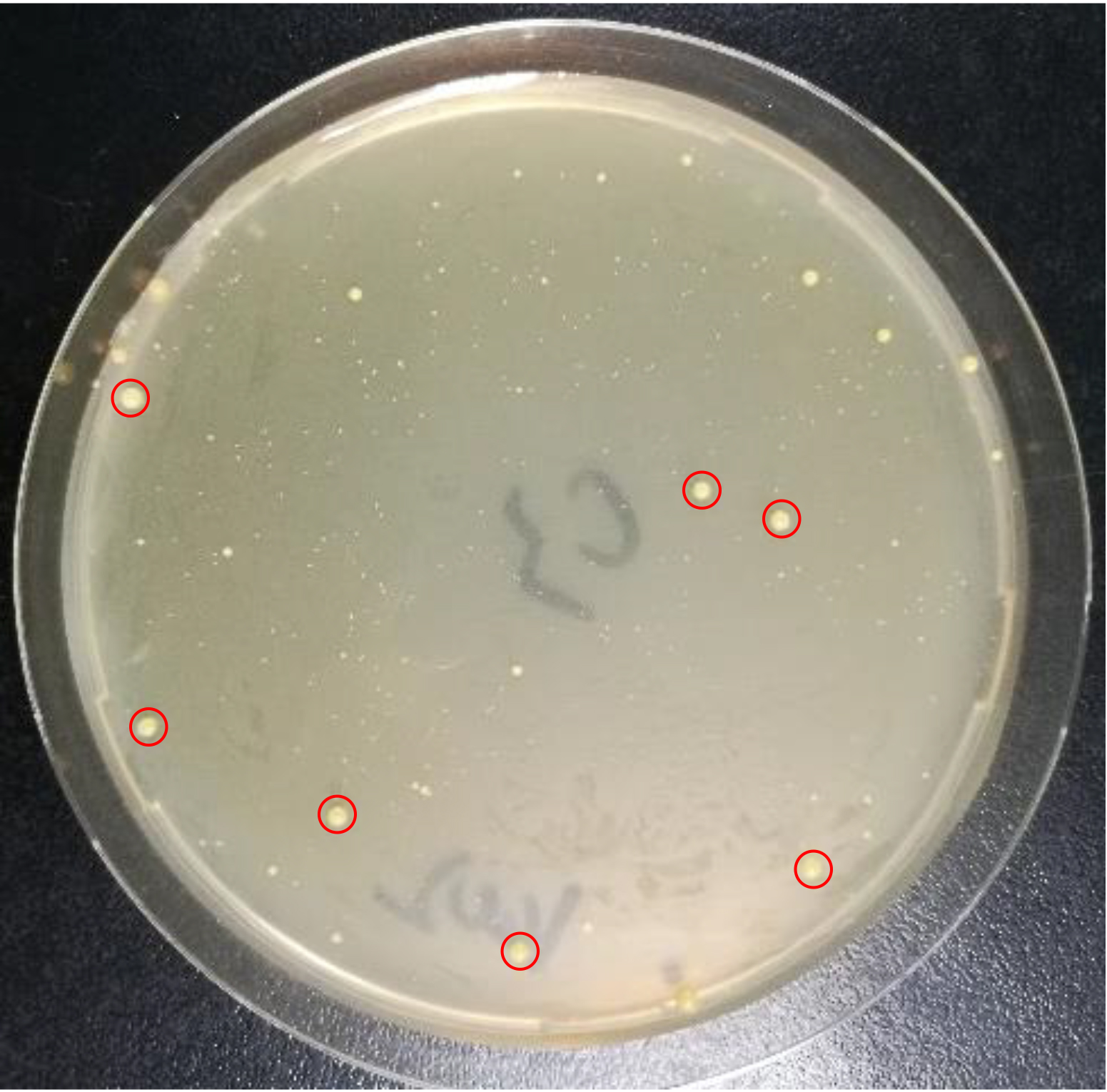
Figure 5. Transformants obtained by CRISPR/Cas9-mediated ssDNA recombineering. Colonies marked in red cycles were the so-called abnormally large colonies.
To assay the loss of editing plasmids pCas9 and pgRNA-target, pick 10 to 20 isolates from LBHIS solid medium without antibiotics and inoculate LBHIS solid media with and without 25 μg/ml kanamycin (or 5 μg/ml chloramphenicol). The isolates that are sensitive to both kanamycin and chloramphenicol have been cured of the editing plasmid. The efficiency of plasmid curing = No. of colonies without plasmid/No. of colonies tested.
Recipes
- 50x TAE electrophoresis buffer
242 g Tris base
57.1 ml glacial acetic acid
100 ml EDTA, pH 8.0
Add Milli-Q H2O to a total volume of 1 L
Store at room temperature - 1x TAE electrophoresis buffer
20 ml 50x TAE
980 ml Milli-Q H2O
Store at room temperature - LB medium
10 g NaCl
10 g tryptone
5 g yeast extract
Add Milli-Q H2O to a total volume of 1 L
Autoclave to sterilize - LB solid medium
1 L LB medium
18 g agar
Autoclave to sterilize
Allow cooling to about 60 °C. If antibiotics are required, they are added at this point - BHI medium
37 g Brain Heart Infusion (BHI)
10 g (NH4)2SO4
0.2 g K2HPO4
0.3 g NaH2PO4
0.5 g MgSO4•7H2O
Adjust pH to 7.2
Add H2O to a total volume of 1 L
Autoclave to sterilize - NCM medium
Containing 0.46% INH, 0.11% Tween 80, 1.11% DL-Thr and 3.59% Gly- NCM medium
17.4 g K2HPO4
11.6 g NaCl
5 g glucose
5 g tryptone
1 g yeast extract
0.3 g trisodium citrate
0.05 g MgSO4•7H2O
91.1 g sorbitol
Adjust pH to 7.2
Add H2O to a total volume of 1 L
Autoclave to sterilize - Use sterilized NCM medium to prepare INH, DL-Thr and Gly solution
23 g isonicotinic acid hydrazide (INH)
5.5 g Tween 80
55.5 g DL-threonine (DL-Thr)
179.5 g glycine (Gly)
Add sterilized NCM medium to a total volume of 0.2 L
Filtration sterilization - Mix 0.8 L NCM medium with 0.2 L INH, DL-Thr and Gly solution
- NCM medium
- BHIS medium
18.5 g BHI
91 g sorbitol
Adjust pH to 7.2
Add H2O to a total volume of 1 L
Autoclave to sterilize - LBHIS solid medium
5 g Tryptone
5 g NaCl
2.5 g yeast extract
18.5 g BHI
91 g sorbitol
18 g agar
Adjust pH to 7.2
Add H2O to a total volume of 1 L
Autoclave to sterilize
Allow cooling to about 60 °C. If antibiotics are required, they are added at this point - 25 mg/ml kanamycin stock
250 mg kanamycin
Add Milli-Q water to 10 ml
Filtration sterilization
Store at -20 °C - 5 mg/ml chloramphenicol stock
50 mg chloramphenicol
Add ethanol to 10 ml
Filtration sterilization
Store at -20 °C - 0.1 M IPTG stock
23.8 mg IPTG
Add Milli-Q water to 10 ml
Filtration sterilization
Store at -20 °C - 500 g/L sodium propionate stock
5 g sodium propionate
Add Milli-Q water to 10 ml
Filtration sterilization
Store at -20 °C
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31700044), the Key Research Program of the Chinese Academy of Sciences (ZDRW-ZS-2016-2), and the first Special Support Plan for Talents Development and High-level Innovation and Entrepreneurship Team of the Tianjin Municipal City.
Competing interests
The authors declare that they have no competing interests.
References
- Becker, J. and Wittmann, C. (2016). Industrial microorganisms: Corynebacterium glutamicum. In: Wittmann, C. and Liao, J.C. (Eds.). Industrial Biotechnology: Microorganisms. pp: 183-220. Wiley-VCH Verlag GmbH & Co. KGaA.
- Binder, S., Siedler, S., Marienhagen, J., Bott, M. and Eggeling, L. (2013). Recombineering in Corynebacterium glutamicum combined with optical nanosensors: a general strategy for fast producer strain generation. Nucleic Acids Res 41(12): 6360-6369.
- Choi, K. R. and Lee, S. Y. (2016). CRISPR technologies for bacterial systems: Current achievements and future directions. Biotechnol Adv 34(7): 1180-1209.
- Jiang, W., Bikard, D., Cox, D., Zhang, F. and Marraffini, L. A. (2013). RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31: 233–239.
- Kalinowski, J., Bathe, B., Bartels, D., Bischoff, N., Bott, M., Burkovski, A., Dusch, N., Eggeling, L., Eikmanns, B. J., Gaigalat, L., Goesmann, A., Hartmann, M., Huthmacher, K., Kramer, R., Linke, B., McHardy, A. C., Meyer, F., Mockel, B., Pfefferle, W., Puhler, A., Rey, D. A., Ruckert, C., Rupp, O., Sahm, H., Wendisch, V. F., Wiegrabe, I. and Tauch, A. (2003). The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J Biotechnol 104(1-3): 5-25.
- Liu, J., Wang, Y., Lu, Y., Zheng, P., Sun, J. and Ma, Y. (2017). Development of a CRISPR/Cas9 genome editing toolbox for Corynebacterium glutamicum. Microb Cell Fact 16(1): 205.
- Ozaki, A., Katsumata, R., Oka, T. and Furuya, A. (1984). Functional expression of the genes of Escherichia coli in gram-positive Corynebacterium glutamicum. Mol Gen Genet 196(1): 175-178.
- Ruan, Y., Zhu, L. and Li, Q. (2015). Improving the electro-transformation efficiency of Corynebacterium glutamicum by weakening its cell wall and increasing the cytoplasmic membrane fluidity. Biotechnol Lett 37(12): 2445-2452.
- Schafer, A., Tauch, A., Jager, W., Kalinowski, J., Thierbach, G. and Puhler, A. (1994). Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145(1): 69-73.
- Vertes, A. A., Inui, M. and Yukawa, H. (2005). Manipulating corynebacteria, from individual genes to chromosomes. Appl Environ Microbiol 71(12): 7633-7642.
- Wang, Y., Liu, Y., Liu, J., Guo, Y., Fan, L., Ni, X., Zheng, X., Wang, M., Zheng, P., Sun, J. and Ma, Y. (2018). MACBETH: Multiplex automated Corynebacterium glutamicum base editing method. Metab Eng 47: 200-210.
- Xie, S., Shen, B., Zhang, C., Huang, X. and Zhang, Y. (2014). sgRNAcas9: a software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PLoS One 9(6): e100448.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Liu, J., Wang, Y., Zheng, P. and Sun, J. (2018). CRISPR/Cas9-mediated ssDNA Recombineering in Corynebacterium glutamicum. Bio-protocol 8(19): e3038. DOI: 10.21769/BioProtoc.3038.
Category
Microbiology > Microbial genetics > DNA
Molecular Biology > DNA > Chromosome engineering
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.