- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Extraction and Quantification of Polyphosphate (polyP) from Gram-negative Bacteria
(*contributed equally to this work) Published: Vol 8, Iss 18, Sep 20, 2018 DOI: 10.21769/BioProtoc.3011 Views: 7251
Reviewed by: Valentine V TrotterAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Polyphosphate (polyP), a universally conserved biomolecule, is composed of up to 1,000 phosphate monomers linked via phosphoanhydride bonds. Reaching levels in bacteria that are in the high nmoles per mg protein range, polyP plays important roles in biofilm formation and colonization, general stress protection and virulence. Various protocols for the detection of polyP in bacteria have been reported. These methods primarily differ in the ways that polyP is extracted and/or detected. Here, we report an improved method, in which we combine polyP extraction via binding to glassmilk with a very sensitive PolyP kinase/luciferase-based detection system. By using this procedure, we significantly enhanced the sensitivity of polyP detection, making it potentially applicable for mammalian tissues.
Keywords: PolyphosphateBackground
Polyphosphate (polyP), a biopolymer composed of linear chains of up to 1,000 inorganic phosphate monomers, is found in cells of all three domains of life. Yet, bacteria are the only organisms for which the enzymes of polyP metabolism have been well studied. Bacterial polyP kinase (PPK), which converts ATP into polyP, catalyzes both forward and reverse reactions. While synthesis of polyP is clearly the favored reaction in the cell, by providing sufficient amounts of ADP in vitro, the enzyme can be used to generate ATP from polyP, making a luciferase-based ATP detection possible (Ault-Riché et al., 1998). Bacteria lacking PPK are defective in biofilm formation, motility, persistence, and various stress responses, and show significantly increased sensitivity towards hypohalous acids (i.e., bleach) stress or phosphate starvation (Figure 1) (Rao et al., 2009; Gray et al., 2014; Maisonneuve and Gerdes, 2014; Gray and Jakob, 2015; Groitl et al., 2017).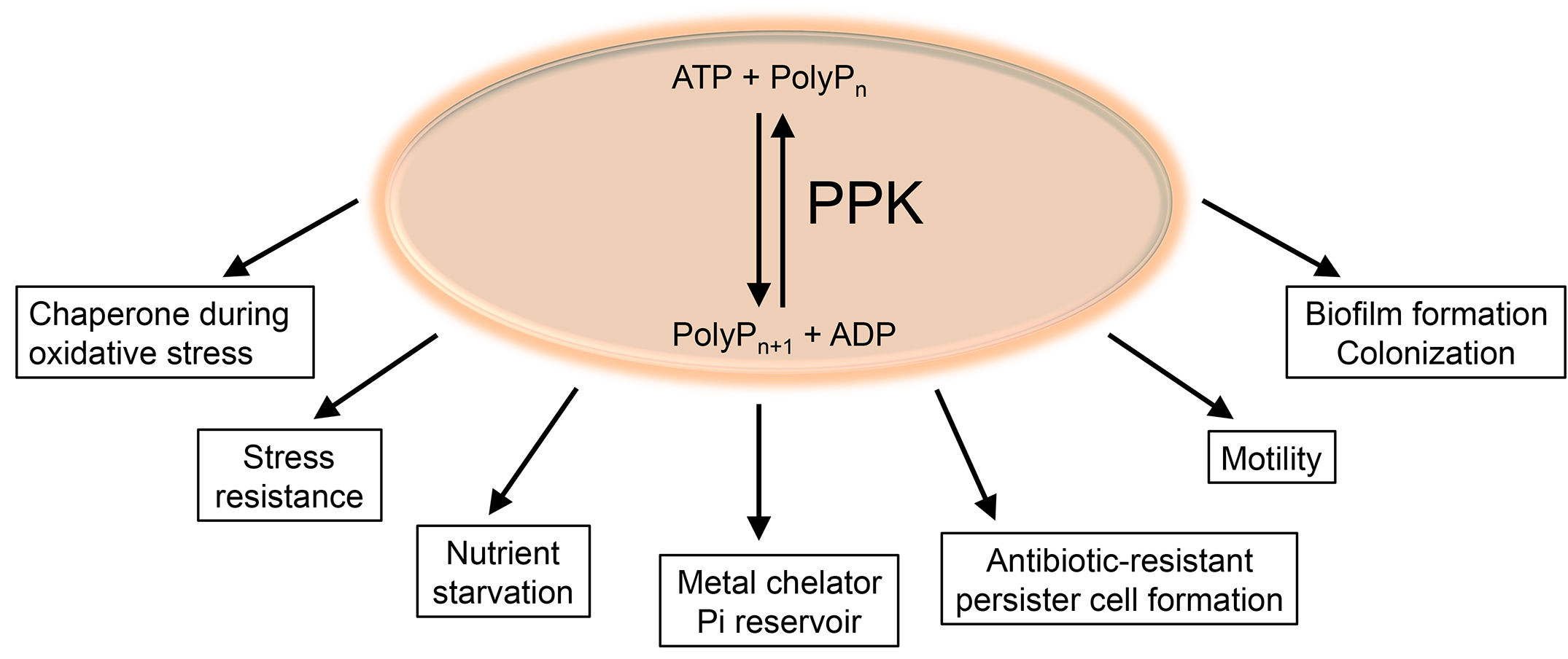
Figure 1. Synthesis of polyP and its role in Gram-negative Bacteria. The bacteria-specific polyphosphate kinase (PPK) reversibly catalyzes the conversion from ATP into polyP and ADP. Various functions for polyP have been described in Gram-negative bacteria, including its involvement in biofilm formation, colonization, motility, and formation of antibiotic-resistant persister cells. PolyP also contributes to the resistance of bacteria towards various stresses, including oxidative stress and starvation, and serves as metal chelator and Pi reservoir.
Given the many roles that polyP plays in Gram-negative bacteria, PPK became attractive as drug target to interfere with biofilm formation, make bacteria less persistent, and sensitize them towards physiological oxidants such as bleach (Dahl et al., 2017). Therefore, reliable and sensitive methods to determine the polyP levels in vivo are necessary. Several methods for the extraction and detection of polyP have been reported in Bio-protocol, including extraction of polyP with (i) perchloric acid, (ii) sodium hypochlorite, and (iii) phenol/chloroform and detection of polyP via visualization with urea-PAGE or colorimetric assays using malachite green or molybdenum blue (Gomez Garcia, 2014; Canadell et al., 2016; Ota and Kawano, 2017). In this protocol, we combined extraction of polyP via binding to glassmilk (Ault-Riché et al., 1998) with a very sensitive two-step enzyme-based detection system. First, the extracted polyP is converted into ATP by E. coli PPK in the presence of ultra-pure ADP. The ATP levels are then quantified using a luciferase-based detection system and corrected for cellular ATP. In comparison to the urea-PAGE or the colorimetric methods, the luciferase-based detection allows the quantification of much lower levels of polyP. This protocol has been successfully applied to quantify polyP levels from Pseudomonas aeruginosa.
Materials and Reagents
- Microcentrifuge tubes, 1.5 ml, clear (BioExpress, catalog number: C-3260-1 )
- Silica membrane spin columns (Epoch Life Science, EconoSpinTM, catalog number: 1920-250 )
- TempPlate non-skirted 96-well PCR plate, low profile, natural (USA Scientific, catalog number: 1402-9500 )
- 96-well plate solid white (Corning, catalog number: 3912 )
- 96-well plate clear flat bottom (Corning, catalog number: 3596 )
- Bradford reagent (Bio-Rad Laboratories, catalog number: 5000006 )
- Bovine Serum Albumin (BSA) (Sigma-Aldrich, catalog number: A3059-100G )
- Tris (Fisher Scientific, catalog number: BP152-5 )
- Ultra-pure ADP (Cell Technology, catalog number: ADP100-2 )
- E. coli polyphosphate kinase (PPK) (for expression and purification of E. coli PPK, see Gray et al., 2014)
- Dithiothreitol (DTT), > 99% pure, protease-free (Gold Biotechnology, catalog number: DTT25 )
- Sodium-dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: L3771-1KG )
- 95% v/v Ethanol
- QuantiLum® Recombinant Luciferase (Promega, catalog number: E1701 ; exact concentration of aliquots may vary)
Notes:- Prepare aliquots of the enzyme and store in small glass vials at -80 °C.
- Thaw and use the enzyme for each experiment.
- Discard all unused product.
- Do not subject to freeze-thaw cycles.
- Guanidine thiocyanate (Sigma-Aldrich, catalog number: G6639-250G )
- Silicon dioxide (Sigma-Aldrich, catalog number: S5631 )
- NaCl (Fisher Scientific, catalog number: S271-10 )
- EDTA (Fisher Scientific, catalog number: BP120-500 )
- HEPES (pH 7.5) (Fisher Scientific, catalog number: BP310-100 )
- Ammonium sulfate (Fisher Scientific, catalog number: A702-500 )
- Tricine buffer (pH 7.8) (Sigma-Aldrich, catalog number: T0377 )
- MgSO4 (Thermo Fisher Scientific, catalog number: M63500 )
- Sodium azide (MP Biomedicals, catalog number: 210289125 )
- Luciferin (Biotium, catalog number: 10101-1 )
- Glycylglycine (pH 7.8) (MP Biomedicals, catalog number: 210185610 )
- ddH2O
- Hydrochloric acid (Fisher Scientific, catalog number: A144212 )
- GITC lysis buffer (see Recipes)
- Glassmilk (see Recipes)
- New Wash (NW-) Buffer (see Recipes)
- Elution Buffer (see Recipes)
- PPK buffer (see Recipes)
- Luciferase reaction buffer (see Recipes)
- Luciferin working solution (see Recipes)
Equipment
- Eppendorf Pipettes: 100-1,000 μl, 20-200 μl, 2-20 μl, 1-10 μl
- Thermomixer (Eppendorf, model: 5350 )
- Microcentrifuge (Eppendorf, model: 5415D )
- Incubator 37 °C (VWR, model: 1555 )
- -80 °C Freezer (Eppendorf, New BrunswickTM, model: Innova® U725 , catalog number: U9440-0002)
- Fluostar Omega Plate Reader with luminescence reading function and injector module (BMG Labtech, catalog number: 0415-102 )
- pH meter
- Vortexer
- Refrigerator (4 °C)
Software
- Fluostar Omega Control and Evaluation Software (BMG Labtech, catalog number: 1300-501)
Procedure
The procedure is ideally suited to extract and quantify polyP from gram-negative PPK-containing bacteria, including P. aeruginosa, which accumulate polyP under stress, e.g., sublethal concentration of hypohalous acids or nutrient starvation (for details regarding growth and stress treatment, see Ault-Riché et al., 1998; Groitl et al., 2017).
- Prepare samples for the extraction of polyP
Note: As GITC is considered as being moderately toxic, keep separate GITC waste container for tips and flow-through.- Collect bacterial cells sufficient to yield 100-300 μg of total cellular protein (approximately 1 ml of P. aeruginosa culture at OD600 = 1), and spin down cells at 13,000 x g, 4 °C.
- Discard supernatant.
- Resuspend cell pellets in 0.25 ml GITC lysis buffer.
- Incubate the samples in a Thermomixer for 10 min at 95 °C without mixing.
- Take 20 μl samples for determining the protein concentration (see Step B2).
- Store samples at -80 °C after cooling down to RT until further procedure. Degradation is no concern as GITC denatures proteins including proteases.
Note: We always processed the samples within 7 days.
- Determine the protein concentration of the samples
- Prepare a standard curve by dissolving BSA in GITC lysis buffer to a final concentration of 0, 0.25, 0.5, 1, 1.5 or 2 μg/μl.
- Mix 195 μl Bradford Reagent with 5 μl of the BSA standard or 5 μl of the lysed samples, and incubate for 5-10 min at room temperature (RT) in microcentrifuge tubes. Use triplicates for each sample.
- Transfer samples into 96-well clear plates with flat bottom and measure absorbance at 595 nm in the Fluostar Omega Plate Reader.
- Calculate the protein concentration as described in the Data analysis section. Samples will be normalized at the end.
- Extract polyP
- Thaw samples from Step A6 and vortex to mix.
- Add subsequently to the samples: 15 μl 10% w/v SDS, 0.5 ml 95% v/v ethanol, and 5 μl glassmilk (see Recipes) and vortex in between.
- Transfer samples to silica membrane spin columns and spin down in microcentrifuge for 3 min at 3,000 x g, RT. Note that we observed a pellet formation in the flow-through at velocities higher than 3,000 x g.
- Discard flow-through.
- Add 0.75 ml NW-Buffer (see Recipes).
- Spin down for 3 min at 3,000 x g at RT and discard flow-through.
- Repeat Steps C5 and C6.
- Dry samples by centrifugation at 11,000 x g for 3 min at RT.
- Transfer column to a clean 1.5 ml microcentrifuge tube.
- Add 50 μl Elution buffer to the column, incubate for 15 min at RT and elute polyP by centrifugation at 11,000 x g for 3 min at RT.
- Samples can be stored at -80 °C.
Note: Processing should be done within 1-3 days.
- Convert polyP into ATP
- Thaw samples from Step C11 and vortex to mix.
- Prepare two PPK Assay Master Mixes according to the number of samples. Master mix I (+PPK) contains 50 nM of E. coil PPK, whereas master mix II (-PPK) does not. The reactions without PPK are used to correct for cellular ATP that was extracted during the process. The following volumes refer to the amount required for one sample.
PPK Assay Master Mix I (+ PPK) (total 20 μl per sample):7.5 μl 4-fold PPK Buffer 0.9 μl ultra-pure ADP [stock concentration of 8.4 mM (may vary among batches); final concentration of 250 μM] 0.7 μl E. coli PPK [stock concentration of 2.4 μM (may vary among batches); final concentration of 50 nM] 10.9 μl H2O
PPK Assay Master Mix II (-PPK) (total 20 μl per sample):7.5 μl 4-fold PPK Buffer 0.9 μl ultra-pure ADP ultra-pure ADP [stock concentration of 8.4 mM (may vary among batches); final concentration of 250 μM] 11.6 μl H2O - Combine 20 μl PPK Assay Master Mix I or II with 10 μl of the sample by pipetting up and down twice.
- Incubate the reaction for 1 h at 37 °C in the incubator to allow PPK to fully convert polyP to ATP.
- Inactivate PPK by incubating samples at 95 °C for 2 min in the thermomixer.
- Cool samples on ice or at 4 °C, then spin 1 min at 11,000 x g and continue with the supernatant.
- Detect ATP
Note: Much of this is specific to the Omega plate reader. Modify as necessary for the instrument you have available.
- Take 10 μl aliquots of the PPK-digested samples (incubated in PPK Assay Master Mix I) and undigested samples (incubated in PPK Assay Master Mix II), and transfer them into wells of white flat bottom 96-well plate.
- Prepare Luciferase Reaction Master Mix as follows and keep it at RT.
Luciferase Reaction Master Mix (total 10 ml):
Note: The reaction is very temperature sensitive, therefore do not store the reaction master mix on ice. Because luciferase loses its activity quickly, use the reaction master mix within 15-20 min of preparation.0.5 ml 20-fold Luciferase Reaction Buffer 10 μl 1 M DTT (final concentration of 1 mM) (Thaw on ice) 1 ml Luciferin working solution (final concentration of 100 μM) 1 μl luciferase [stock concentration of 244 μM (may vary among batches), final concentration of 25 nM] 8.5 ml H2O - Prime injector of Omega plate reader with 2 ml Luciferase Reaction Master Mix.
- Set the temperature of the Omega plate reader to 25 °C and the gain to 3,000 (This is specific to the OMEGA plate reader and may vary in other plate readers).
- Inject 90 μl of Luciferase Reaction Master Mix to each sample, mix for 10 sec fast mixing and measure luminescence over 2-3 min. The length of each cycle depends on the amount of samples.
- Measure the PPK-digested (incubated with PPK-Assay Master Mix I) samples in the same order as the undigested samples (incubated with PPK-Assay Master Mix II). Measure luminescence over 2-3 min.
- When finished, wash injector by priming with 4 ml dH2O.
Data analysis
- Calculate protein amount in each sample:
- Standard Curve: plot the measured absorbances against the respective BSA concentrations of the standard curve.
- Determine the trend line and calculate a linear equation.
- Calculate the protein amount for each sample using the equation of the trend line.
Note: Protein content of the samples should be in the range of 100-300 μg. Dilute if necessary.
- Calculate fold-change of polyP in treated relative to untreated cells
- Subtract the relative luminescence units (RLU) values of samples incubated in the absence of PPK (incubated with PPK-Assay Master Mix II) from the values of the same samples incubated in the presence of PPK (incubated with PPK-Assay Master Mix I), usually of cycle 2.
Note: The RLU values of the undigested samples represent the baseline. - Normalize the resulting RLU for each sample to the respective protein amount.
- Calculate the fold-change in polyP accumulation in each sample by normalizing the values of samples after treatment (i.e., treatment with HOCl, HOBr and HOSCN, respectively) to those without treatment.
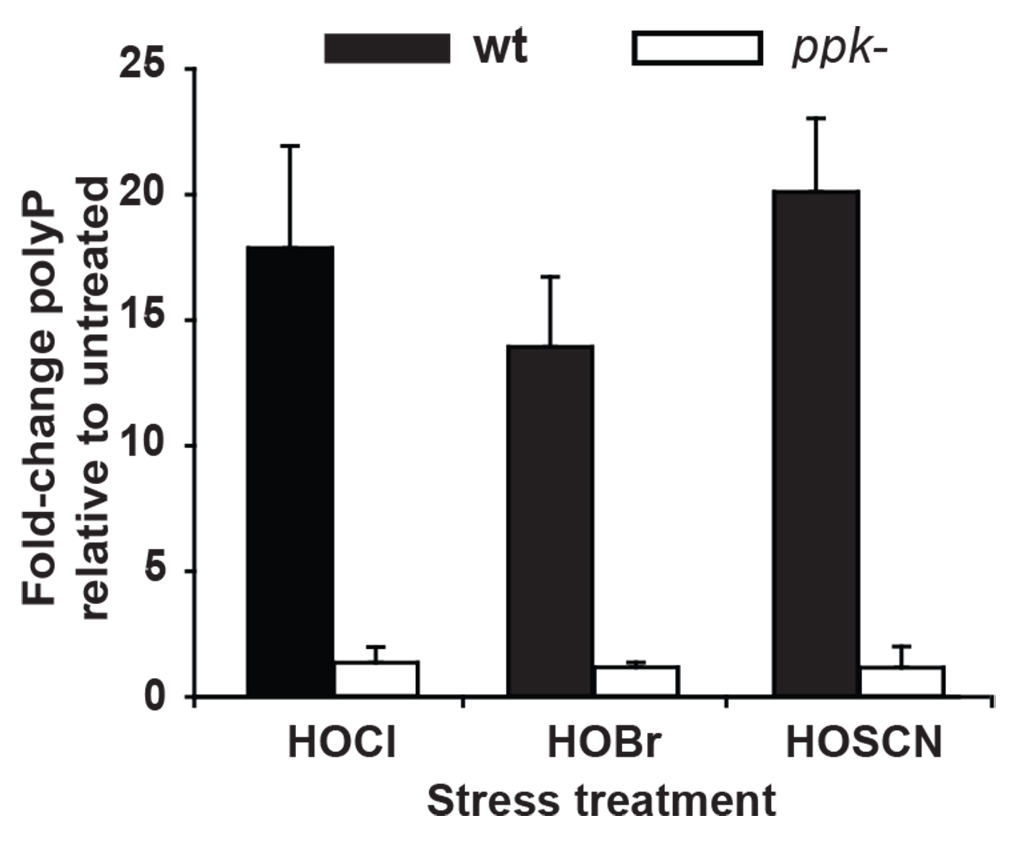
Figure 2. Extraction and quantification of polyP from P. aeruginosa PA14 wild-type and ∆ppk cells. PA14 wild-type (WT) and ∆ppk cells were grown to mid-log phase and incubated with sublethal concentrations of HOCl (0.5 mM), HOBr (0.15 mM) or HOSCN (0.25 mM) for 2.5 h. Then, polyP was extracted by binding to glassmilk and quantified via the luciferase-based detection system after conversion of the extracted polyP to ATP. The resulting amount was normalized to the respective protein amount in each sample. Fold-change in polyP levels was calculated relative to the untreated cells. Experiments were performed independently for at least three times. Data represent means ± S.D. [Copyright© 2017, Wiley Online Library (Groitl et al., 2017)].
- Subtract the relative luminescence units (RLU) values of samples incubated in the absence of PPK (incubated with PPK-Assay Master Mix II) from the values of the same samples incubated in the presence of PPK (incubated with PPK-Assay Master Mix I), usually of cycle 2.
Notes
- Short chains of polyP (< 60 Pi units) are considered to bind less efficiently to glassmilk than longer chains and most likely are lost during this extraction method. While bacteria produce polyP of up to 1,000 Pi molecules per chain, the average chain length of polyP in eukaryotes is considerably shorter. For commercially available polyP45, which is a mix of different polyP with enrichment in polyP chains with 45 phosphate residues, we determined a recovery rate of polyP between 24% and 38% using polyP concentrations in a range between 500 nM and 10 mM. In comparison to previously described methods, the previously described methods allow a similar recovery, however, the PPK-based detection system appears to be more sensitive (Canadell et al., 2016; Ota and Kawano, 2017).
- The present protocol was designed for the extraction and quantification of polyP from bacteria after treatment with sublethal concentrations of hypohalous acids or after nutrient starvation (shift from rich media into minimal media with low phosphate and lack of amino acids). These conditions are known to result in the accumulation of high amounts of long-chain polyP (Ault-Riché et al., 1998; Gray et al., 2014). For samples that contain lower polyP levels, two additional steps can be incorporated after extraction of polyP by glassmilk to reduce the risk of interference during PPK reaction and/or ATP detection: (i) Treatment with apyrase to decrease the background ATP level, and (ii) Benzonase treatment to remove DNA/RNA (Ault-Riché et al., 1998).
Recipes
- GITC lysis buffer (500 ml)
4 M guanidine thiocyanate
50 mM Tris-HCl (pH 7.0)
Store at RT for up to several months - Glassmilk (25 ml)
Note: Binding capacity ≥ 100 nmol polyphosphates (in Pi equivalents) per μl. Glassmilk will also bind DNA and RNA.- Suspend 5 g of silicon dioxide in 40 ml of 0.1 M HCl and mix thoroughly
- Pellet the glassmilk by centrifugation at 2,000 x g for 5 min at RT
- Remove supernatant and repeat Steps a and b
- Wash the pellet with 10 ml ddH2O
- Repeat Step d at least once and check pH (pH has to be neutral)
- Store at room temperature for up to several months
- New Wash (NW) Buffer (500 ml)
5 mM Tris-HCl (pH 7.5)
50 mM NaCl
5 mM EDTA
50% v/v ethanol
Store at RT for up to several months - Elution Buffer (25 ml)
50 mM Tris-HCl (pH 8)
Store at RT for up to several months - PPK Buffer (4-fold) (25 ml)
200 mM HEPES (pH 7.5)
200 mM ammonium sulfate
20 mM MgCl2
Store at RT for up to several months - Luciferase Reaction Buffer (20-fold) (25 ml, storage time several months at RT)
500 mM Tricine buffer (pH 7.8)
100 mM MgSO4
2 mM EDTA
2 mM Sodium azide
2 mg/ml BSA
Store at RT for up to several months - Luciferin working solution (50x 1 ml)
1 mM luciferin dissolved in 25 mM glycylglycine (pH 7.8)
Aliquot in 1 ml aliquots and store at -80 °C for up to several months
Acknowledgments
This work was funded by the National Institute of Health grants GM065318 and GM116582 to U.J. J.-U. D. was supported by a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft (DA 1697/1-1). We thank Dr. Michael J. Gray (University of Alabama) for helping to establish the assay and fruitful discussions.
Competing interests
The authors declare no conflict of interest.
References
- Ault-Riché, D., Fraley, C. D., Tzeng, C. M. and Kornberg, A. (1998). Novel assay reveals multiple pathways regulating stress-induced accumulations of inorganic polyphosphate in Escherichia coli. J Bacteriol 180(7): 1841-1847.
- Canadell, D., Bru, S., Clotet, J. and Ariño, J. N. (2016). Extraction and quantification of polyphosphate in the budding yeast Saccharomyces cerevisiae. Bio-protocol 6(14): e1874.
- Dahl, J. U., Gray, M. J., Bazopoulou, D., Beaufay, F., Lempart, J., Koenigsknecht, M. J., Wang, Y., Baker, J. R., Hasler, W. L., Young, V. B., Sun, D. and Jakob, U. (2017). The anti-inflammatory drug mesalamine targets bacterial polyphosphate accumulation. Nat Microbiol 2: 16267.
- Gomez Garcia, M. R. (2014). Extraction and quantification of poly P, poly P analysis by Urea-PAGE. Bio-protocol 4(9): e1113.
- Gray, M. J. and Jakob, U. (2015). Oxidative stress protection by polyphosphate--new roles for an old player. Curr Opin Microbiol 24: 1-6.
- Gray, M. J., Wholey, W. Y., Wagner, N. O., Cremers, C. M., Mueller-Schickert, A., Hock, N. T., Krieger, A. G., Smith, E. M., Bender, R. A., Bardwell, J. C. and Jakob, U. (2014). Polyphosphate is a primordial chaperone. Mol Cell 53(5): 689-699.
- Groitl, B., Dahl, J. U., Schroeder, J. W. and Jakob, U. (2017). Pseudomonas aeruginosa defense systems against microbicidal oxidants. Mol Microbiol 106(3): 335-350.
- Maisonneuve, E. and Gerdes, K. (2014). Molecular mechanisms underlying bacterial persisters. Cell 157(3): 539-548.
- Ota, S. and Kawano, S. (2017). Extraction and molybdenum blue-based quantification of total phosphate and polyphosphate in Parachlorella. Bio-protocol 7(17): e2539.
- Rao, N. N., Gomez-Garcia, M. R. and Kornberg, A. (2009). Inorganic polyphosphate: essential for growth and survival. Annu Rev Biochem 78: 605-647.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Dahl, J. U., Xie, L. and Jakob, U. (2018). Extraction and Quantification of Polyphosphate (polyP) from Gram-negative Bacteria. Bio-protocol 8(18): e3011. DOI: 10.21769/BioProtoc.3011.
Category
Microbiology > Microbial biochemistry > Other compound
Microbiology > Microbial physiology > Stress response
Biochemistry > Other compound > Polyphosphate
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.