- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Preparation of Cerebellum Granule Neurons from Mouse or Rat Pups and Evaluation of Clostridial Neurotoxin Activity and Their Inhibitors by Western Blot and Immunohistochemistry


Published: Vol 8, Iss 13, Jul 5, 2018 DOI: 10.21769/BioProtoc.2918 Views: 11832
Reviewed by: Andrea PuharXuecai GeAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Cerebellar Granule Neurons (CGN) from post-natal rodents have been widely used as a model to study neuronal development, physiology and pathology. CGN cultured in vitro maintain the same features displayed in vivo by mature cerebellar granule cells, including the development of a dense neuritic network, neuronal activity, neurotransmitter release and the expression of neuronal protein markers. Moreover, CGN represent a convenient model for the study of Clostridial Neurotoxins (CNT), most notably known as Tetanus and Botulinum neurotoxins, as they abundantly express both CNT receptors and intraneuronal substrates, i.e., Soluble N-ethylmaleimide-sensitive factor activating protein receptors (SNARE proteins). Here, we describe a protocol for obtaining a highly pure culture of CGN from postnatal rats/mice and an easy procedure for their intoxication with CNT. We also illustrate handy methods to evaluate CNT activity and their inhibition.
Keywords: Cerebellar granule neuronsBackground
The large family of Clostridial Neurotoxins (CNT) is formed by Tetanus Neurotoxin (TeNT) and the many variants of Botulinum Neurotoxins (BoNT) which are the neuroparalytic toxins responsible for tetanus and botulism, respectively (Schiavo et al., 2000; Johnson and Montecucco, 2008; Rossetto et al., 2014). TeNT, the seven BoNT serotypes (BoNT/A to /G) and their many subtypes are metalloproteases that cause neuroparalysis by blocking neurotransmitter release via the cleavage of SNARE proteins (Soluble N-ethylmaleimide-sensitive factor activating protein receptors), the three essential proteins governing the fusion of synaptic vesicle with the presynaptic plasma membrane (Rossetto et al., 2014; Montecucco and Rasotto, 2015; Pirazzini et al., 2017). In addition, some putative novel serotypes (BoNT/X and BoNT/En aka eBoNT/J) (Zhang et al., 2017; Brunt et al., 2018; Zhang et al., 2018) and a BoNT-like toxin (BoNT/Wo) (Zornetta et al., 2016) displaying metalloprotease activity against SNARE proteins have been recently identified (Azarnia Tehran and Pirazzini, 2018). Yet, whether they are naturally produced and can be considered true BoNT still requires validation. Each toxin has a selective action against one specific protein that is cleaved at a distinct peptide bond (Binz, 2013; Pantano and Montecucco, 2014; Zornetta et al., 2016; Zhang et al., 2017; Zhang et al., 2018): BoNT/B, /D, /F /G, /Wo and /X hydrolyze VAMP-1/2 (vesicle-associated membrane protein) while BoNT/A and BoNT/E cleave the membrane protein SNAP-25 (synaptosomal-associated protein of 25 kDa). BoNT/C and BoNT/En are unique as they cleave more than one SNARE type: BoNT/C cleaves SNAP-25 and many isoforms of syntaxin (Pirazzini et al., 2017; Zanetti et al., 2017); instead BoNT/En cleaves different members of VAMP family and SNAP-25/23 (Zhang et al., 2018). To reach their intraneuronal substrates, CNT carry out a very sophisticated mechanism of intoxication aimed at delivering the catalytic part of the toxin within the cytosol of nerve terminals (reviewed in [Montal, 2010; Rossetto et al., 2014; Pirazzini et al., 2017]).
This process relies on cardinal functions of neuron physiology which are exploited by BoNT to enter the neuron: the expression of appropriate glycolipid and protein receptors for binding (Binz and Rummel, 2009; Rummel, 2017), the recycling of synaptic vesicles for internalization (Matteoli et al., 1996; Harper et al., 2011; Colasante et al., 2013), the generation of an electrochemical gradient across synaptic vesicle membrane to translocate the catalytic domain in the cytosol (Montal, 2010; Pirazzini et al., 2016) and the presence of a redox-chaperone system to enable SNARE proteins’ cleavage (Pirazzini et al., 2018). These features are fully preserved by cultured Cerebellar Granule Neurons (CGN), a primary culture of cerebellar granule cells from post-natal rodent cerebellum. The cerebellar cortex is composed of a few neuronal types like Purkinje cells, inhibitory interneurons and granule cells that form a highly organized tissue with well-characterized neuronal circuitries (Bilimoria and Bonni, 2008). Cerebellar granule cells constitute the most numerous and homogeneous neuronal population and can be easily isolated (Messer, 1977). Cultured CGN recapitulate many characteristics of development and maturation observed in vivo and have been extensively used as a useful model to study basic molecular and biological processes of neuron physiology like apoptosis, migration and differentiation (Contestabile, 2002).
Many neuronal models, including spinal cord neurons, hiPSC derived neurons, mES derived neurons, hippocampal neurons, cortical neurons and several methods have been developed to study BoNT activity in vitro (Pellett, 2013).In our laboratory, we choose CGN as their preparation is relatively simple, rapid and very reliable and it provides a highly pure (more than 95%) and homogeneous (mostly granule cells) neuronal culture model to conveniently evaluate CNT activity by monitoring the cleavage of SNARE proteins via Western blotting or immunocytochemistry (Pirazzini et al., 2011; Eleopra et al., 2013; Pirazzini et al., 2013b and 2013c). Moreover, CGN can be adapted to the investigation of putative inhibitors and can be used as a solid platform for screening anti-BoNT antitoxins (Pirazzini et al., 2013a and 2014; Azarnia Tehran, et al., 2015; Zanetti et al., 2015; Pirazzini and Rossetto, 2017). Here, we describe a simple protocol for fast isolation of CGN, an easy procedure for their intoxication with CNT, and two methods (Western blot and immunocytochemistry) to evaluate their toxicity and inhibition.
Materials and Reagents
- 0.22 µm filters (33 mm – Merck, Millex, catalog number: SLGP033RS )
- Culture plates
6 wells (Corning, Falcon®, catalog number: 353046 )
24 wells (Corning, Falcon®, catalog number: 353047 )
96 wells (Corning, Falcon®, catalog number: 353072 ) - 35 mm Petri dishes (Corning, Falcon®, catalog number: 353001 )
- 50 ml sterile conical plastic tubes (Corning, Falcon®, catalog number: 352070 )
- Coverslips 13 mm #1 (Thermo Fisher Scientific, catalog number: 1014355113NR1 )
- Micropipette tips
1,000 µl tips (SARSTEDT, catalog number: 70.762.211 )
200 µl tips (SARSTEDT, catalog number: 70.760.211 )
10 µl tips (SARSTEDT, catalog number: 70.1130.210 ) - Nitrocellulose membranes (Sartorius, Stedim Biotech, catalog number: 11306-41BL )
- Plastic Pasteur pipettes (LP ITALIANA, catalog number: 133030 )
- Serological plastic pipettes:
5 ml plastic pipettes (Corning, Falcon®, catalog number: 357543 )
10 ml plastic pipettes (Corning, Falcon®, catalog number: 357551 )
25 ml plastic pipettes (Corning, Falcon®, catalog number: 357525 ) - Single-use stericups 0.22 µm filters ExpressTM Plus (Merck, catalog number: SCGPU0SRE )
- Sterile scalpel (CHEMIL, catalog number: 11230028 )
- Mice or rats (any gender) postnatal day 4-6
- Distilled H2O
- Ethanol (Sigma-Aldrich, catalog number: 46139 )
- Poly-L-lysine hydrobromide solution (Sigma-Aldrich, catalog number: P8920 )
- Phenol red (Sigma-Aldrich, catalog number: P3532 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P5405 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S3014 )
- D-(+)-Glucose (Sigma-Aldrich, catalog number: G7528 )
- Sodium phosphate monobasic monohydrate (NaH2PO4) (Sigma-Aldrich, catalog number: S3522 )
- KH2PO4 (Sigma-Aldrich, catalog number: P5655 )
- HEPES (Sigma-Aldrich, catalog number: H3375 )
- Calcium chloride dihydrate (CaCl2·2H2O) (Sigma-Aldrich, catalog number: C3306 )
- Magnesium sulfate heptahydrate (MgSO4·7H2O) (Sigma-Aldrich, catalog number: 63138 )
- Fatty acid free bovine serum albumin (Sigma-Aldrich, catalog number: A7030 )
- Trypsin from porcine pancreas (Sigma-Aldrich, catalog number: T4799 )
- Trypsin inhibitor from soybean (Sigma-Aldrich, catalog number: T9003 )
- CompleteTM Mini, EDTA-free Protease Inhibitor Cocktail (Sigma-Aldrich, Roche Diagnostics, catalog number: 04693159001 )
- Trypan blue solution, 0.4% (Thermo Fisher Scientific, catalog number: 15250061 )
- Deoxyribonuclease I from bovine pancreas (Sigma-Aldrich, catalog number: D5025 )
- Basal Medium Eagle (BME) (Thermo Fischer Scientific, GibcoTM, catalog number: 21010046 )
- GlutamaxTM Supplement (Thermo Fischer Scientific, GibcoTM, catalog number: 35050061 )
- Gentamicin solution (Sigma-Aldrich, catalog number: G1272 )
- Cytosine β-D-arabinofuranoside (AraC) (Sigma-Aldrich, catalog number: C1768 )
- Fetal Bovine Serum (FBS) (EUROCLONE, catalog number: EUS 028877 )
Note: Comparative testing for culture optimization is needed when changing serum lot or supplier. - Trizma® base (Sigma-Aldrich, catalog number: T1503 )
- Trizma® hydrochloride (Sigma-Aldrich, catalog number: T3253 )
- Glycine (Sigma-Aldrich, catalog number: G8898 )
- Sodium dodecyl sulfate (SDS, Sigma-Aldrich, catalog number: L3771 )
- Bromophenol Blue (Sigma-Aldrich, catalog number: B0126 )
- Glycerol (Sigma-Aldrich, catalog number: G9012 )
- 2-mercaptoethanol (Sigma-Aldrich, catalog number: M6250 )
- NuPAGETM 12% Bis-Tris Gels (Thermo Fisher Scientific, catalog number: NP0341BOX )
- NuPAGETM 4-12% Bis-Tris Gels (Thermo Fisher Scientific, catalog number: NP0321BOX )
- NuPAGETM MES SDS Running Buffer (20x) (Thermo Fisher Scientific, catalog number: NP0002 )
- NuPAGETM MOPS SDS Running Buffer (20x) (Thermo Fisher Scientific, catalog number: NP0001 )
- Methanol (Sigma-Aldrich, catalog number: 322415 )
- Ponceau S (Sigma-Aldrich, catalog number: P3504 )
- Acetic acid (Sigma-Aldrich, catalog number: 45726 )
- Tween 20® (Sigma-Aldrich, catalog number: P9416 )
- Anti Syntaxin-1A/1B antibody (homemade)
- Anti-BoNT/A-cleaved SNAP-25 antibody (homemade)
- Anti-BoNT/B-TeNT-cleaved VAMP2 antibody (homemade)
- Anti-BoNT/E-cleaved SNAP-25 antibody (homemade)
- Anti-SNAP-25 antibody (SMI81, Abcam, catalog number: ab24737 )
- Anti-Syntaxin1 antibody (Synaptic System, catalog number: 110 011 )
- Anti-VAMP-2 antibody (Synaptic System, catalog number: 104 211 )
- Paraformaldehyde (Sigma-Aldrich, catalog number: P6148 )
- Ammonium chloride (Sigma-Aldrich, catalog number: A9434 )
- Fluorescence mounting medium (Agilent Technologies, Dako, catalog number: S3023 )
- HRP-conjugated or fluorescent-conjugated secondary antibodies (any supplier)
- Sodium dodecyl sulfate (Sigma-Aldrich, catalog number: 74255 )
- Tetanus neurotoxin and botulinum neurotoxins. The neurotoxins used in our laboratory are purified as previously described (Schiavo and Montecucco, 1995; Shone and Tranter, 1995) or produced recombinantly (Bade et al., 2004; Zanetti et al., 2017)
- Krebs solution (10x) (see Recipes)
- 155 mM MgSO4 stock solution (see Recipes)
- 12.2 mM CaCl2 stock solution (see Recipes)
- Solution A (see Recipes)
- Solution B (see Recipes)
- Solution C (see Recipes)
- Solution D (see Recipes)
- Solution E (see Recipes)
- Poly-L-lysine solution (see Recipes)
- BME complete medium, CGN culture medium (see Recipes)
- Cytosine β-D-arabinofuranoside (AraC) stock solution (see Recipes)
- PBS (see Recipes)
- PBST (see Recipes)
- SDS-PAGE Sample loading buffer (4x) (see Recipes)
- Transfer buffer (see Recipes)
- Ponceau S solution (see Recipes)
- Blocking buffer (see Recipes)
- 4% Paraformaldehyde solution (see Recipes)
- Quenching solution (see Recipes)
- Permeabilization solution (see Recipes)
- Blocking solution (see Recipes)
Equipment
- Micropipettes (any supplier)
- Pipette controller (any supplier)
- Centrifuge for conical tubes (Eppendorf, model: 5804 R )
- Dissecting hood (any supplier)
- Dissecting stereomicroscope (OPTIKA Microscopes, model: SZM-LED2 )
- Sterile laminar flow hood (any supplier)
- Hemacytometer (any supplier)
- Humidified incubator (5% CO2 at 37 °C) (any supplier)
- Ice bucket and ice
- Scissors #1 (Rudolf Medical GmbH, catalog number: RU 2675-18 )
- Forceps #2 (Rudolf Medical GmbH, catalog number: RU 7584-16 )
- Scissors #3 (Rudolf Medical GmbH, catalog number: RU 2246-09 )
- Tweezers #4 (Rudolf Medical GmbH, catalog number: RU 4240-05 )
- Mini gel tank (Thermo Fisher Scientific catalog number: A25977 )
- Mini Trans-Blot® Cell (Bio-Rad Laboratories, catalog number: 1703930 )
- Thermoblock (any supplier)
- Thermosettable water-bath (37 °C) (any supplier)
- Vortex mixer (any supplier)
- Epifluorescence or confocal microscope: Leica CTR6000 equipped with X5 N PL AN (Leica Microsystems, model: Leica CTR6000 )
- 0.12, x20 N PL AN 0.40 objectives or Leica SP5 equipped with 100x HCX PL APO NA 1.4 objective, respectively (Leica Microsystems, Wetzlar, Germany)
Procedure
- Culturing Cerebellar Granule neurons (CGN)
- Prepare poly-L-lysine coated 24 well-plates at least 1 h before preparation.
Notes:- According to the experimental design, different cell cultures plates (6, 12, 24, 96 wells) or dishes can be used.
- Cover with 1,000, 500, 250, 50 µl of poly-L-lysine solution each well of 6, 12, 24, 96 wells cell cultures plates respectively. Incubate at room temperature for at least 2 h.
- Poly-L-lysine solution has to be aspirated, and dry the plate/dish/coverslip before cell plating (see Step A8, Note b).
- According to the experimental design, different cell cultures plates (6, 12, 24, 96 wells) or dishes can be used.
- Wipe down with 70% ethanol the dissecting area of the hood and the stereomicroscope to be used for dissection. Under a dissection hood, add 2.5 ml of cold Solution A into two 35 mm plastic Petri dish and keep them on ice in an ice bucket. The surgical tools to be used hereafter must be sterile.
- Wipe the head of the pup with 70% ethanol and euthanize it by decapitation with scissors #1. Use forceps #2 to grab the head of the pup (Figure 1A) and with scissors #3 remove the skin covering the skull and the muscles at the base of the head (Figure 1B). Note: Skin incision should be performed starting from the forehead (roughly between eyes, black star in Figure 1A) and should continue straight toward the neck (green dotted traces of Figure 1A and of Figure 1B top left). The result of this step is shown in Figure 1B (top right). Then, starting from the sagittal incision (few millimeters above the original incision site), cut the skin perpendicularly toward the jaw (red dotted trace of Figure 1A and of Figure 1B bottom left). Repeat the step for the other side and pull away the skin. If necessary cut the muscles at the base of the skull (white star in Figure 1B, bottom right panel). Muscles can be chopped roughly but avoiding damage of the skull because the cerebellum is underneath this area. The final result of these steps is shown in Figure 1C.
- With a second pair of scissors #3 (clean, i.e. not used for skinning to avoid contamination), incise the skull over the hemispheres area (white circle in Figure 1C) and cut the cranial bone along the sagittal suture (black dotted lines in Figures 1C and 2A) until the interception with the interaural line (white dotted traces of Figure 1C and Figure 2A). Once there, cut perpendicularly in both directions following the interaural lines until the acoustic meati (white arrow in Figure 2A bottom panel) to generate a coronal craniotomy of the parietal bone. Lift up the bone to spot the mid brain, the cerebellum and part of the brain stem (cyan dotted lines in top left Figure 2B). Cut sagittal (black dotted line in Figure 2B top right) until the foramen magnum without damaging the cerebellum. Pull out the two “bone-lids” with a pair of tweezers #4 (left bottom panel of Figure 2B). As a result, mid brain, cerebellum and brainstem are fully exposed (right bottom panel of Figure 2B).
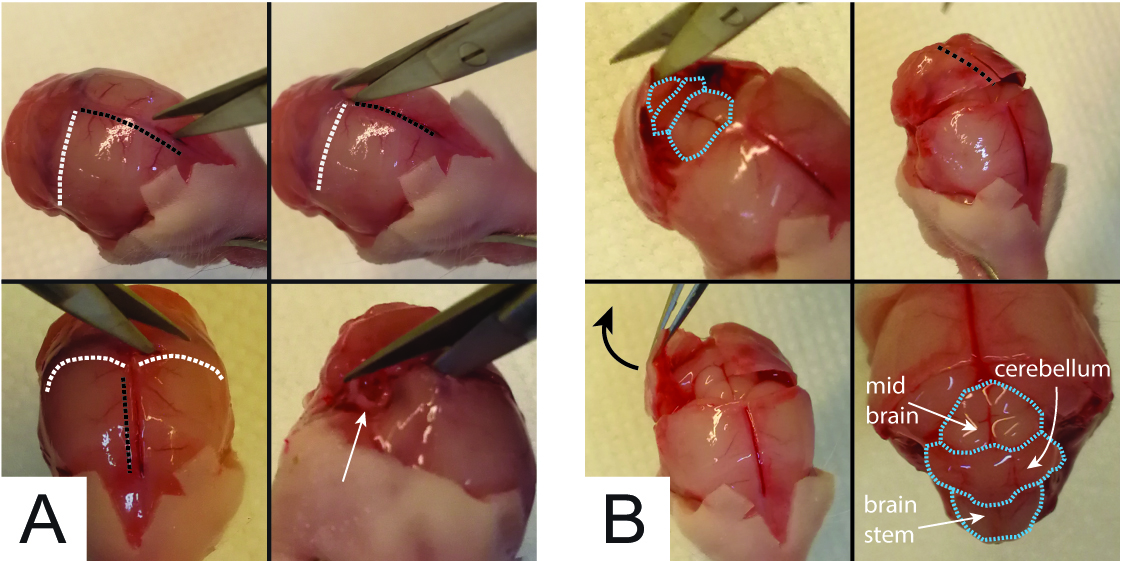
Figure 2. Craniotomy to expose the cerebellar area - Using a second pair of forceps #2 (clean, i.e., different from that used to grab the pup face), gently detach the mid brain from the hemispheres (Figure 3A). This step is achieved by introducing forceps #2 in between the midbrain and the hemispheres (Figure 3A) and cutting the connections between them by tightening forceps endings. Then, using the same forceps as a spoon, gently grab the midbrain, cerebellum and brainstem (Figure 3B) and transfer them into a 35 mm plastic Petri dish filled with 2.5 ml of ice cold Solution A (Figure 3C).
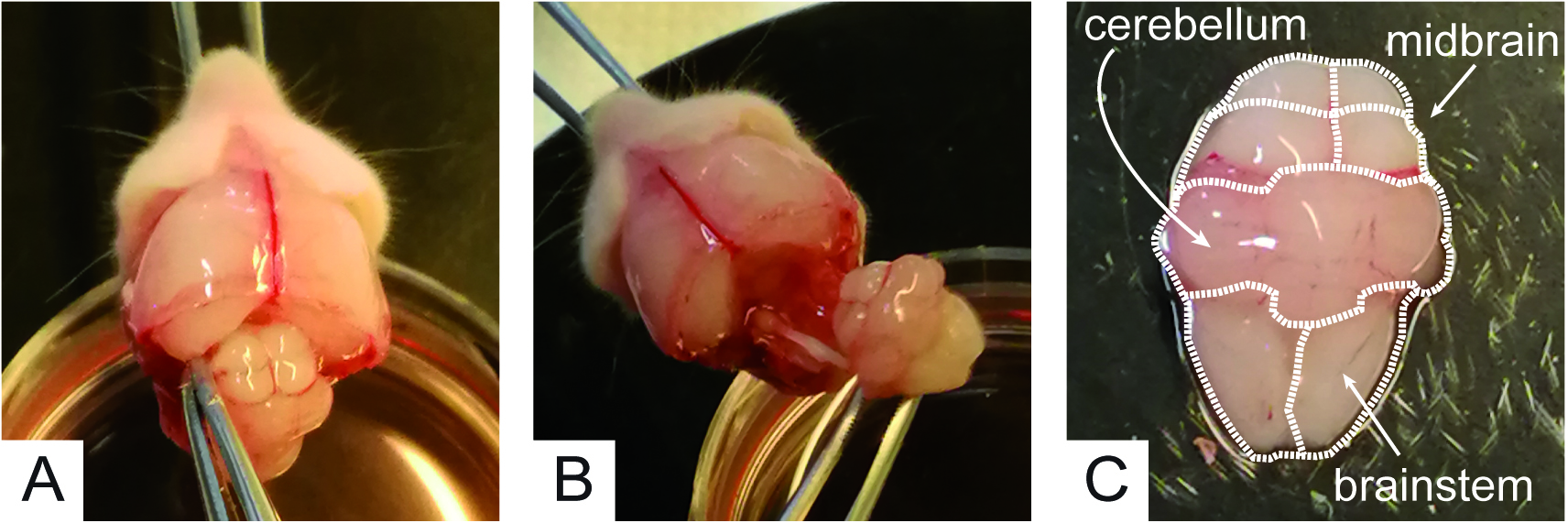
Figure 3. Isolation of midbrain, cerebellum and brainstem. Dotted lines spot the cerebellum the mid brain and the brainstem. - Under the dissection microscope, use one pair of tweezers #4 to hold the brainstem and a second pair of tweezers #4 to remove the meninges from the cerebellum until it takes on a matted white appearance. With tweezers #4, gently separate the cerebellum from the brainstem and move it into a fresh 35 mm plastic Petri dish filled with 2.5 ml of ice cold fresh Solution A to be kept on ice (Figure 4A). Repeat this procedure for 5 cerebella maximum.
Notes:- Decapitation must be performed at the base of the neck to avoid cerebellum damage.
- Dissect one pup per time and try to reduce the dissection time to avoid neuronal death.
- Blood vessels of the meningeal branches are very useful to spot meninges as they are red-colored and run along the entire wrapping, forming a visible network. Larger vessels can be grasped with the tweezers and pulled to remove the meninges. Once skilled, a user can take advantage of this trick to rapidly peel of very large portions of the meningeal wrapping to speed up this step.
- Use fresh Solution A for each cerebellum.
- Decapitation must be performed at the base of the neck to avoid cerebellum damage.
- Remove Solution A, gather (Figure 4A) and mince cerebella using a sterile scalpel (Figure 4B).
Note: Mincing is carried out by repetitive up-and-down movements of the scalpel. Scalpel blade must be sharp to produce clear incisions to the tissue, thus limiting neuron damage. A “mortar-pestle” mincing (non-sharp scalpel) must be avoided as it may cause neuronal death. - Using a small volume of pre-warmed Solution B (~1 ml) and a sterile plastic Pasteur pipette, collect minced cerebella and transfer them into a 50 ml plastic tube with the remaining Solution B and incubate for 15-30 min at 37 °C, gently swirling every 2-3 min (Figure 4C).
Notes:- For 3 cerebella, 15 min at 37 °C is sufficient; with more cerebella (4-5) 25-30 min is recommended.
- During this incubation period, remove poly-L-lysine by aspiration under a sterile laminar flow hood and let the plates to dry.
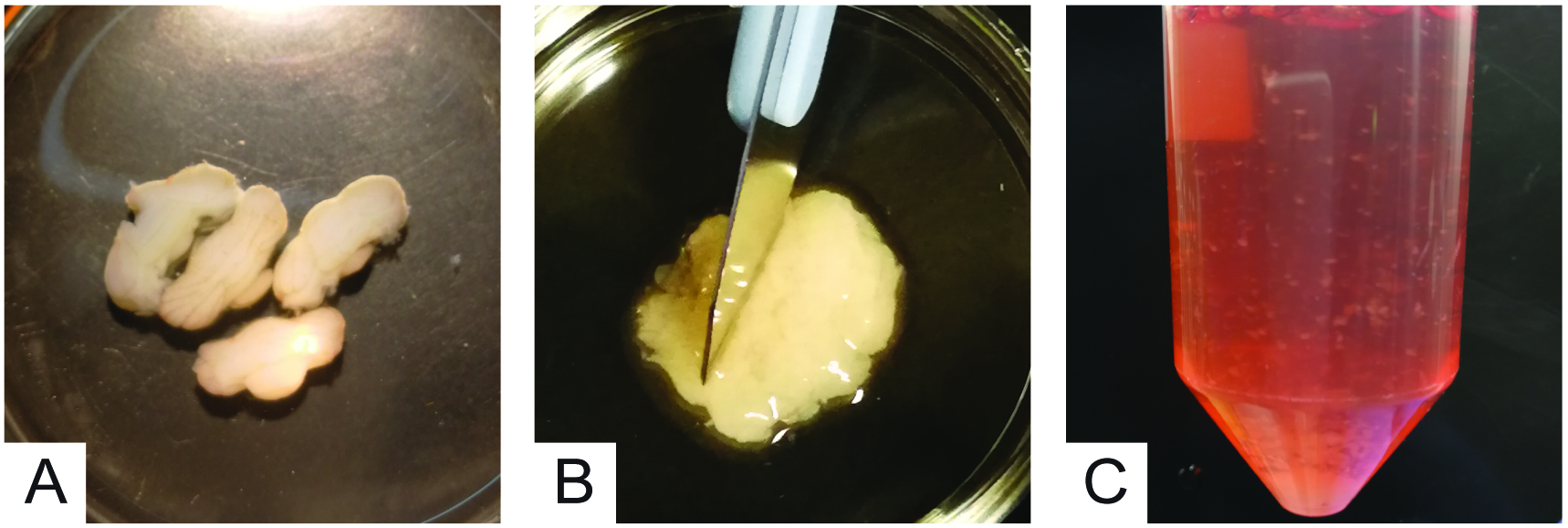
Figure 4. Cerebella mincing and trypsinization - For 3 cerebella, 15 min at 37 °C is sufficient; with more cerebella (4-5) 25-30 min is recommended.
- Add 10 ml of Solution D and centrifuge at 300 x g for 2 min at RT.
- In a sterile laminar flow hood, carefully discard the supernatant without losing cerebellar tissue.
- Add 3 ml of Solution C to the pelleted cerebella and use a sterile P1000 (1,000 µl) micropipette to mechanically dissociate the tissue with 20-30 pipette-strokes.
Notes:- Before pipetting, the solution is transparent with big tissue aggregates floating around (Figure 5A). After pipetting, the solution gets cloudy due to dissociated cells going in suspension (Figure 5B).
- Some undigested tissue fragments may be present. Leave them to settle down before proceeding.
- Before pipetting, the solution is transparent with big tissue aggregates floating around (Figure 5A). After pipetting, the solution gets cloudy due to dissociated cells going in suspension (Figure 5B).
- With a P1000 (1,000 µl) micropipette, collect the cell suspension (900 µl per time) avoiding undissociated tissue fragments (Figure 5C) and transfer it into a new sterile 50 ml conical tube, adding an equal volume of Solution E.

Figure 5. Mechanical disruption and recovery of neurons - Repeat Steps A11-A12 a second time.
- Centrifuge the cell suspension at 300 x g for 8 min at RT.
- Discard the supernatant and resuspend the cell pellet in 10 ml of pre-warmed (37 °C) culture medium.
- Count dissociated neurons with a hemocytometer and plate at the desired density.
Notes:- A typical yield is about 20-25 million (rat) and 10-15 million (mouse) neurons per cerebellum.
- Lower yields may be due to i) inefficient trypsinization: prolong incubation time of Step A6; ii) neuronal death due to excessive trypsinization and/or too harsh mechanical tissue disruption: shorten incubation time of Step A8 and/or reduce trypsin concentration, and/or be gentler during mechanical dissociation in Step A11.
- Trypan blue (permeable to dead cells, not taken up by healthy cells) can be used for a more accurate cell counting.
- Plate neurons with a minimal density of 1.5 x 105 cells/cm2 (i.e., 3.0 x 105 cells in a 24-well culture plate). This density is suitable for imaging experiments. For biochemistry (Western blotting), a recommended density is of 2 x 105 cells/cm2 (i.e., 4.0 x 105 cells in a 24-well culture plate), yet optimal densities should be set according to planned experiments.
- At lower density, maturation of neuronal networks is slower, viability is lower and cells tend to group forming clumps.
- A typical yield is about 20-25 million (rat) and 10-15 million (mouse) neurons per cerebellum.
- Incubate at 37 °C and 5% CO2. Eighteen to twenty-four hours after plating, add Cytosine β-D-arabinofuranoside (araC) to a final concentration of 10 µM to block the growth of non-neuronal cells.
Notes:- araC allows the culture of CGNs as an almost pure neuronal culture by blocking the proliferation of non-neuronal cells.
- The concentration of araC suggested blocks proliferation of non-neuronal cells without causing detectable stress/death of CGNs.
- araC-free medium can be used but CGN tend to be overrun by non-neuronal cells at longer DIVs, even if cultured in a serum-free medium (Wong et al., 2001).
- araC allows the culture of CGNs as an almost pure neuronal culture by blocking the proliferation of non-neuronal cells.
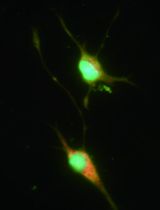
- Incubate at 37 °C and 5% CO2. Depending on cell density, neuron maturation takes 5 to 6 days in vitro (DIV). Starting from DIV 5-6, neuronal culture is suitable for testing clostridial neurotoxins. Figure 6 shows mature neurons at DIV 6.
Notes:- The culture has to be considered mature when neurons have formed a thick network of neuronal processes. Typically, the culture is suitable for use at DIV 5-6.
- The culture should not be used when neurons get grouped forming clasps. In our experience, this is a sign of cellular stress.
- Variability in the serum lot for culture medium strongly influences culture maturation and neuron viability. Comparative trials of multiple serum lots are recommended for culture optimization.
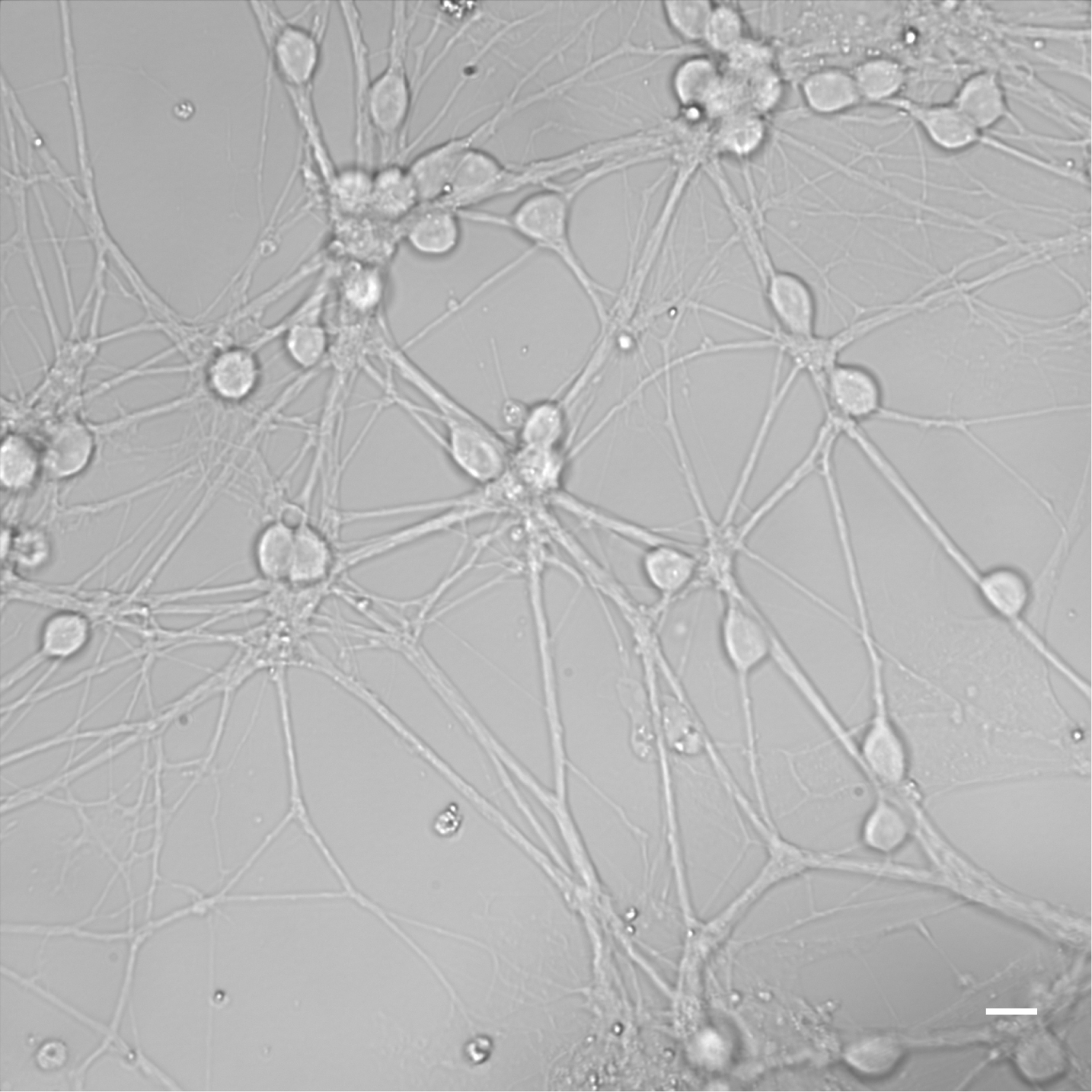
Figure 6. Mature CGN at DIV 6. Scale bar = 10 µm. - The culture has to be considered mature when neurons have formed a thick network of neuronal processes. Typically, the culture is suitable for use at DIV 5-6.
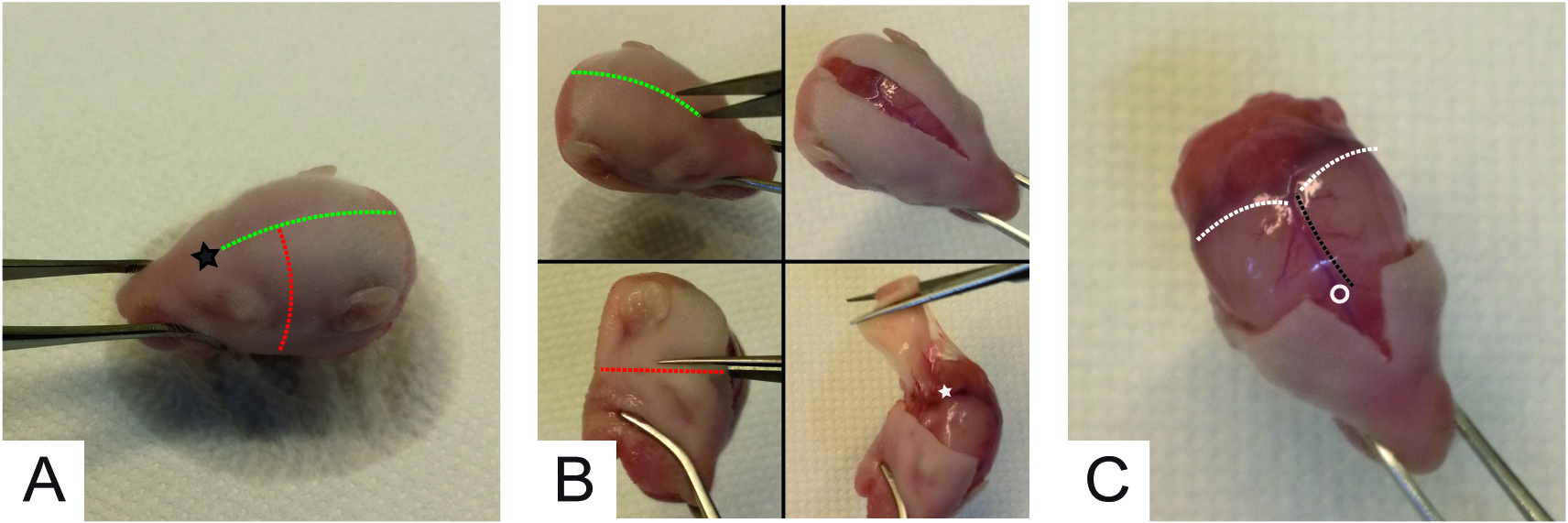
Figure 1. Skinning of pups’ head to expose the skull for cerebellum dissection - Prepare poly-L-lysine coated 24 well-plates at least 1 h before preparation.
- Investigation of CNT activity by Western blotting
- Prepare the clostridial neurotoxin at the intended concentration(s) in culture medium. Typical concentrations range from low pM to nM, depending on experimental design. Intoxicate cultured CGNs by replacing the culture medium with fresh pre-warmed medium supplemented with the toxin.
Notes:- CNT concentration, time and incubation protocol hardly impacts on the number of toxin molecules entering the neurons, thus on the overall cleavage of SNARE. In general, a pulse incubation allows the entry of less molecules than leaving the toxins for the entire duration of the experiment. As a consequence, the cleavage of SNARE proteins with pulse incubation is slower.
- Pulse experiments can be done by incubating neurons in ice-cold toxin medium to allow CNT binding to neurons not followed by internalization. Addition of fresh, 37 °C pre-warmed, medium will trigger toxin internalization in a synchronized fashion.
- Media replacements (removal followed by addition of fresh medium) is critical and must be performed carefully to avoid neuron damage or detachment.
- If using different media for intoxication, note that CNT entry into neurons is strongly influenced by neuronal activity that strictly depends on calcium (at least 1-2 mM). Depolarizing conditions, like high K+, significantly increase toxin entry (and toxicity). The culture medium of CGN contains ~1.6 mM calcium and is slightly depolarizing (25 mM K+), allowing CNT internalization by itself. For short pulse-intoxication (5-10 min at 37 °C), K+ concentration can be risen up to 90 mM. Intoxication with basal potassium in CGN can be performed but for short time incubations as these neurons deprived of high potassium in culture undergo apoptosis (D'Mello et al., 1993).
- Although not essential for toxin activity in vitro, extracellular fluids in vivo contain serum. Thus, intoxication using media supplemented with serum can be considered more similar to the environment where CNT naturally act.
- CNT concentration, time and incubation protocol hardly impacts on the number of toxin molecules entering the neurons, thus on the overall cleavage of SNARE. In general, a pulse incubation allows the entry of less molecules than leaving the toxins for the entire duration of the experiment. As a consequence, the cleavage of SNARE proteins with pulse incubation is slower.
- Prolong the incubation time according to the experimental design.
Note: CNT are metalloproteases (enzymes) thus the longer the incubation time the more efficient will be the cleavage of SNARE substrates. - Aspirate culture medium, rinse the neurons with a saline solution (PBS is fine) and directly lyse the cells into the wells using LSB (Laemmli Sample Buffer 1x).
Note: LSB volume must be set according to neuron density and plate surface. We typically use 25 µl/4.0 x 105 cells (24-well culture plate). - Collect cell lysates into 1.5 ml tubes and heat-denature proteins for at least 15 min at 100 °C.
- Separate proteins by SDS-PAGE.
Notes:- We routinely use SDS-PAGE buffers and reagents from Thermo Fischer Scientific following manufacturer’s instructions. For BoNT/A and BoNT/C, we use NuPAGETM 12% Bis-Tris gels and 1x MOPS running buffer (70 V until the running front exits the gel). For BoNT serotypes and TeNT cleaving VAMP, NuPAGETM 4-12% Bis-Tris gels and 1x MES running buffer (200 V, ~1 h) are recommended. These parameters may vary and may need optimization depending on the SDS-PAGE gel used, in particular to separate BoNT/A- and BoNT/C-truncated SNAP-25 from intact SNAP-25 whose molecular weights differ for less than 2 kDa.
- A pre-stained protein standard can be loaded to estimate the progression of band separation.
- We routinely use SDS-PAGE buffers and reagents from Thermo Fischer Scientific following manufacturer’s instructions. For BoNT/A and BoNT/C, we use NuPAGETM 12% Bis-Tris gels and 1x MOPS running buffer (70 V until the running front exits the gel). For BoNT serotypes and TeNT cleaving VAMP, NuPAGETM 4-12% Bis-Tris gels and 1x MES running buffer (200 V, ~1 h) are recommended. These parameters may vary and may need optimization depending on the SDS-PAGE gel used, in particular to separate BoNT/A- and BoNT/C-truncated SNAP-25 from intact SNAP-25 whose molecular weights differ for less than 2 kDa.
- Transfer proteins onto a matrix suitable for Western blotting.
Note: We use nitrocellulose and a transfer buffer containing 20% methanol. - Check protein transfer by Ponceau S staining (optional).
- De-stain Ponceau S with extensive washes with PBST.
- Block the membrane for 1 h at room temperature using blocking buffer.
- Incubate the membrane overnight with indicated dilutions of primary antibodies in blocking buffer.
Note: We routinely use the following primary antibodies for SNARE proteins: anti VAMP-2 (1:2,000, Synaptic System, 104 211), anti SNAP-25 (1:5,000, SMI81, Abcam, ab24737), anti syntaxin-1A (1:2,000, Synaptic System, 110 011), anti BoNT/B-TeNT-cleaved VAMP-2 (1:200, homemade), anti BoNT/A-cleaved SNAP-25 (1:5,000, homemade), anti BoNT/E-cleaved SNAP-25 (1:5,000, homemade) and anti syntaxin-1A/1B (1:2,000, homemade). Characterization of homemade antibodies have been previously reported (Antonucci et al., 2008; Antonucci et al., 2009; Pirazzini et al., 2014; Azarnia Tehran et al., 2015). - Wash the membrane three times with PBST 5 min each. Incubate the membrane with appropriate secondary antibodies in PBST for 1 h.
Note: We routinely use HRP-conjugated secondary antibodies. However, comparable results have been obtained using different developing system (and appropriate secondary antibodies) like the Odyssey imaging system from LI-COR Biosciences. - Wash the membrane three times using PBST and twice with PBS, 5 min each. Develop the membrane with the appropriate developing system (according to secondary antibodies used).
- Prepare the clostridial neurotoxin at the intended concentration(s) in culture medium. Typical concentrations range from low pM to nM, depending on experimental design. Intoxicate cultured CGNs by replacing the culture medium with fresh pre-warmed medium supplemented with the toxin.
- Investigation of CNT activity by imaging
Alternatively to Western blot analysis, CNT activity can be evaluated using CGN seeded on glass coverslips via immunofluorescence analysis. Although this method is sensitive, this analysis has to be considered more qualitative than quantitative.- Following intoxication (Steps B1-B2 previous paragraph), aspirate culture medium, rinse the neurons with PBS and fix neurons for 15 min at RT using 4% paraformaldehyde solution.
- Wash neurons two times with PBS and quench paraformaldehyde for 20 min using quenching solution.
- Remove quenching solution and permeabilize neurons with permeabilization solution for 20 min at -20 °C.
- Remove permeabilization solution and saturate cells with blocking solution for 30 min at RT.
- Add primary antibodies in blocking solution and incubate using a humid chamber at 37 °C for at least 2 h or at 4 °C overnight.
Notes:- Primary antibodies used for Western blotting can also be used for imaging.
- Non-treated (with a CNT) samples must always be included in the experimental design.
- A control using only secondary antibodies is recommended to evaluate unspecific binding of secondary antibodies.
- Primary antibodies used for Western blotting can also be used for imaging.
- Wash neurons at least three times with PBS.
- Detect primary antibodies using fluorescently labeled secondary antibodies in blocking solution. Perform the incubation using a humid chamber at 37 °C for 45 min.
- Wash neurons three times with PBS.
- Mount coverslips using Fluorescence Mounting Medium.
Collect images with a fluorescence microscope with appropriate optical filters. Confocal microscopy is more suitable, yet satisfactory results can also be obtained with epifluorescence microscopy.
- Following intoxication (Steps B1-B2 previous paragraph), aspirate culture medium, rinse the neurons with PBS and fix neurons for 15 min at RT using 4% paraformaldehyde solution.
- Use CGNs as a platform for screening inhibitors of CNT
- Use CGNs at 5-6 DIV seeded in 24-well plates with a density of at least 4.0 x 105 neurons.
- Prepare a stock solution of the compound to be tested and then make serial dilutions in culture medium.
Notes:- We usually prepare 500 µl of compound solution per well and test each concentration in triplicate.
- Different media can be used to prepare compound dilution. We prefer to use culture medium to keep the neurons in the same culture conditions where they grew avoiding extra stress. In addition, culture medium contains serum which helps in solubilizing poorly water soluble compounds avoiding the need of excessive amounts of organic solvents.
- We usually prepare 500 µl of compound solution per well and test each concentration in triplicate.
- Replace the culture medium with the inhibitor solution.
Notes:- The control condition is carried out with fresh culture medium supplemented with an equal amount of the vehicle used to prepare the inhibitor stock solution (e.g., ethanol, dimethyl sulfoxide, etc.).
- Preincubation (addition of the inhibitor before intoxication) is recommended to figure out if the tested compound is actually effective as an inhibitor. This preliminary assay should be followed by further experiments aimed at understanding which stage of the intoxication process is inhibited. This can be carried out by adding the putative inhibitor at distinct stages of intoxication: i) preincubation with the compound before intoxication; ii) addition of the compound to the cells together with the toxin; iii) addition of the compound after intoxication; iv) addition of the compound at different stages after intoxication.
- Preincubation time may vary depending on different parameters of the inhibitors: physico-chemical properties, neuronal membrane permeability, molecular target(s) of the compound, etc.
- The range of effective concentrations and possible intrinsic toxicity on neurons must be taken into account.
- A control condition evaluating whether the diluent (e.g., ethanol, dimethyl sulfoxide, etc.) of putative inhibitor blocks CNT by itself must be included.
- Chemical compounds can alter neuron physiology and block toxin activity/uptake due to an indirect effect. This should be tested by an appropriate experiment which depends on the nature of the putative inhibitor.
- The control condition is carried out with fresh culture medium supplemented with an equal amount of the vehicle used to prepare the inhibitor stock solution (e.g., ethanol, dimethyl sulfoxide, etc.).
- Add the toxin at the desired concentration(s) and prolong the incubation time according to the experimental design.
Notes:- Several concentrations of inhibitors should be tested to achieve a dose-response curve.
- The toxin can be added as a pulse or left for the entire duration of the experiment (see Steps B1 and B2).
- Multiple CNT (targeting different SNARE substrates) can be added simultaneously to investigate the possibility that the mechanism of inhibition is shared.
- Several concentrations of inhibitors should be tested to achieve a dose-response curve.
- If the toxin is given as a pulse, after toxin washed out restore the culture medium with the desired concentration of inhibitor and prolong the incubation according to the experimental design.
Notes:- In this case, prepare (in Step D2) a double amount of culture medium containing the desired concentration of inhibitor or vehicle.
- Incubation time significantly impact on SNARE proteins cleavage and on inhibitor efficacy as well.
- In this case, prepare (in Step D2) a double amount of culture medium containing the desired concentration of inhibitor or vehicle.
- Continue from Step B1 (WB protocol) or from Step C1 (imaging protocol) of previous paragraphs.
- Use CGNs at 5-6 DIV seeded in 24-well plates with a density of at least 4.0 x 105 neurons.
Data analysis
CNT cleave SNARE proteins with very high selectivity. Not only does each toxin hydrolyze one specific SNARE, but it also cleaves a very specific peptide bond (Pirazzini et al., 2017) (Figure 7A). This is due to multiple and selective interactions taking place between CNT protease and the SNARE protein involving the active site and many exosites (Binz, 2013; Rossetto et al., 2014; Pirazzini et al., 2017). Cleavage sites within SNARE proteins are remarkably conserved in vertebrates, including humans (Carle et al., 2017). At the same time, even a single mutation in the peptide bond or in exosites mediating toxin-substrate recognition, make the SNARE protein non-cleavable and the vertebrate carrying this alteration non-sensitive to that particular toxin (Patarnello et al., 1993; Eleopra et al., 2013; Peng et al., 2014).
This specificity makes easy to interpret the results of intoxicated CGN both via Western blotting and via imaging using specific antibodies. Figure 7B shows a schematization of a Western blot membrane in which the use of the most popular antibodies is simulated. Depending on the CNT, toxin activity can be clearly evaluated by following the staining of SNARE proteins’ bands.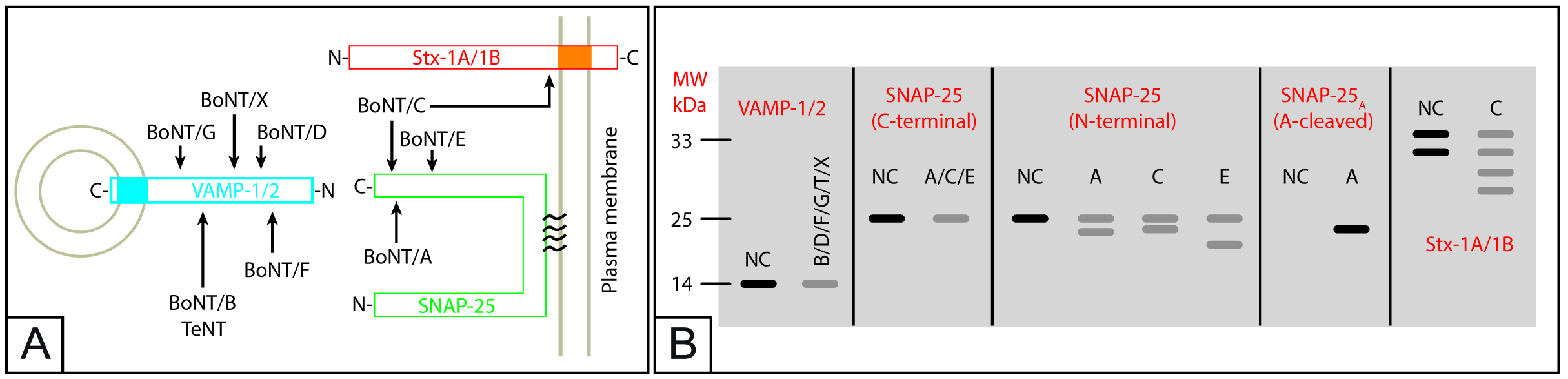
Figure 7. Schematization of CNT activity on SNARE proteins (A) and simulation of Western blotting results with indicated antibodies (B). Note that these results would be achieved by ideally cleaving 50% of the considered SNARE protein(s). NC is “negative control” and corresponds to the normal electrophoretic mobility and band intensity of indicated SNARE proteins in samples not treated with CNT. Bands in grey represent a decreased intensity (~50%) of the band. Syntaxins-1A/1B are simultaneously expressed by CGNs and are shown as a doublet because syntaxin-1A and syntaxin-1B, in spite of very similar molecular weights (~33 kDa), run slightly different when separated by SDS-PAGE. Since BoNT/C cleaves both stx-1A and stx-1B, it generates the 4-bands pattern shown in the last panel on the right.
Figure 8 shows typical Western blotting results. Although Western blotting is a semiquantitative assay, with modern developing techniques and instruments, CNT activity (or inhibition) can be accurately determined by band densitometry.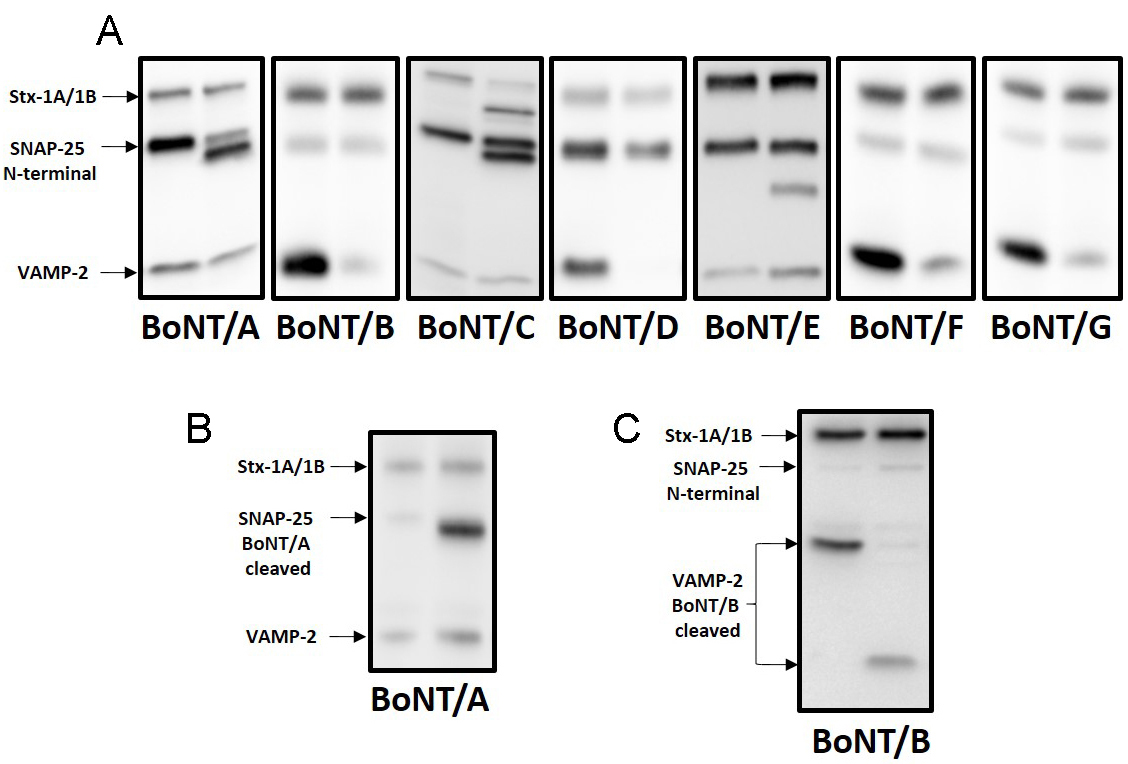
Figure 8. Investigation of CNT activity by Western blotting. The image shown are representative experiments in which CGN were treated with different serotypes of BoNT. A. In each panel are shown two samples: a not-treated with the toxin (toxin free, untreated cells, left) and another treated with the indicated serotype of BoNT (right). The antibodies used were: anti-syntaxin1A, anti-SNAP-25 and anti-VAMP-2. BoNT/B, /D, /F and /G cleave VAMP-2 leading to the disappearance of VAMP-2 signal. BoNT/A and BoNT/E cleave SNAP-25 leading to the appearance of the SNAP-25 C-terminal fragment. Instead, BoNT/C hydrolyze both Syntaxin 1A/1B and SNAP-25 leading to appearance of both generated fragments. B. SNAP-25-BoNT/A-cleaved antibody recognize the generated C-terminal fragment after BoNT/A cleavage. This antibody is raised against the C-terminus newly generated after BoNT/A cleavage and does not cross-react with intact SNAP-25. C. VAMP-2 BoNT/B cleaved antibody recognize the generated N-terminal fragment after BoNT/B cleavage. Quantitative interpretation critically depends on the developing techniques, the linear dynamic ranges of the type of secondary antibodies and of the detection systems used.
VAMP-2 BoNT/B cleaved antibody and SNAP-25-BoNT/A-cleaved antibody can also be used to stain neurons for imaging CNT activity in fixed neurons. Figure 9 shows some examples.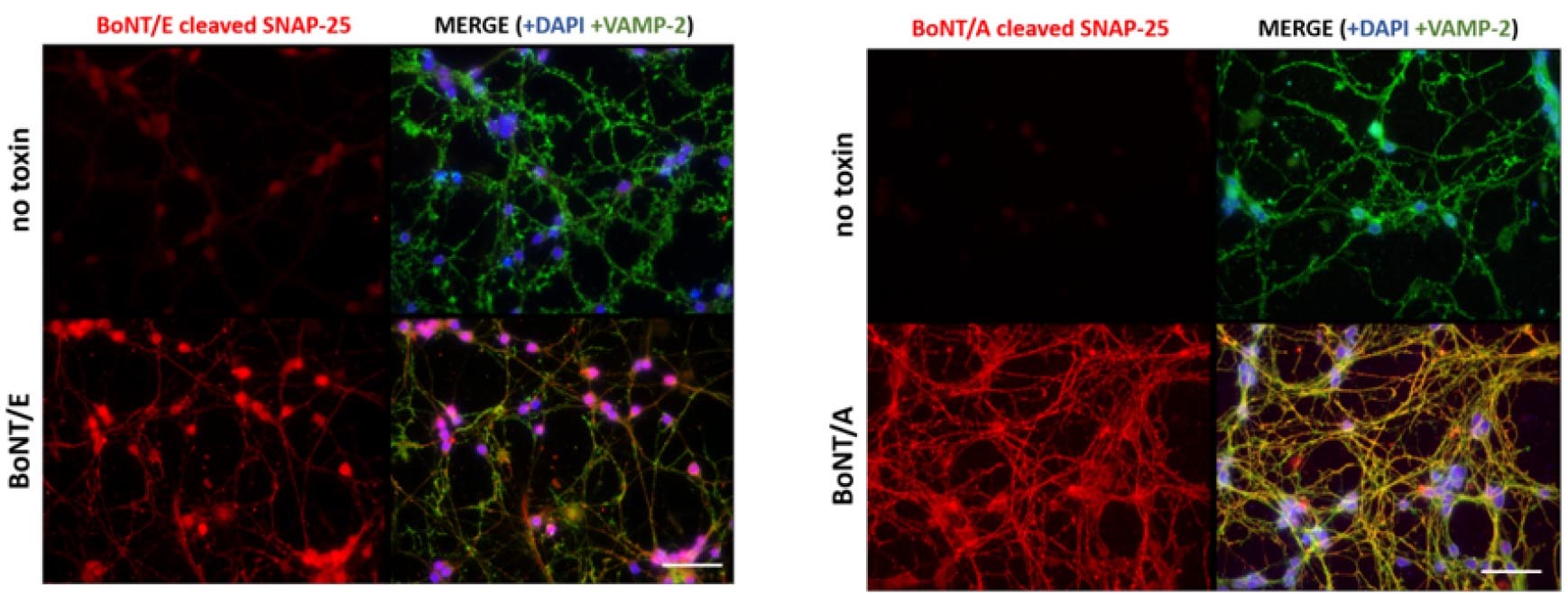
Figure 9. Investigation of CNT activity by imaging. CGNs were treated with vehicle (no toxin) or with the indicated serotype of BoNT for 2 h. Neurons were fixed and stained with an antibody against BoNT/E-cleaved SNAP-25 (left panel) or BoNT/A-cleaved SNAP-25 (right panel) and VAMP2 as housekeeping staining. Both BoNT/A and BoNT/E cleaved-SNAP25 are detected with an Alexa555 conjugated secondary antibody while VAMP2 with an Alexa488 secondary antibody. DAPI is used to stain nuclei. Scale bars = 10 µm.
Recipes
- Krebs solution (10x)
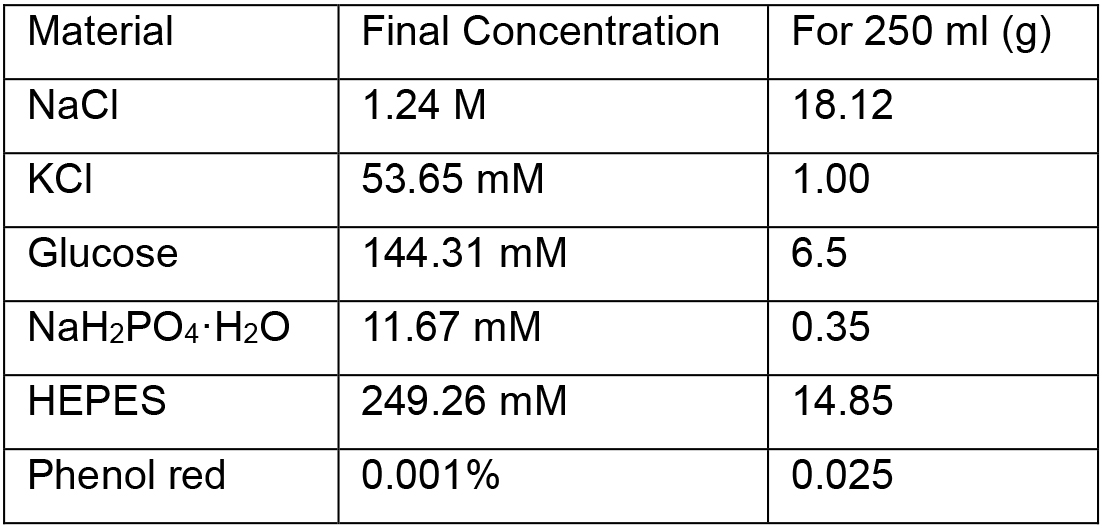
Adjust to pH 7.4 and filter using a 0.20 µm filter. This solution can be aliquoted (50 ml each) and stored at -20 °C until further usage (stable for at least three months) - 155 mM MgSO4 stock solution
To make 155 mM MgSO4 solution:- Dissolve 1.91 g of MgSO4·7H2O in 50 ml of distilled H2O
- Filter the solution in the laminar flow hood using a 0.20 µm filter and keep at 4 °C (stable for at least two months)
- Dissolve 1.91 g of MgSO4·7H2O in 50 ml of distilled H2O
- 12.2 mM CaCl2 stock solution
To make 12.2 mM CaCl2 solution:- Dissolve 0.09 g of CaCl2·2H2O in 50 ml of distilled H2O
- Filter the solution in the laminar flow hood using a 0.20 µm filter and keep at 4 °C (stable for at least two months)
- Dissolve 0.09 g of CaCl2·2H2O in 50 ml of distilled H2O
- Solution A
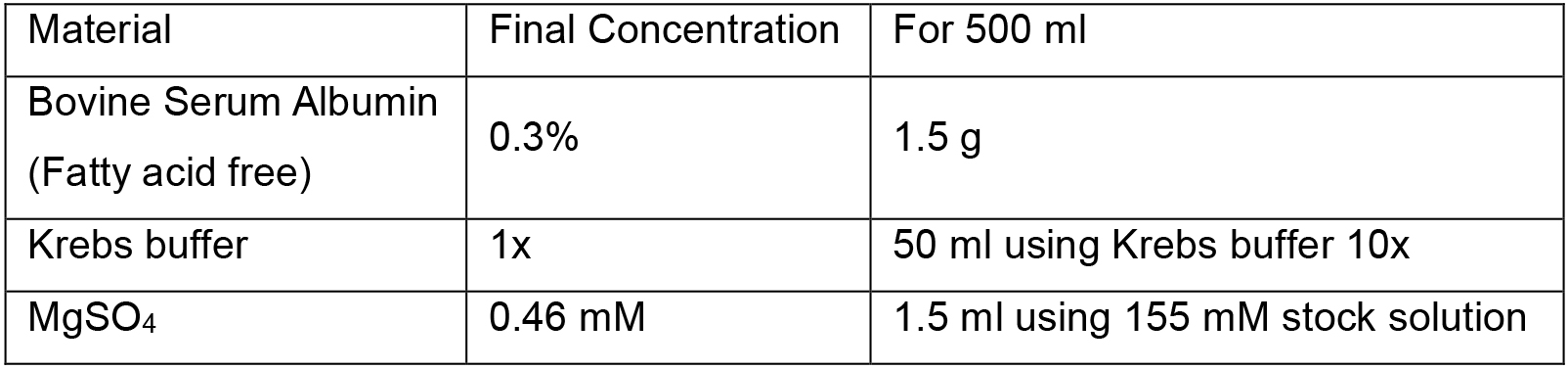
Filter the solution in the laminar flow hood using single-use stericups. This solution can be aliquoted (50 ml each) and stored at -20 °C until further usage (stable for at least three months) - Solution B
- Add 0.01 g of Trypsin in 12.5 ml of Solution A
- Filter the solution in the laminar flow hood using a 0.20 µm filter
- Add 0.01 g of Trypsin in 12.5 ml of Solution A
- Solution C
- Add 120 µl of DNase (10 mg/ml, stock solution), 7.8 mg of trypsin inhibitor and 150 µl of 155 mM MgSO4 stock solution in 15 ml of Solution A
- Filter the solution in the laminar flow hood using a 0.20 µm filter
- Add 120 µl of DNase (10 mg/ml, stock solution), 7.8 mg of trypsin inhibitor and 150 µl of 155 mM MgSO4 stock solution in 15 ml of Solution A
- Solution D
Mix 2.4 ml of Solution C with 12.6 ml Solution A
Note: Always prepare fresh. - Solution E
- Add 120 µl of 12.2 mM CaCl2 stock solution and 120 µl of 155 mM MgSO4 stock solution in 15 ml of Solution A
- Filter the solution in the laminar flow hood sing a 0.20 µm filter
- Add 120 µl of 12.2 mM CaCl2 stock solution and 120 µl of 155 mM MgSO4 stock solution in 15 ml of Solution A
- Poly-L-lysine solution
Prepare a stock solution of Poly-L-lysine in the range of 5-10 mg/ml
Working concentration is 50 µg/ml - BME complete medium, CGN culture medium
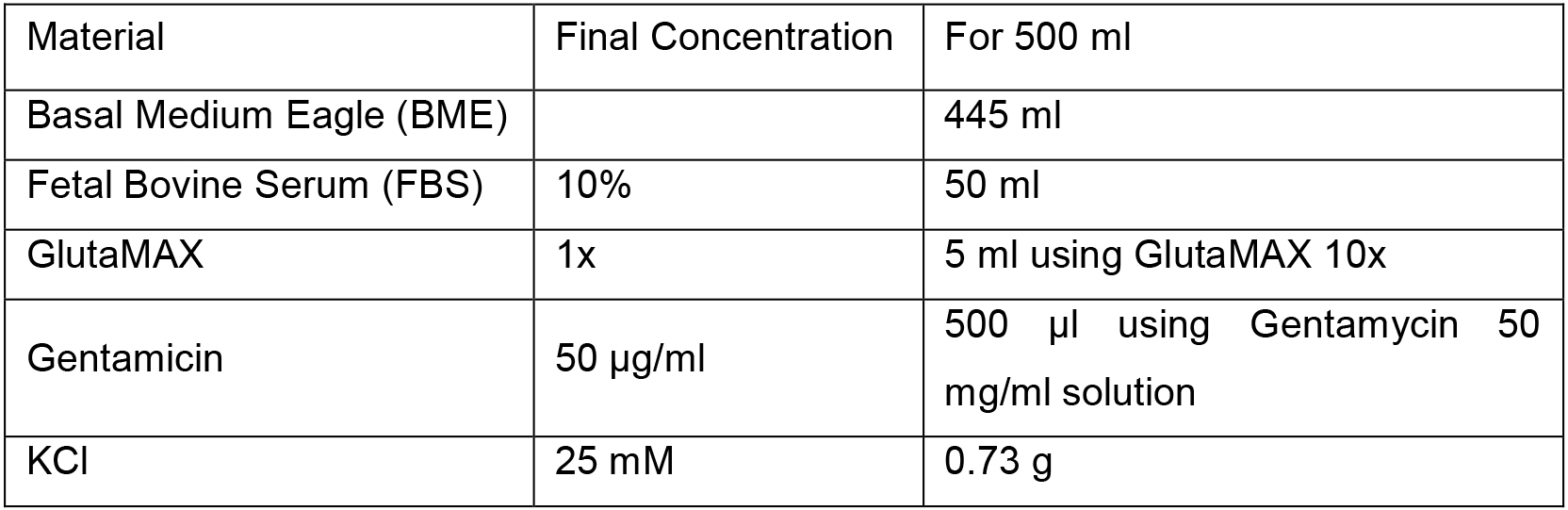
Filter the medium in the laminar flow hood using single-use stericups. The medium can be stored at 4 °C (stable for at least one month) - Cytosine β-D-arabinofuranoside (AraC) stock solution
- Add 4.9 mg of Cytosine β-D-arabinofuranoside in 5 ml of distilled H2O (1 mg/ml, 4 mM)
- Incubate at 37 °C for a few minutes and vortex until the solution appears clear and colorless
- Filter in the laminar flow hood using a 0.20 µm filter
- Add 4.9 mg of Cytosine β-D-arabinofuranoside in 5 ml of distilled H2O (1 mg/ml, 4 mM)
- PBS
NaCl 137 mM
KCl 2.7 mM
Na2HPO4 10 mM
KH2PO4 1.8 mM - PBST
PBS supplemented with 0.1% (v/v) Tween 20 - Laemmli Sample Buffer. Recipe for 10 ml (4x)
2.5 ml of 1 M Tris-HCl (pH 6.8):
1 g of SDS
0.8 ml of 0.1% Bromophenol Blue
4 ml of 100% glycerol
2 ml of 14.3 M β-mercaptoethanol
4 tablets of protease inhibitors
Adjust the final volume to 10 ml with distilled H2O - Transfer buffer
To make a 10x transfer buffer:- Dissolve 288 g of glycine and 60.4 g of Tris base and add distilled H2O to a final volume of 2 L
- To make 1x transfer buffer mix 700 ml distilled H2O, 200 ml methanol and 100 ml 10x transfer buffer
- Dissolve 288 g of glycine and 60.4 g of Tris base and add distilled H2O to a final volume of 2 L
- Ponceau S solution
0.5% (w/v) Ponceau S, 1% acetic acid in distilled H2O - Blocking buffer
5% nonfat dry milk in PBST - 4% Paraformaldehyde solution
- Place 450 ml of distilled H2O in a glass beaker and heat at 60 °C using a hot plate with stirring
- Add 20 g of paraformaldehyde
- Then, add 5 drops of 2 N NaOH and wait until the solution becomes clear (few minutes)
- Remove from the heat and add 50 ml of 10x PBS and adjust pH to 7.4
- Filter using a 0.2 µm filter and aliquots
- Place 450 ml of distilled H2O in a glass beaker and heat at 60 °C using a hot plate with stirring
- Quenching solution
50 mM ammonium chloride in PBS - Permeabilization solution
5% acetic acid in pure ethanol
Store at -20°C - Blocking solution
2.5% BSA in PBS
Acknowledgments
This work was supported by the University of Padova with the grant DOR to MP. DAT is currently supported by an Alexander von Humboldt (AvH) Research Fellowship for Postdoctoral Researcher. All the procedures have been optimized in the laboratory of ‘Neurotoxins, Neuroparalysis and Regeneration’ headed by Prof. Cesare Montecucco at the Department of Biomedical Sciences (University of Padova). The method for the preparation of CGN was adapted from the previous work of Anne Messer and Parizad M. Bilimoria et al. The authors declare no competing interests. MP and DAT wrote the paper with equal contribution.
References
- Antonucci, F., Bozzi, Y. and Caleo, M. (2009). Intrahippocampal infusion of botulinum neurotoxin E (BoNT/E) reduces spontaneous recurrent seizures in a mouse model of mesial temporal lobe epilepsy. Epilepsia 50(4): 963-966.
- Antonucci, F., Rossi, C., Gianfranceschi, L., Rossetto, O. and Caleo, M. (2008). Long-distance retrograde effects of botulinum neurotoxin A. J Neurosci 28(14): 3689-3696.
- Azarnia Tehran, D., Zanetti, G., Leka, O., Lista, F., Fillo, S., Binz, T., Shone, C. C., Rossetto, O., Montecucco, C., Paradisi, C., Mattarei, A. and Pirazzini, M. (2015). A novel inhibitor prevents the peripheral neuroparalysis of botulinum neurotoxins. Sci Rep 5: 17513.
- Bade, S., Rummel, A., Reisinger, C., Karnath, T., Ahnert-Hilger, G., Bigalke, H. and Binz, T. (2004). Botulinum neurotoxin type D enables cytosolic delivery of enzymatically active cargo proteins to neurones via unfolded translocation intermediates. J Neurochem 91(6): 1461-1472.
- Bilimoria, P. M. and Bonni, A. (2008). Cultures of cerebellar granule neurons. CSH Protoc 2008: pdb prot5107.
- Binz, T. (2013). Clostridial neurotoxin light chains: devices for SNARE cleavage mediated blockade of neurotransmission. Curr Top Microbiol Immunol 364: 139-157.
- Binz, T. and Rummel, A. (2009). Cell entry strategy of clostridial neurotoxins. J Neurochem 109(6): 1584-1595.
- Brunt, J., Carter, A. T., Stringer, S. C. and Peck, M. W. (2018). Identification of a novel botulinum neurotoxin gene cluster in Enterococcus. FEBS Lett 592(3): 310-317.
- Carle, S., Pirazzini, M., Rossetto, O., Barth, H. and Montecucco, C. (2017). High conservation of tetanus and botulinum neurotoxins cleavage sites on human SNARE proteins suggests that these pathogens exerted little or no evolutionary pressure on humans. Toxins (Basel) 9(12).
- Colasante, C., Rossetto, O., Morbiato, L., Pirazzini, M., Molgo, J. and Montecucco, C. (2013). Botulinum neurotoxin type A is internalized and translocated from small synaptic vesicles at the neuromuscular junction. Mol Neurobiol 48(1): 120-127.
- Contestabile, A. (2002). Cerebellar granule cells as a model to study mechanisms of neuronal apoptosis or survival in vivo and in vitro. Cerebellum 1(1): 41-55.
- D'Mello, S. R., Galli, C., Ciotti, T. and Calissano, P. (1993). Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci U S A 90(23): 10989-10993.
- Eleopra, R., Montecucco, C., Devigili, G., Lettieri, C., Rinaldo, S., Verriello, L., Pirazzini, M., Caccin, P. and Rossetto, O. (2013). Botulinum neurotoxin serotype D is poorly effective in humans: an in vivo electrophysiological study. Clin Neurophysiol 124(5): 999-1004.
- Harper, C. B., Martin, S., Nguyen, T. H., Daniels, S. J., Lavidis, N. A., Popoff, M. R., Hadzic, G., Mariana, A., Chau, N., McCluskey, A., Robinson, P. J. and Meunier, F. A. (2011). Dynamin inhibition blocks botulinum neurotoxin type A endocytosis in neurons and delays botulism. J Biol Chem 286(41): 35966-35976.
- Johnson, E. A. and Montecucco, C. (2008). Botulism. Handb Clin Neurol 91: 333-368.
- Matteoli, M., Verderio, C., Rossetto, O., Iezzi, N., Coco, S., Schiavo, G. and Montecucco, C. (1996). Synaptic vesicle endocytosis mediates the entry of tetanus neurotoxin into hippocampal neurons. Proc Natl Acad Sci U S A 93(23): 13310-13315.
- Messer, A. (1977). The maintenance and identification of mouse cerebellar granule cells in monolayer culture. Brain Res 130(1): 1-12.
- Montal, M. (2010). Botulinum neurotoxin: a marvel of protein design. Annu Rev Biochem 79: 591-617.
- Montecucco, C. and Rasotto, M. B. (2015). On botulinum neurotoxin variability. MBio 6(1).
- Pantano, S. and Montecucco, C. (2014). The blockade of the neurotransmitter release apparatus by botulinum neurotoxins. Cell Mol Life Sci 71(5): 793-811.
- Patarnello, T., Bargelloni, L., Rossetto, O., Schiavo, G. and Montecucco, C. (1993). Neurotransmission and secretion. Nature 364(6438): 581-582.
- Pellett, S. (2013). Progress in cell based assays for botulinum neurotoxin detection. Curr Top Microbiol Immunol 364: 257-285.
- Peng, L., Adler, M., Demogines, A., Borrell, A., Liu, H., Tao, L., Tepp, W. H., Zhang, S. C., Johnson, E. A., Sawyer, S. L. and Dong, M. (2014). Widespread sequence variations in VAMP1 across vertebrates suggest a potential selective pressure from botulinum neurotoxins. PLoS Pathog 10(7): e1004177.
- Pirazzini, M., Azarnia Tehran, D., Leka, O., Zanetti, G., Rossetto, O. and Montecucco, C. (2016). On the translocation of botulinum and tetanus neurotoxins across the membrane of acidic intracellular compartments. Biochim Biophys Acta 1858(3): 467-474.
- Pirazzini, M., Azarnia Tehran, D., Zanetti, G., Megighian, A., Scorzeto, M., Fillo, S., Shone, C. C., Binz, T., Rossetto, O., Lista, F. and Montecucco, C. (2014). Thioredoxin and its reductase are present on synaptic vesicles, and their inhibition prevents the paralysis induced by botulinum neurotoxins. Cell Rep 8(6): 1870-1878.
- Pirazzini, M., Azarnia Tehran, D., Zanetti, G., Rossetto, O. and Montecucco, C. (2018). Hsp90 and Thioredoxin-Thioredoxin Reductase enable the catalytic activity of Clostridial neurotoxins inside nerve terminals. Toxicon 147: 32-37.
- Pirazzini, M., Bordin, F., Rossetto, O., Shone, C. C., Binz, T. and Montecucco, C. (2013a). The thioredoxin reductase-thioredoxin system is involved in the entry of tetanus and botulinum neurotoxins in the cytosol of nerve terminals. FEBS Lett 587(2): 150-155.
- Pirazzini, M., Henke, T., Rossetto, O., Mahrhold, S., Krez, N., Rummel, A., Montecucco, C. and Binz, T. (2013b). Neutralisation of specific surface carboxylates speeds up translocation of botulinum neurotoxin type B enzymatic domain. FEBS Lett 587(23): 3831-3836.
- Pirazzini, M. and Rossetto, O. (2017). Challenges in searching for therapeutics against botulinum neurotoxins. Expert Opin Drug Discov 12(5): 497-510.
- Pirazzini, M., Rossetto, O., Bertasio, C., Bordin, F., Shone, C. C., Binz, T. and Montecucco, C. (2013c). Time course and temperature dependence of the membrane translocation of tetanus and botulinum neurotoxins C and D in neurons. Biochem Biophys Res Commun 430(1): 38-42.
- Pirazzini, M., Rossetto, O., Bolognese, P., Shone, C. C. and Montecucco, C. (2011). Double anchorage to the membrane and intact inter-chain disulfide bond are required for the low pH induced entry of tetanus and botulinum neurotoxins into neurons. Cell Microbiol 13(11): 1731-1743.
- Pirazzini, M., Rossetto, O., Eleopra, R. and Montecucco, C. (2017). Botulinum neurotoxins: Biology, pharmacology, and toxicology. Pharmacol Rev 69(2): 200-235.
- Rossetto, O., Pirazzini, M. and Montecucco, C. (2014). Botulinum neurotoxins: genetic, structural and mechanistic insights. Nat Rev Microbiol 12(8): 535-549.
- Rummel, A. (2017). Two feet on the membrane: Uptake of clostridial neurotoxins. Curr Top Microbiol Immunol 406: 1-37.
- Schiavo, G., Matteoli, M. and Montecucco, C. (2000). Neurotoxins affecting neuroexocytosis. Physiol Rev 80(2): 717-766.
- Schiavo, G. and Montecucco, C. (1995). Tetanus and botulism neurotoxins: isolation and assay. Methods Enzymol 248: 643-652.
- Shone, C. C. and Tranter, H. S. (1995). Growth of clostridia and preparation of their neurotoxins. Curr Top Microbiol Immunol 195: 143-160.
- Tehran, D. A. and Pirazzini, M. (2018). Novel botulinum neurotoxins: Exploring underneath the iceberg tip. Toxins (Basel) 10(5).
- Wong, J. K., Kennedy, P. R. and Belcher, S. M. (2001). Simplified serum- and steroid-free culture conditions for high-throughput viability analysis of primary cultures of cerebellar granule neurons. J Neurosci Methods 110(1-2): 45-55.
- Zanetti, G., Azarnia Tehran, D., Pirazzini, M., Binz, T., Shone, C. C., Fillo, S., Lista, F., Rossetto, O. and Montecucco, C. (2015). Inhibition of botulinum neurotoxins interchain disulfide bond reduction prevents the peripheral neuroparalysis of botulism. Biochem Pharmacol 98(3): 522-530.
- Zanetti, G., Sikorra, S., Rummel, A., Krez, N., Duregotti, E., Negro, S., Henke, T., Rossetto, O., Binz, T. and Pirazzini, M. (2017). Botulinum neurotoxin C mutants reveal different effects of syntaxin or SNAP-25 proteolysis on neuromuscular transmission. PLoS Pathog 13(8): e1006567.
- Zhang, S., Lebreton, F., Mansfield, M. J., Miyashita, S. I., Zhang, J., Schwartzman, J. A., Tao, L., Masuyer, G., Martinez-Carranza, M., Stenmark, P., Gilmore, M. S., Doxey, A. C. and Dong, M. (2018). Identification of a botulinum neurotoxin-like toxin in a commensal strain of Enterococcus faecium. Cell Host Microbe 23(2): 169-176 e166.
- Zhang, S., Masuyer, G., Zhang, J., Shen, Y., Lundin, D., Henriksson, L., Miyashita, S. I., Martinez-Carranza, M., Dong, M. and Stenmark, P. (2017). Identification and characterization of a novel botulinum neurotoxin. Nat Commun 8: 14130.
- Zornetta, I., Azarnia Tehran, D., Arrigoni, G., Anniballi, F., Bano, L., Leka, O., Zanotti, G., Binz, T. and Montecucco, C. (2016). The first non Clostridial botulinum-like toxin cleaves VAMP within the juxtamembrane domain. Sci Rep 6: 30257.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Azarnia Tehran, D. and Pirazzini, M. (2018). Preparation of Cerebellum Granule Neurons from Mouse or Rat Pups and Evaluation of Clostridial Neurotoxin Activity and Their Inhibitors by Western Blot and Immunohistochemistry. Bio-protocol 8(13): e2918. DOI: 10.21769/BioProtoc.2918.
Category
Neuroscience > Cellular mechanisms > Cell isolation and culture
Cell Biology > Cell isolation and culture > Cell isolation
Biochemistry > Protein > Electrophoresis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.