- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

An in vitro Co-culture System for the Activation of CD40 by Membrane-presented CD40 Ligand versus Soluble Agonist

Published: Vol 8, Iss 13, Jul 5, 2018 DOI: 10.21769/BioProtoc.2907 Views: 9483
Reviewed by: HongLok LungTomas AparicioPornima Phatak
Original research article
The authors used this protocol in:

May 2017
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
One fundamental property of the TNR receptor (TNFR) family relates to how ‘signal quality’ (the extent of receptor ligation or cross-linking) influences the outcome of receptor ligation, for instance the induction of death in tumour cells. It is unequivocal that membrane-presented ligand (delivered to target cells via cell-surface presentation by co-culture with ligand-expressing third-party cells) induces a greater extent of carcinoma cell death in vitro in comparison to non-cross-linked agonists (agonistic antibodies and/or recombinant ligands). The CD40 receptor epitomises this fundamental property of TNF receptor-ligand interactions, as the extent of CD40 cross-linking dictates cell fate. Membrane-presented CD40 ligand (mCD40L), but not soluble agonists (e.g., agonistic anti-CD40 antibody), induces high level of pro-inflammatory cytokine secretion and causes extensive cell death (apoptosis) in malignant (but not normal) epithelial cells. In this article, we describe a co-culture system for the activation of CD40 by mCD40L and subsequent detection of various features of apoptosis (including cell membrane permeabilisation, DNA fragmentation, caspase activation) as well as detection of intracellular mediators of cell death (including adaptor proteins, pro-apoptotic kinases and reactive oxygen species, ROS).
Keywords: TNF receptors (TNFRs)Background
The role of the TNFRs and their ligands in regulating cell proliferation or death in lymphoid tissues as well as in epithelial (and particularly carcinoma) cells has been under extensive research, as their ability to induce cell death (mainly via apoptosis) represents a promising target for cancer therapy. Importantly, however, there is a clear difference in the ability of TNFR agonists to trigger cell death when presented in soluble versus membrane-bound form. Soluble agonists often demonstrate relatively low cytotoxic potency when administrated as a sole treatment, whereas membrane-presented ligands appear to be superior (Albarbar et al., 2015).
In this context, CD40 represents the most prominent TNFR family member. The receptor is expressed on a variety of epithelial cells and the effect of CD40 activation is exquisitely contextual (Young and Eliopoulos, 2004). Most importantly, the ability of CD40 to induce cytostasis or cell death (apoptosis) is highly dependent on the ‘quality’ of receptor engagement (degree of receptor cross-linking). Soluble CD40 agonists (recombinant soluble CD40L or agonistic antibody) are only cytostatic or weakly pro-apoptotic and only rendered pro-apoptotic by pharmacological intervention (Bugajska et al., 2002). By contrast, membrane-presented CD40L (mCD40L) is highly pro-apoptotic and induces extensive apoptosis in carcinoma cells, when presented to target carcinoma cells on the surface of third-party cells (Georgopoulos et al., 2006 and 2007) or by mCD40L-expressing, naturally-activated immunocytes (Hill et al., 2008).
The ability of mCD40L (but not soluble agonists) to efficiently kill malignant cells, and in a tumour cell-specific fashion, reflects the two most remarkable properties of the CD40-mCD40L dyad and our recent studies have deciphered these two fundamental properties of CD40. We utilised a co-culture system that involved culture of target, carcinoma (or normal) cells with growth-arrested, third-party, effector cells engineered to express the CD40L on their surface, in order to achieve presentation of mCD40L. This allowed us to study the ability of mCD40L to induce a number of different morphological and biochemical features of apoptosis, as well as define the intracellular mediators of cell death (Dunnill et al., 2017). Here, we provide a detailed protocol for the preparation of the co-culture system for mCD40L delivery to epithelial target cells (in comparison to soluble agonist, i.e., agonistic anti-CD40 antibody) and methodologies to assess mCD40L-induced apoptosis and detection of its intracellular mediators.
Materials and Reagents
- Materials
- T75 tissue culture flasks with vent (SARSTEDT, catalog number: 83.1813.002 )
- T25 tissue culture flasks with vent (SARSTEDT, catalog number: 83.1810.002 )
- 96 well Nunc, white, flat bottom tissue culture multi-well plates (Thermo Fisher Scientific, catalog number: 136101 )
- 96 well, flat bottom, Costar transparent tissue culture plates (Corning, catalog number: 3595 )
- 96 well ELISA microplates (Greiner Bio One International, catalog number: 655101 )
- 24 well plates (Corning, catalog number: 3526)
- 6 well plates (Corning, catalog number: 3516)
- Tissue culture dishes, 10 cm diameter, Nunclon with lid (Thermo Fisher Scientific, catalog number: 150350 )
- Bijou tubes 5 ml sterile (x2,000) (SARSTEDT, catalog number: 60.9921.532 )
- Cryopure tubes, 2.0 ml white cap (SARSTEDT, catalog number: 72.380 )
- 5 ml serological pipettes (SARSTEDT, catalog number: 86.1253.001 )
- 10 ml serological pipette (SARSTEDT, catalog number: 86.1254.001 )
- 25 ml serological pipettes (SARSTEDT, catalog number: 86.1685.001 )
- 120 ml sterile container graduated (x250) (SARSTEDT, catalog number: 75.9922.420 )
- 250 ml SterilinTM containers (Thermo Fisher Scientific, catalog number: 190A )
- 30 ml SterilinTM universals (Thermo Fisher Scientific, catalog number: 128AFS )
- 2 ml aspiration pipette individually wrapped sterile Non Pyrogenic (SARSTEDT, catalog number: 86.1252.011 )
- 5.0 ml TipOne® Repeat Dispenser Tip (Sterile) (STARLAB, catalog number: S4761-0500 )
- 1.25 ml TipOne Repeat Dispenser Tip (Sterile), Ind. Wrapped (STARLAB, catalog number: S4786-0125 )
- Cell scrapers (Fisher Scientific, catalog number: FB55199 )
- 20 ml disposable sterile syringe (BD, catalog number: 300296 )
- 10 ml disposable sterile syringe (BD, catalog number: 302188 )
- 1.5 ml tubes (SARSTEDT, catalog number: 72.690.001 )
- 0.5 ml tubes (5,000) (SARSTEDT, catalog number: 72.699 )
- 0.2 µm syringe filter sterile (SARSTEDT, catalog number: 83.1826.102 )
- Syringe filter 0.4 μm (x50) (GE Healthcare, Whatman, catalog number: 6896-2504 )
- Haemocytometer (Fisher Scientific, catalog number: MNK-420-010N )
- Haemocytometer cover slips (Fisher Scientific, catalog number: MNK-504-030M )
- X2500 Countess Chamber Slides (2,500) (Thermo Fisher Scientific, catalog number: C10314 )
- Blue loose pipette tips (1,000 µl) (SARTEDT, catalog number: 70.762 )
- 200 µl yellow pipette tips (SARTEDT, catalog number: 70.760.002 )
- Neutral pipette tips (10 µl) (SARTEDT, catalog number: 70.1130 )
- T75 tissue culture flasks with vent (SARSTEDT, catalog number: 83.1813.002 )
- Cells
- Human bladder carcinoma-derived EJ cells and colorectal carcinoma-derived HCT116 cells (obtained from the ATCC)
Note: They were cultured in a 1:1 (v/v) mixture of DMEM and RPMI 1640 containing 5% FBS, referred to as ‘DR/5%’ medium. - Normal human urothelial (NHU) cells
Note: They were established and cultured in complete KSFM as described (Bugajska et al., 2002). - 3T3neo and 3T3CD40L fibroblasts – stably transfected NIH3T3 derivatives generated as previously described (Bugajska et al., 2002)
Note: They were cultured in DMEM supplemented with 10% FBS and containing 0.5 mg/ml G418, with omission of antibiotic in co-culture experiments (Georgopoulos et al., 2006).
- Human bladder carcinoma-derived EJ cells and colorectal carcinoma-derived HCT116 cells (obtained from the ATCC)
- Reagents
- Purified water (double distilled ddH2O)
- D-PBS, 10x, no calcium, no magnesium (Thermo Fisher Scientific, catalog number: 14200067 )
- Mikrozid AF liquid, 10 L canister (LAVABIS, catalog number: SF000301 )
- G418, 100 mg/ml solution (InvivoGen, supplied by Source BioScience LifeSciences, catalog number: ant-gn-1 )
- Keratinocyte Serum Free Medium (KSFM) and supplements (Thermo Fisher Scientific, GibcoTM, catalog number: 17005075 )
- RPMI-1640 Medium, with sodium bicarbonate, without L-glutamine (Sigma-Aldrich, catalog number: R0883-6X500ML )
- Dulbecco’s Modified Eagle’s Medium (DMEM) (high glucose) with sodium bicarbonate, without L-glutamine (Sigma-Aldrich, catalog number: D6546-6X500ML )
- Hanks' Balanced Salt Solution (HBSS) (Sigma-Aldrich, catalog number: H9394-6X500ML )
- L-Glutamine solution (Sigma-Aldrich, catalog number: G7513-100ML )
- Fetal bovine serum (FBS) (qualified fetal bovine serum, 500 ml) (Sigma-Aldrich, catalog number: F7524-500ML )
- Ethylenediaminetetraacetic acid (EDTA) (Santa Cruz Biotechnology, catalog number: sc-29092 )
- Trypsin-EDTA (Sigma-Aldrich, catalog number: T4174 )
- Dimethyl Sulphoxide (DMSO) (Sigma-Aldrich, catalog number: D2650-100ML )
- Mr. FrostyTM Freezing Container (Thermo Fisher Scientific, catalog number: 5100-0001 )
- Isopropanol (Fisher Scientific, catalog number: A415-4 )
- MycoProbeTM Mycoplasma detection assay (R&D Systems, catalog number: CUL001B )
- Mitomycin C (10 mg) (Santa Cruz Biotechnology, catalog number: sc-3514B )
- CK18 (cytokeratin 18) monoclonal antibody clone CY-90 (Sigma-Aldrich, catalog number: C8541-.2ML )
- Goat anti Rabbit IgG IRDYE800 secondary antibody (tebu-bio, catalog number: 039611-132-122 )
- Goat anti-Mouse IgG Alexa Fluor® 680 secondary antibody (Thermo Fisher Scientific, InvitrogenTM, catalog number: A-21057 )
- TRAF-3 antibody (Santa Cruz Biotechnology, catalog number: sc-949 )
- Phospho-ASK1 (Thr845) antibody (Cell Signalling Technology, catalog number: 3765 )
- Agonistic anti-CD40 mAb G28-5 (used at 10 µg/ml), purified from culture supernatants of the HB-9110 hybridoma line (purchased from the ATCC)
- Affinity-purified human serum protein-adsorbed goat anti-mouse IgG (X-linker) (used at 5 µg/ml) (Sigma-Aldrich, catalog number: M8645 )
- Staurosporine from Streptomyces sp. (Sigma-Aldrich, catalog number: S4400-.1MG )
- Docetaxel (Sigma-Aldrich, catalog number: 01885-5MG-F )
- CellTiter 96® AQueous One Solution Assay (5,000 assays) (Promega, catalog number: G3581 )
- Cellular DNA Fragmentation ELISA kit (for up to 500 tests) (Roche Diagnostics, catalog number: 11585045001 )
- Cytotox-GloTM cytotoxicity assay (5 x 10 ml) (Promega, catalog number: G9291 )
- Sensolyte® Homogeneous AFC Caspase-3/7 Assay kit (Cambridge Bioscience, catalog number: ANA71114 )
- CM-H2DCFDA (chloromethyl derivative of 5-(and-6)-chloromethyl-2',7'-dichlorodihydrofluorescein diacetate) (Thermo Fisher Scientific, catalog number: C6827 )
- Glycerol (Fisher Scientific, catalog number: 10795711 )
- Sodium dodecyl sulphate (SDS) (Thermo Fisher Scientific, InvitrogenTM, catalog number: NP0002 )
- Tris-HCl, powder, 1 KG (Melford, catalog number: T1513 )
- Sodium fluoride (Acros Organics, catalog number: 424325000 )
- Sodium pyrophosphate tetrabasic (Sigma-Aldrich, catalog number: P8010-500G )
- Sodium orthovanadate (Sigma-Aldrich, catalog number: S6508-10G )
- Protease inhibitor (PI) cocktail (Merck, catalog number: 535140-1 )
- PI cocktail (New England Biolabs, catalog number: 5872S )
- AQUAGUARD-1 solution for disinfection of water baths and CO2 incubators (Biological Industries, catalog number: 01-867-1B ) (see Recipes)
- 1x PBS (see Recipes)
- PBS/EDTA solution (see Recipes)
- Freezing medium (see Recipes)
- FACS buffer (see Recipes)
- 70% ethanol (see Recipes)–Ethanol, Absolute (Fisher Scientific, catalogue number: E/0650DF/P17 )
- DR medium (see Recipes)
- SSB buffer (see Recipes)
- Lysis buffer (see Recipes)
- Purified water (double distilled ddH2O)
Equipment
- Gilson Pipettes
P1000 (200-1,000 μl)
P200 (50-200 μl)
P20 (2-20 μl)
P10 (1-10 μl) - Gilson REPETMAN electronic pipette 0.1-50 ml (Gilson, catalog number: F164503 )
- Water bath (Memmert)
- Boiling water bath (Grant)
- -80 °C freezer
- Sparkfree Refrigerator and Freezer (Labcold)
- Electrophoresis Power Supply
- Refrigerated centrifuge (PRISM R)
- Universal 320 benchtop centrifuge (Hettich Zentrifugen)
- Vortex mixer
- Ultrasonic Homogenizer Sonicator
- NuAire CellGard ES Biological Safety Cabinet (TripleRed)
- Iso Class 5 Nuaire Autoflow IR direct heat CO2 incubator with a HEPA filtration system at 37 °C and 5% CO2 (TripleRed)
- OdysseyTM Infra-red Imaging system (Li-Cor)
- Guava EasyCyteTM Flow Cytometer (Millipore)
- Countess II Automated Cell Counter (Thermo Fisher Scientific, catalog number: AMQAX1000 )
- EVOSTM XL Core Imaging System (Fisher Scientific)
- FLUOstar OPTIMA (BMG Labtech)
Software
- MARS software (BMG Labtech), Version 2.0
- Guava EasyCyte flow cytometry software (Millipore), Guavasoft Version 2.6
- Image Studio Life, Version 4.0
- Adobe Photoshop CS, Version 8.0
Procedure
- General cell culture
- Cell maintenance and passage
- Cells were routinely maintained at 37 °C in a 5% CO2 humidified atmosphere provided by autoclaved ddH2O, supplemented with 1% AQUAGUARD-1 (see Recipes). All class II cabinet surfaces as well as CO2 incubators were routinely disinfected with either Mikrozid (weekly) or 70% ethanol (daily) solution.
- Cultured cells were routinely observed by phase contrast microscopy. All cell lines were maintained in T75 flasks with 12-14 ml medium or T25 flasks in 4-5 ml medium. All cell lines were sub-cultured every 2-3 days or when ~80-95% confluent. Epithelial cells were routinely passaged at 1:10 ratios and effector (3T3Neo and 3T3CD40L) cells at 1:6 ratios.
- For routine passaging, cells were collected by washing with a solution of 0.1% (w/v) EDTA in phosphate buffered saline (EDTA/PBS) (see Recipes) for 5 min and then addition of Trypsin-EDTA solution, until cells detached from the culture flasks. Trypsin was inactivated by the addition of the respective serum-supplemented culture medium when cells were re-suspended, followed by centrifugation to aspirate medium and resuspension in fresh culture medium.
- Cultured cells were medium-changed every two days, unless otherwise stated. 3T3 cells were harvested and passaged as with carcinoma cells with the exemption of a very short EDTA/PBS treatment, as extended periods risked rapid cell detachment.
- Cells were routinely maintained at 37 °C in a 5% CO2 humidified atmosphere provided by autoclaved ddH2O, supplemented with 1% AQUAGUARD-1 (see Recipes). All class II cabinet surfaces as well as CO2 incubators were routinely disinfected with either Mikrozid (weekly) or 70% ethanol (daily) solution.
- Cryopreservation and recovery
- For long-term storage, following growth and appropriate passage, cell ‘banks’ and ‘sub-banks’ were cryo-preserved in liquid nitrogen as described below.
- For cryopreservation of cell lines, cultures were collected by routine passaging and precipitated by centrifugation. The cell pellet was re-suspended in Freezing medium (see Recipes) at a cell density not less than 1 x 106 cells/ml.
- Cells were aliquoted in a total of 1-1.5 ml to polypropylene cryovials and then transferred to an ice-cold ‘Mr Frosty freezing container’ containing 250 ml of isopropanol to control the cooling rate to 1 °C per minute. Cells were then placed within a -80 °C freezer for 4-6 h prior to transfer to liquid nitrogen.
- Cells were recovered by thawing rapidly at 37 °C, before 5-10 ml of pre-warmed growth medium was added. Cells were centrifuged at 210 x g for 5 min and then seeded to tissue culture flasks as required (T25 flasks for routine maintenance and T75 flasks for preparation of co-cultures).
Notes:- We recommend that experiments be carried out for cultures maintained for a maximum of 10-15 passages. To avoid any artefacts due to possible culture-associated genetic drift, new cell cultures should be established following that number of passages. The usage of cell ‘banks’ and ‘sub-banks’ allows all experimentation to be performed with consistent results.
- For cell lines maintained in culture medium supplemented with FBS, the serum batch used was previously tested (‘batch-testing’) to ensure it was optimal for cell growth. Cell growth/proliferation was tested using the CellTiter 96® AQueous One Solution Assay.
- We recommend that experiments be carried out for cultures maintained for a maximum of 10-15 passages. To avoid any artefacts due to possible culture-associated genetic drift, new cell cultures should be established following that number of passages. The usage of cell ‘banks’ and ‘sub-banks’ allows all experimentation to be performed with consistent results.
- For long-term storage, following growth and appropriate passage, cell ‘banks’ and ‘sub-banks’ were cryo-preserved in liquid nitrogen as described below.
- Other
All cell lines were routinely tested to confirm absence of Mycoplasma contamination using the MycoProbeTM Mycoplasma detection assay (see Reagents).
- Cell maintenance and passage
- Co-culture of effector (3T3Neo and 3T3CD40L) and target (epithelial) cells (Figure 1)
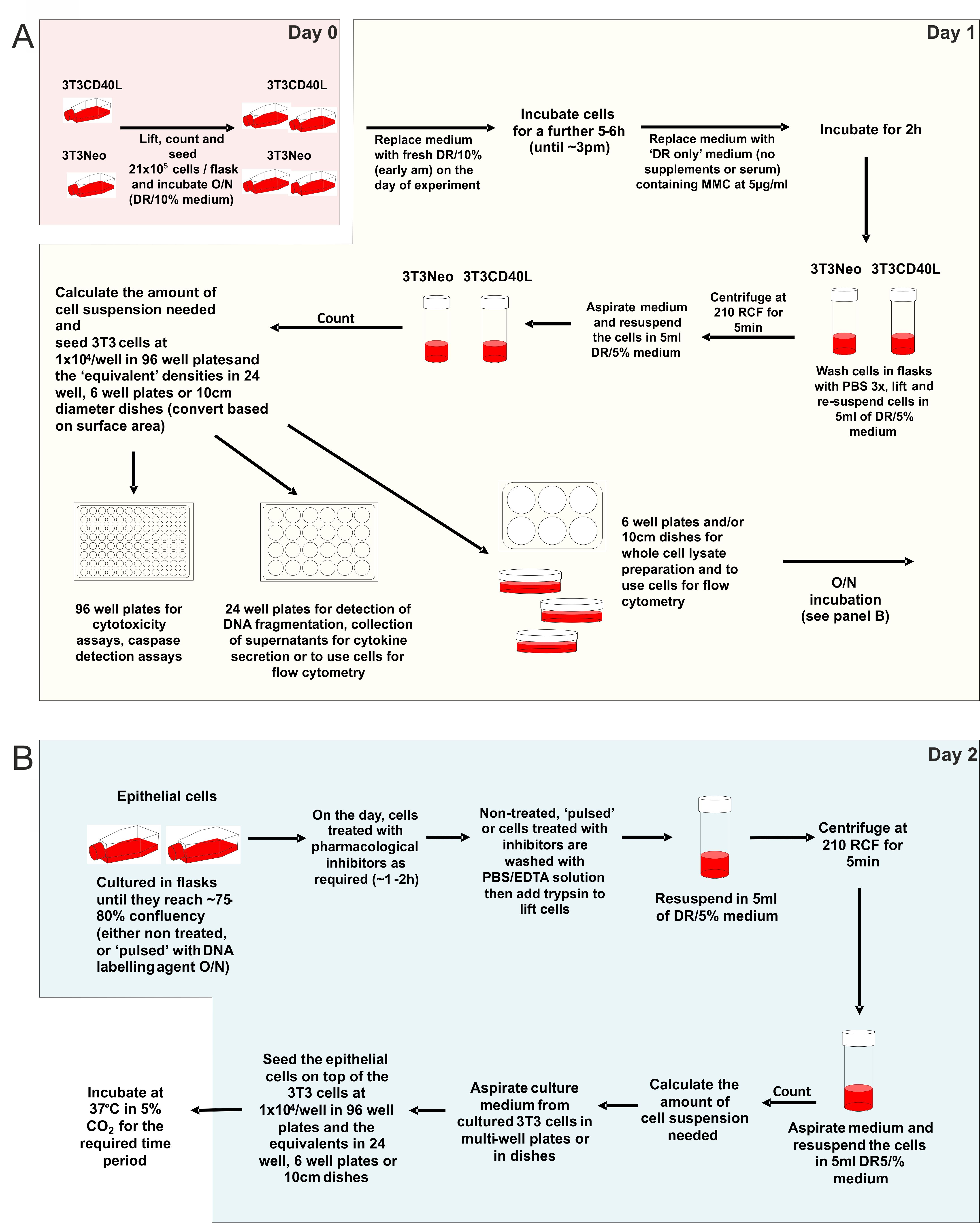
Figure 1. Step-by-step guide for the preparation of co-cultures for the delivery of mCD40L to target epithelial cells. Effector (3T3) cells are prepared for co-cultures (‘Day 0’ and ‘Day 1’) as detailed (A) and seeded in appropriate multi-well formats (dependent on the readout/assay). Thereafter, target epithelial cells are prepared (‘Day 2’) and seeded on top of the effector cells (B) to set up the co-cultures. Following completion of the procedure, the co-cultures are incubated for the time period required in order to detect biochemical markers of apoptosis or intracellular signaling mediators of cell death (as indicated in A).- Preparation of effector and target cells (‘Day 0’)
- The day before establishing the co-cultures, the effector cells (3T3Neo and 3T3CD40L) should be in exponential growth phase and no more than 85% confluent.
- Passage 3T3Neo and 3T3CD40L effector cells and seed in T75 flasks at a density of 21 x 105 cells per flask and incubate overnight (O/N).
- In parallel ensure that target cells (epithelial cells, e.g., HCT116 and EJ) are passaged appropriately in order to ensure they are in exponential growth phase (the ‘splitting’ ratio depends on the growth characteristics of the cell line).
- The following day, i.e., the day when co-culture experiments are initiated (‘Day 1’ – see below), replenish the culture medium for all cultures (i.e., medium-change 3T3s and target epithelial cells).
Note: Ensure this takes place first thing am (particularly for 3T3 cells, before the next step is performed).
- The day before establishing the co-cultures, the effector cells (3T3Neo and 3T3CD40L) should be in exponential growth phase and no more than 85% confluent.
- Growth-arresting of effector 3T3 cells by Mitomycin C (MMC) treatment (‘Day 1’)
- Approximately 5-6 h (i.e., mid-afternoon) after the effector 3T3 cells have been medium-changed, remove the medium from 3T3Neo and 3T3CD40L flasks and add 9 ml per T75 flask of DR only medium (without supplements) containing 5 µg/ml of Mitomycin C (MMC), and incubate for 2 h at 37 °C. Figure 2A shows representative cultures of 3T3 cells before and after MMC treatment.
- Aspirate the medium and wash the cells with PBS 3-times to remove any traces of MMC. Add 0.8 ml of trypsin-EDTA to each flask and incubate for 1-2 min until cells have detached, then inactivate with complete culture medium. Centrifuge at 210 x g for 5 min, aspirate the supernatant and resuspend in complete culture medium.
- Count cells (3T3Neo and 3T3CD40L) and calculate the required volume of cell suspension. Add 100 µl containing 1 x 104 cells per well in 96 well plates (for cytotoxicity assays), 6 x 104 cells per well in 24 well plates (for supernatant collection), 3 x 105 cells per well in 6 well plates, or 30 x 105 cells per well in 10 cm diameter dishes (for cultures to be used for flow cytometry experiments and preparation of whole cell lysates).
Note: The number of cells and minimal volume per well for different types of multi-well plates is based on a conversion according to the differences in culture surface area. - Once the effector 3T3 cells have been seeded, incubate plates O/N at 37 °C (Figure 1A).
Note: For DNA fragmentation assays, the epithelial cells can be pre-pulsed with DNA labelling agent O/N (see below).
- Approximately 5-6 h (i.e., mid-afternoon) after the effector 3T3 cells have been medium-changed, remove the medium from 3T3Neo and 3T3CD40L flasks and add 9 ml per T75 flask of DR only medium (without supplements) containing 5 µg/ml of Mitomycin C (MMC), and incubate for 2 h at 37 °C. Figure 2A shows representative cultures of 3T3 cells before and after MMC treatment.
- Seeding of target (epithelial cells) onto 3T3 effector cells (co-culture) (‘Day 2’)
- The following day (first thing in the morning), the target cells are washed with EDTA/PBS (following medium removal), then 0.8 ml of trypsin-EDTA are added per flask and cells are incubated at 37 °C for 2-3 min until cells have detached. The trypsin is inactivated with addition of complete culture medium.
Note: If necessary, target cells can be pre-treated with any pharmacological inhibitors (for functional experiments) as required, before trypsinisation and counting. - Centrifuge at 210 x g for 5 min and then aspirate the supernatant and resuspend in complete culture medium. Then, count the cells and calculate the required volume of cell suspension.
- Remove the culture medium from each well or culture dishes containing the effector cells (3T3s) from the previous day (Step B2c above) using a small yellow tip and gentle aspiration (it helps to ensure the multi-well plate is tilted and the tip touches just the edge at the bottom of the well to avoid removal of adhered cells).
- Add 100 µl of suspension containing 1 x 104 cells per well of epithelial cells on top of the 3T3 effectors (or the equivalent volume and number of cells based on the conversion described in Step B2 above).
Note: By seeding the above-stated number of epithelial cells, a 1:1 (effector:target) ratio is utilised (e.g., HCT116 cells); however in some cases a 1:0.8 ratio might be optimal (e.g., EJ cells). - The co-cultures are now established and multi-well plates (or dishes) are incubated at 37 °C (Figure 1B).
- Incubation period varies depending on the assay or procedure to be performed (e.g., cell death assays, collection of supernatants or to lyse the cells). Figure 2B provides an example of representative co-cultures at 72 h.
Notes:- The same steps are followed for the co-culture of normal epithelial cells with 3T3 effectors, however for such co-cultures normal (NHU) cells are seeded in their own (serum-free, KSFM medium).
- For functional inhibition experiments, co-cultures can take place in the continued presence of any pharmacological inhibitor, as required.
- The same steps are followed for the co-culture of normal epithelial cells with 3T3 effectors, however for such co-cultures normal (NHU) cells are seeded in their own (serum-free, KSFM medium).
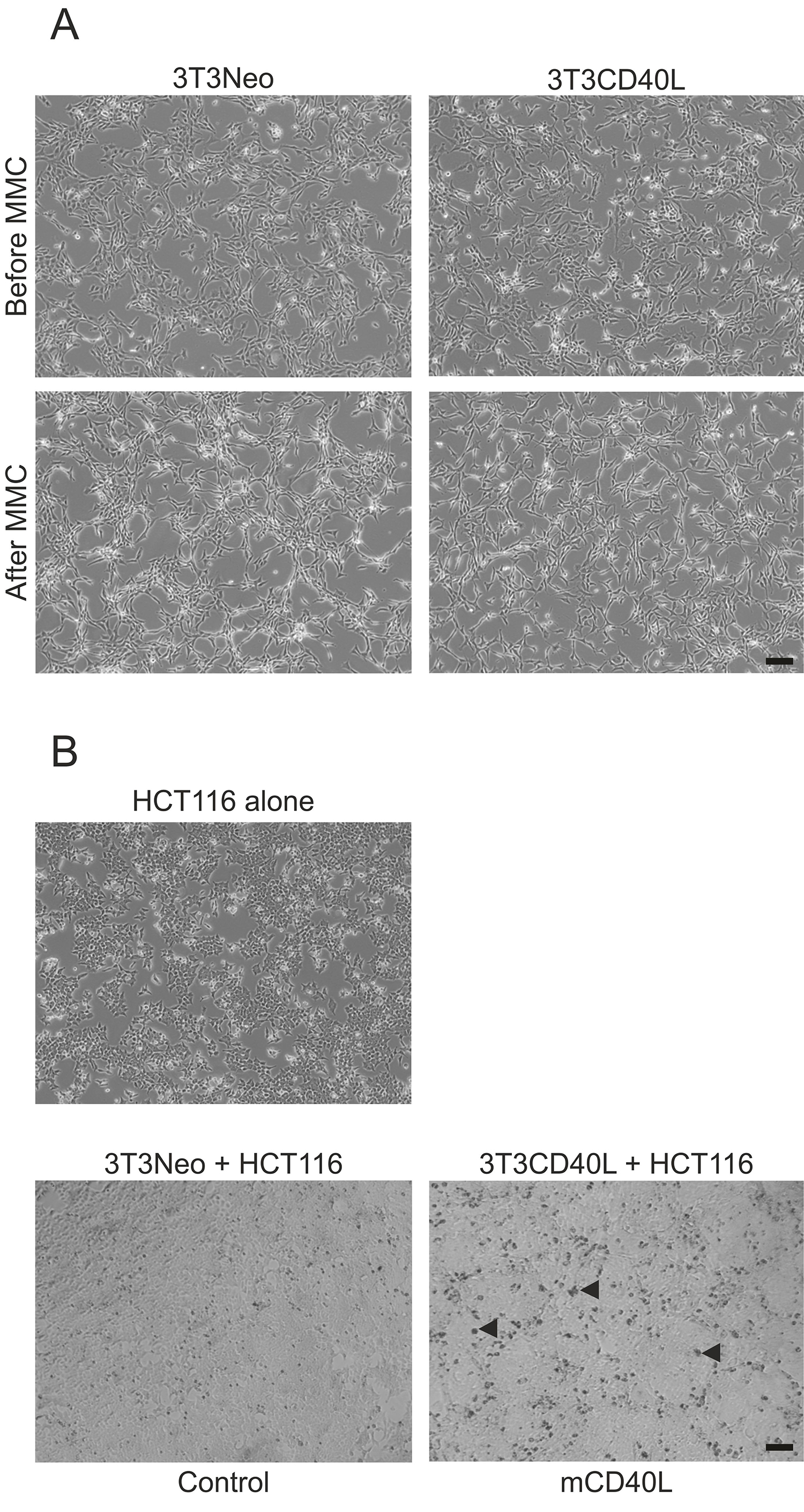
Figure 2. Images of effector (3T3Neo and 3T3CD40L) cells alone or in co-culture with epithelial (HCT116) cells. A. Representative phase contrast images of 3T3Neo and 3T3CD40L effector cells before and after MMC treatment, which is carried out in order to mediate 3T3 cell growth arrest. The images confirm how 3T3 cells remain viable and healthy following treatment. B. Phase contrast microscopy images of representative co-cultures of growth-arrested 3T3Neo and 3T3CD40L effector cells with the carcinoma cell line HCT116 for 72 h. Co-culture of HCT116 cells with 3T3CD40L fibroblasts represents delivery of ‘mCD40L’, whilst co-culture with 3T3Neo serves as ‘Control’. Note how cytotoxicity is evident by the presence of dark, phase-non-bright apoptotic cells (representative dead cells, or aggregates of such cells, are indicated by solid triangles). Scale bars = 100 μm. - The following day (first thing in the morning), the target cells are washed with EDTA/PBS (following medium removal), then 0.8 ml of trypsin-EDTA are added per flask and cells are incubated at 37 °C for 2-3 min until cells have detached. The trypsin is inactivated with addition of complete culture medium.
- Preparation of effector and target cells (‘Day 0’)
- Activation of CD40 ligation by soluble agonist
- In order to compare the effects of mCD40L to those of soluble agonists, epithelial cells seeded at the same densities described above, are treated routinely with the well-characterized agonistic anti-CD40 mAb G28-5 (Bugajska et al., 2002).
- Add G28-5 monoclonal antibody (mAb) during seeding of the epithelial cells at a concentration of 10 µg/ml (mAb was purified from culture supernatants of the HB-9110 hybridoma line).
- To increase the cross-linking capacity of the mAb, crosslink the antibody with the affinity-purified human serum protein-adsorbed goat anti-mouse IgG (X-linker) used at 2.5 µg/ml. Add the X-linker to the cultures 30 min following addition of the G28-5 mAb.
Note: The differences in the pro-apoptotic capabilities of soluble agonists versus mCD40L have been presented and detailed elsewhere (Dunnill et al., 2017).
- In order to compare the effects of mCD40L to those of soluble agonists, epithelial cells seeded at the same densities described above, are treated routinely with the well-characterized agonistic anti-CD40 mAb G28-5 (Bugajska et al., 2002).
- Detection of biochemical markers of cell death (apoptosis)
- Loss of cell membrane integrity (permeabilisation)
- Seed carcinoma cells on top of growth-arrested 3T3s in 96 well white, tissue culture-treated plates (as detailed above) and incubate at 37 °C for 48 h (or 72 h).
- Add 50 µl of CytoTox-GloTM substrate (see Reagents) and incubate for 10 min at room temperature in the dark (therefore the final volume is 150 µl/well).
- Use a FLUOstar OPTIMA (BMG Labtech) plate reader to measure luminescence (RLU).
- Acquire data using MARS software (BMG Labtech) and analyse by Microsoft Excel appropriately to correct for background associated with 3T3 fibroblasts (as detailed in the Data analysis section); representative data are provided in Figure 3.
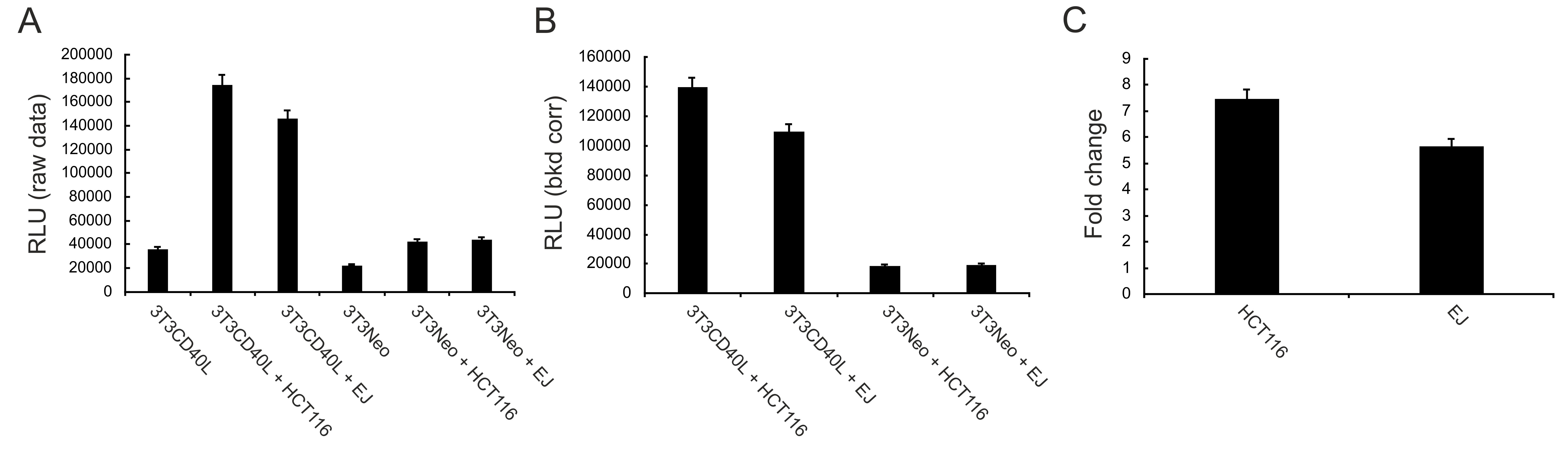
Figure 3. Detection of loss of plasma membrane integrity as a marker of cell death. 3T3Neo and 3T3CD40L fibroblasts were co-cultured with HCT116 and EJ tumour cells as detailed in the main text for 48 h. Loss of membrane integrity was detected using the Cytotox-Glo reagent. Raw data (relative luminescence units, RLU) for all cultures (co- and mono-cultures) are shown in panel A. Data for co-cultures only, obtained following pairwise subtraction of 3T3Neo (Control) and 3T3CD40L (mCD40L) culture RLU readings from the RLU readings obtained for the respective co-cultures (‘bkgd corr’), are presented in panel B (see also Data analysis). Finally, data are expressed as fold change (i.e., ‘mCD40L RLU’/‘Control RLU’ for each co-culture) in panel C. - Seed carcinoma cells on top of growth-arrested 3T3s in 96 well white, tissue culture-treated plates (as detailed above) and incubate at 37 °C for 48 h (or 72 h).
- Effector caspase activation
- Seed carcinoma cells on top of growth-arrested 3T3s in 96 well white, tissue culture-treated plates (as detailed above) and incubate at 37 °C for 48 h or 72 h (the latter time-point often provides more intense caspase activity).
- Top up all wells with 50 µl of medium, then add 50 µl of SensoLyte® Homogenous AFC Caspase-3/7 substrate (see Reagents), so the total volume is 200 µl/well.
- Incubate the plates at room temperature (RT) overnight in the dark, then measure fluorescence (RFU) using a FLUOstar OPTIMA (BMG Labtech) plate reader, using Excitation/Emission 355 nm/520 nm filters, respectively.
- Acquire data using MARS software (BMG Labtech) and analyse using Microsoft Excel appropriately in order to correct for background related to 3T3 fibroblasts (as detailed in the Data analysis section); representative data are provided in Figure 4.

Figure 4. Detection of effector caspase activation. 3T3Neo and 3T3CD40L fibroblasts were co-cultured with HCT116 and EJ tumour cells as in Figure 3 for 72 h. Effector caspase 3/7 activity was detected using the Sensolyte caspase 3/7 reagent. Relative fluorescence unit (RFU) data for co-cultures were obtained following pairwise subtraction of 3T3Neo (Control) and 3T3CD40L (mCD40L) mono-culture RFU readings from the RFU readings obtained for the respective co-cultures (‘bkgd corr’) and shown in panel A. Data are also expressed as fold caspase activity (i.e., ‘mCD40L RFU’/‘Control RFU’ for each co-culture) in panel B. - Seed carcinoma cells on top of growth-arrested 3T3s in 96 well white, tissue culture-treated plates (as detailed above) and incubate at 37 °C for 48 h or 72 h (the latter time-point often provides more intense caspase activity).
- DNA fragmentation
- For this assay, and as shown in Figure 1B, on the day when 3T3 fibroblasts are treated with MMC and seeded, the epithelial cells can be treated (‘pulsed’) with DNA labelling agent BrdU at a concentration of 10 µM (according to the manufacturer’s instructions, see Reagents) overnight (O/N).
- The next day, epithelial cells are washed once with EDTA/PBS, lifted by trypsinisation, counted and then added as a 200 µl suspension on top of the growth-arrested 3T3 cells (following medium removal from the wells and as described in the co-culture protocol above). Cell cultures are then incubated for 48 h (or 72 h).
- As positive control and for the purposes of calculating % DNA fragmentation (below), treat carcinoma cells alone with 0.5 µM of Staurosporine (STS) (or, alternatively, 3 µg/ml Doxorubicin) for 48 h.
- The day before DNA fragmentation is measured, prepare (‘coat’) ELISA plates for the detection of fragmented DNA according to the manufacturer’s instructions.
- On the day of the experiment, collect supernatants from co-cultures and add 100 µl of culture supernatant to the appropriate wells in the coated 96 well ELISA plates; perform ELISA as described by the manufacturer and when complete, incubate the 96 well ELISA plate(s) O/N at 4 °C.
- On the following day, complete the ELISA assay (as described in the manufacturer’s instructions) and using a FLUOstar OPTIMA (BMG Labtech) plate reader measure absorbance (Abs) at 455 nm.
- Acquire data using MARS software (BMG Labtech) and analyse by Microsoft Excel.
- Calculate % DNA fragmentation as follows: % apoptotic cells = (Abs of co-cultured cells) x 100/(Abs of target cells treated with STS); representative data are provided in Figure 5.
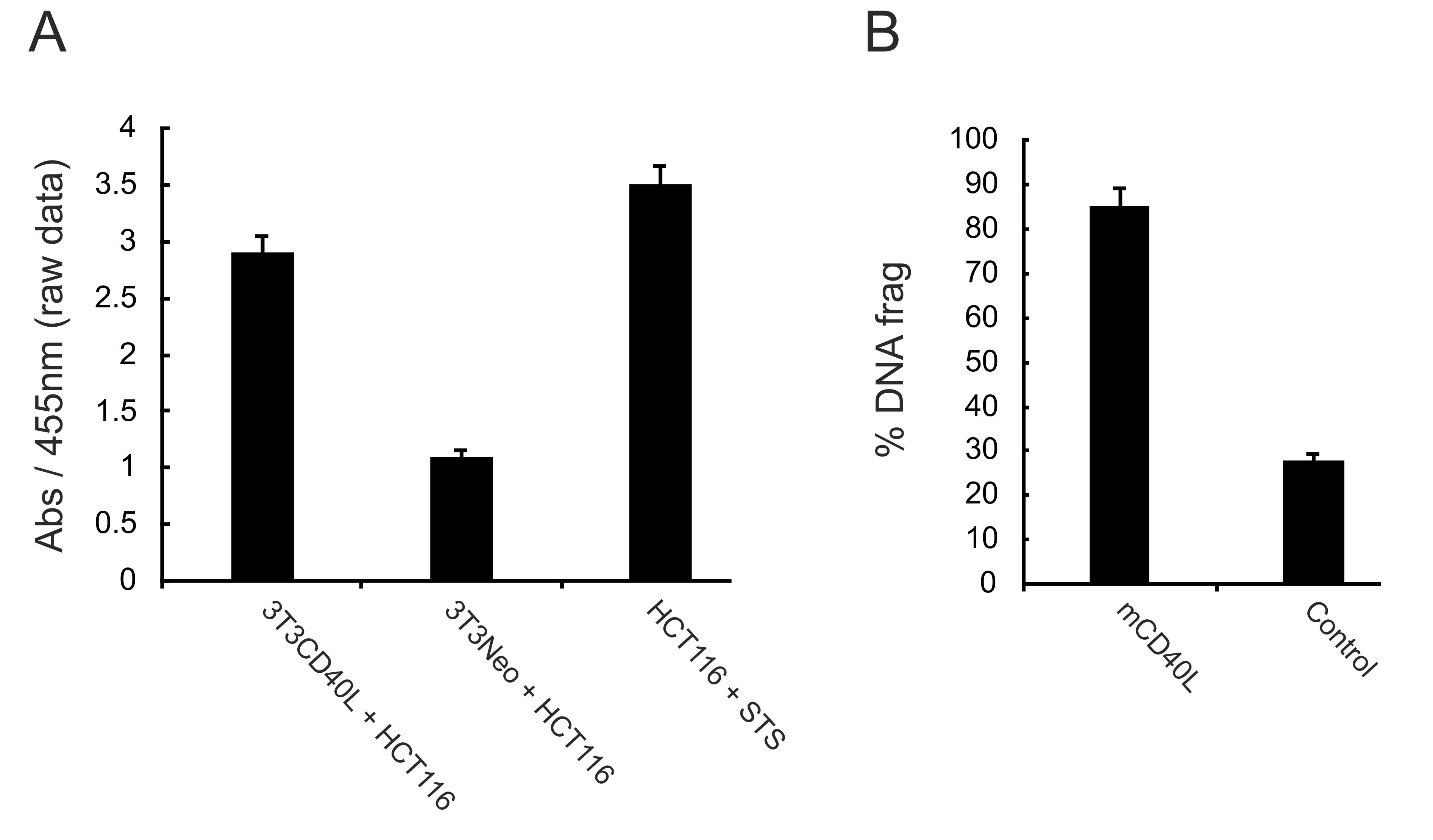
Figure 5. Detection of DNA fragmentation. 3T3Neo and 3T3CD40L fibroblasts were co-cultured with DNA labelling agent (BrdU)-pulsed HCT116 cells for 48 h and DNA fragmentation was detected using an ELISA assay as detailed in the main text. Raw absorbance readings (at 455 nm wavelength) are presented in panel A for both co-cultures, whilst treatment of HCT116 cells with staurosporine (STS) at 0.5 μM served as a positive control for maximal DNA fragmentation. Values obtained for co-cultures were expressed as % DNA fragmentation (% DNA frag) relative to STS (panel B), calculated as detailed in the appropriate section in the main text. - For this assay, and as shown in Figure 1B, on the day when 3T3 fibroblasts are treated with MMC and seeded, the epithelial cells can be treated (‘pulsed’) with DNA labelling agent BrdU at a concentration of 10 µM (according to the manufacturer’s instructions, see Reagents) overnight (O/N).
- Loss of cell membrane integrity (permeabilisation)
- Detection of intracellular mediators of cell death
- Reactive oxygen species
Measurement of fluorescence by spectrophotometry- Epithelial cells are co-cultured with 3T3Neo and 3T3CD40L cells as described above (in 96 well plates). Notably, however, when such co-cultures are performed, 3T3s are not growth-arrested with MMC to avoid high background fluorescence readings due to interaction with CM-H2DCFDA.
Note: The duration of the experiments is short and therefore any growth of 3T3 cells does not affect overall cell viability in such short-term co-cultures. - Incubate co-cultures for the required period of time (from 30 min to 3 h).
- Wash cells with PBS to remove any culture medium then add 100 µl/well of pre-warmed (at 37 °C) PBS containing 1 µM of CM-H2DCFDA and incubate for 30 min at 37 °C in 5% CO2. After treatment, remove the CM-H2DCFDA reagent and add 100 µl of pre-warmed (at 37 °C) PBS.
- Measure fluorescence on a FLUOstar OPTIMA (BMG Labtech) plate reader at Excitation 485 nm/Emission 520 nm.
- Acquire data using MARS software (BMG Labtech) and analyse using Microsoft Excel appropriately in order to correct for background related to 3T3 fibroblasts (as detailed in the Data analysis section); representative data are provided in Figure 6A.
Measurement of fluorescence by flow cytometry- Treat (label) the required number of epithelial cells (e.g., EJ) with pre-warmed PBS containing 1 µM of H2DCFDA and incubate for 30 min at 37 °C in 5% CO2. Epithelial cells can be labelled whilst adherent or in suspension.
- Co-culture CM-H2DCFDA labelled epithelial cells with 3T3Neo and 3T3CD40L effectors as described above in 6 well plates for 3 h.
- Following co-culture, add 200 µl of trypsin to lift cells, and when the cells have detached, inactivate by adding 500 µl of PBS containing 5% FBS.
- Collect cells in 1.5 ml sample tubes and centrifuge for 10 min at 210 x g at 4 °C.
- Discard the supernatant, resuspend in cold PBS (or FACS buffer) and measure fluorescence by flow cytometry using a Guava EasyCyte instrument. Analyse data post-acquisition using Guava EasyCyte software; Figure 6B provides an example of such analysis and illustrates the ‘gating’ strategy followed to measure ROS induction in epithelial cells only (and to exclude any 3T3 cell associated auto-fluorescence).
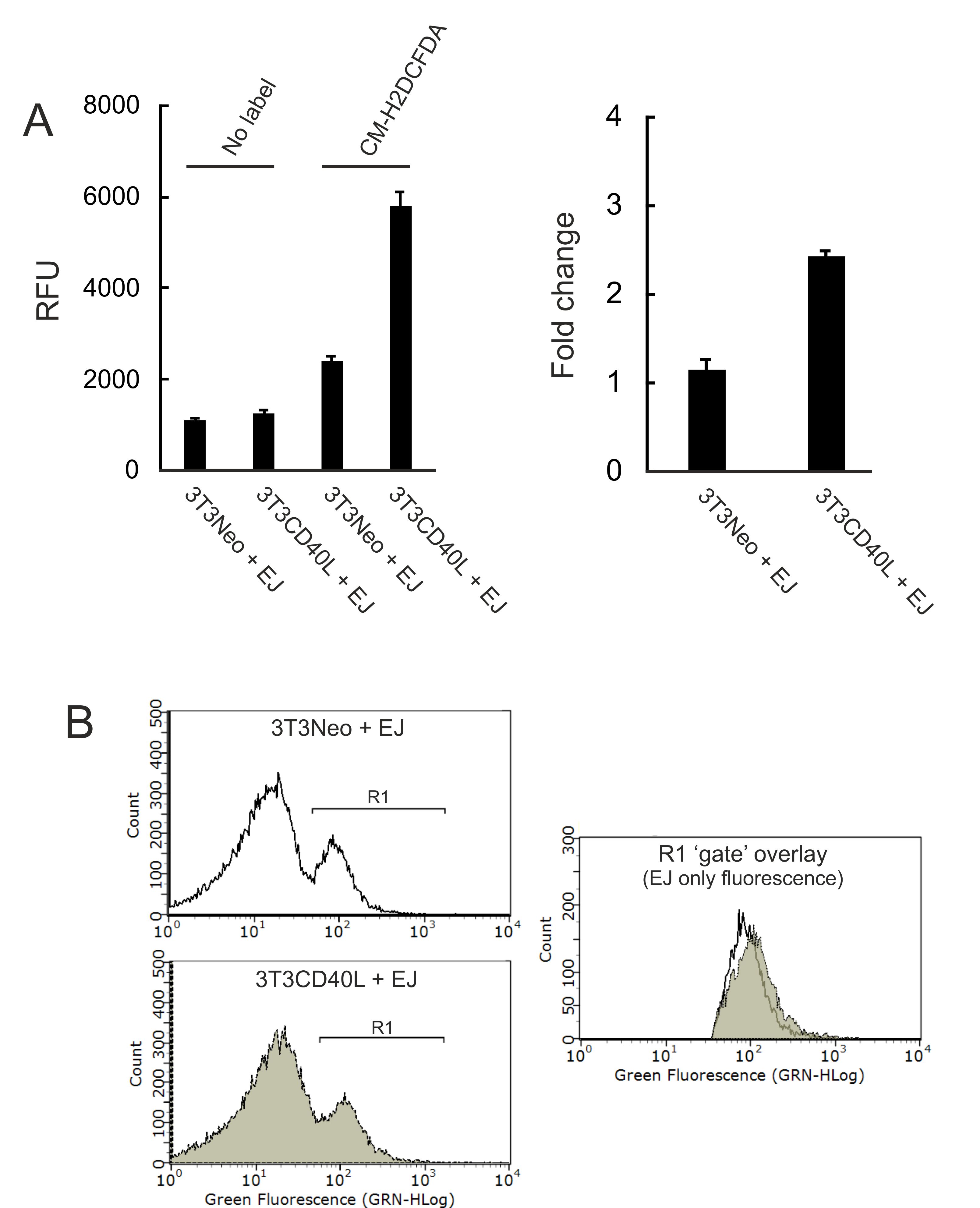
Figure 6. Detection of ROS by spectrophotometry (A) and flow cytometry (B) analysis. A. 3T3Neo and 3T3CD40L fibroblasts were seeded in 96 well plates before being co-cultured with EJ cells for 30 min. Cell cultured were labelled with CM-H2DCFDA as described in the main text. RFU data were obtained following pairwise background subtraction from 3T3Neo and 3T3CD40L alone mono-cultures. Unlabelled co-cultures (No label) served as negative controls (left). Data are also presented as fold change in RFU readings relative to controls (right). B. 3T3Neo and 3T3CD40L fibroblasts were seeded in 24 well plates before being co-cultured with EJ cells for 3 h (EJ were labelled with CM-H2DCFDA before co-culture). Green fluorescence intensity histograms provided a pattern of fluorescence indicative of two populations (‘peaks’), 3T3 and epithelial cells, which were unlabelled and labelled with CM-H2DCFDA, respectively. Appropriate ‘gating’ on the epithelial cell population (right peak) was performed as indicated (gate R1) for 3T3Neo/EJ (upper histogram) and 3T3CD40L/EJ co-cultures (lower histogram). Data from both were overlaid (right panel) following gating, in order to identify changes in fluorescence intensity in EJ cells only. The shift in intensity (grey versus empty) indicates ROS induction mediated by mCD40L.
Note: For both spectrophotometric and flow cytometric detection of ROS, 3T3 cells were not growth-arrested with MMC to ensure lack of significant background fluorescence following labelling with CM-H2DCFDA. - Epithelial cells are co-cultured with 3T3Neo and 3T3CD40L cells as described above (in 96 well plates). Notably, however, when such co-cultures are performed, 3T3s are not growth-arrested with MMC to avoid high background fluorescence readings due to interaction with CM-H2DCFDA.
- Adaptor proteins and pro-apoptotic kinases (immunoblotting)
- Seed 3T3Neo and 3T3CD40L cells following treatment with MMC (above) in 10 cm diameter dishes at 30 x 105 cells/dish in 10 ml of DR 5%, or in 6 well plates at 3 x 105/well in 2 ml of medium (prepare duplicate dishes or wells for every condition). Incubate dishes/plates at 37 °C O/N.
- Following O/N attachment, remove supernatants and add 10 ml of target cell suspension in DR/5% medium containing 30 x 105 cells/dish or 2 ml containing 3 x 105 cells/well in 6 well plates.
- Incubate the dishes/plates at 37 °C and 5% CO2 for the required time period (can vary from 1.5 h to 36 h).
- Upon completion of the incubation period, prepare lysates by adding 150 µl of complete lysis buffer/dish or 70 µl/well for 6-well plates (see Recipes for lysis buffer preparation) and quantify protein concentration in all lysates as described (Dunnill et al., 2017).
- Perform SDS-Polyacrylamide gel Electrophoresis (SDS-PAGE) and immunoblotting for 40 µg of protein lysate for the detection of intracellular proteins expression in target cells; representative data are shown in Figure 7B.
Note: Although expression of β-actin can be used to demonstrate overall equal loading, because lysates are prepared from 3T3neo/3T3CD40L and epithelial cell co-cultures (and thus are a mixture of cell types – epithelial and non-epithelial cells), it is essential to ensure that sample loading is confirmed (and adjusted if necessary) for epithelial cells; this can be carried out according to reactivity with human-specific anti-cytokeratin antibodies e.g., anti-CK8 or anti-CK18 – see Figure 7A for an example and elsewhere (Dunnill et al., 2017).

Figure 7. Detection of intracellular signalling proteins activated by mCD40L using immunoblotting. HCT116 cells were co-cultured with 3T3Neo (‘–’) and 3T3CD40L (‘mCD40L’) fibroblasts for the indicated time periods and whole cell protein lysates prepared as described in the main text. Forty microgram of total protein were analysed by SDS-PAGE and immunoblotting. β-actin expression demonstrated overall equal loading, whilst correct loading for epithelial cell lysate was confirmed according to reactivity with human-specific anti-CK18 antibody (see main text) (panel A). Representative experiments for detection of TRAF3 and phospho-ASK1 are shown in panel B. Note how the inclusion of lysate from 3T3CD40L culture alone confirmed the specificity of the antibodies for human (epithelial) cell protein detection. - Seed 3T3Neo and 3T3CD40L cells following treatment with MMC (above) in 10 cm diameter dishes at 30 x 105 cells/dish in 10 ml of DR 5%, or in 6 well plates at 3 x 105/well in 2 ml of medium (prepare duplicate dishes or wells for every condition). Incubate dishes/plates at 37 °C O/N.
- Reactive oxygen species
Data analysis
- Analysis was carried using the software listed above for the generation of raw data and images. Data was analysed on Excel and images were processed using Adobe Photoshop CS.
- To calculate cell death or measure ROS induction, it was essential to ensure that any background ‘noise’ caused due to the presence of effector 3T3 cells was accounted for by subtraction from the readings of co-cultured cells. Therefore, background fluorescence or luminescence readings were subtracted pairwise as appropriately; i.e., RFU or RLU readings for 3T3CD40L alone cultures were subtracted from 3T3CD40L + HCT116 co-culture readings (and similarly for 3T3Neo and 3T3Neo + HCT116 co-culture readings). The exemption was DNA fragmentation, as this was unnecessary due to the pre-labelling of target epithelial cells only. Moreover, in all experiments blank controls were included as appropriate.
- Mean values and standard deviation (SD) were used as descriptive statistics. Two-tailed, paired or non-paired Student’s t-tests, and where appropriate ANOVA, were used for evaluation of statistical significance. For all experiments a minimum of 5-6 technical replicates were included and experiments were performed in triplicate (n = 3).
Notes
- In order to ensure that all cells used for the preparation of the co-cultures are healthy, it is important to work reasonably fast and ensure that cells are provided with adequate CO2 and are kept at 37 °C when not being used.
- All reagents used for the detection of markers of apoptosis (and unless otherwise stated by the manufacturer) must be thawed and brought to room temperature adequately before addition to cells.
Recipes
- AQUAGUARD-1 solution
10 ml AQUAGUARD-1
1,000 ml ddH2O - 1x PBS
900 ml ddH2O
100 ml 10x D-PBS - PBS/EDTA
1 g Ethylenediaminetetraacetic acid (EDTA)
1,000 ml Phosphate-buffered saline (PBS) (1x) - Freezing medium
10% (v/v) FBS
10% (v/v) DMSO
80% (v/v) standard culture medium - 70% ethanol
700 ml ethanol
300 ml ddH2O - DR medium
250 ml DMEM
250 ml RPMI
Supplement either 5% FBS and 1% L-Glutamine for maintenance of carcinoma cells or 10% FBS and 1% L-Glutamine for the maintenance of 3T3 cells - FACS buffer
9 ml 1x PBS
1 ml FBS - SSB buffer
Make up to a final volume of 50 ml using ddH2OGlycerol 10 ml Sodium dodecyl sulphate (SDS) 1 g Tris-HCl 6.25 ml (stock solution 1 M, pH 6.8) Sodium fluoride (NaF) 0.42 g Sodium pyrophosphate tetrabasic 0.446 g Sodium orthovanadate 0.0184 g
Note: The solution requires stirring whilst being heated at 50 °C. - Lysis buffer (for preparation of cell lysates)
1 ml SSB buffer
13 µl DTT solution (2 mg/ml in PBS)
2 µl protease inhibitor (PI) cocktail (VWR) or 10 µl PI cocktail (New England Biolabs)
Acknowledgments
The co-culture protocol is a refined and further optimised version based on our reviously published study (Dunnill et al., 2017). KI was supported by a Ph.D. scholarship from the Iraqi Ministry of Higher Education and Scientific Research. AM and BA were supported by overseas Ph.D. scholarships from the Libyan Ministry of Higher Education. MI is a Ph.D. student currently being supported by the Jacquie Roeder Research Fund from the Laura Crane Youth Cancer Trust. All authors declare no conflicts of interest.
References
- Albarbar, B., Dunnill, C. and Georgopoulos, N. T. (2015). Regulation of cell fate by lymphotoxin (LT) receptor signalling: Functional differences and similarities of the LT system to other TNF superfamily (TNFSF) members. Cytokine Growth Factor Rev 26(6): 659-671.
- Bugajska, U., Georgopoulos, N. T., Southgate, J., Johnson, P. W., Graber, P., Gordon, J., Selby, P. J. and Trejdosiewicz, L. K. (2002). The effects of malignant transformation on susceptibility of human urothelial cells to CD40-mediated apoptosis. J Natl Cancer Inst 94(18): 1381-1395.
- Dunnill, C. J., Ibraheem, K., Mohamed, A., Southgate, J. and Georgopoulos, N. T. (2017). A redox state-dictated signalling pathway deciphers the malignant cell specificity of CD40-mediated apoptosis. Oncogene 36(18): 2515-2528.
- Georgopoulos, N. T., Merrick, A., Scott, N., Selby, P. J., Melcher, A. and Trejdosiewicz, L. K. (2007). CD40-mediated death and cytokine secretion in colorectal cancer: a potential target for inflammatory tumour cell killing. Int J Cancer 121(6): 1373-1381.
- Georgopoulos, N. T., Steele, L. P., Thomson, M. J., Selby, P. J., Southgate, J. and Trejdosiewicz, L. K. (2006). A novel mechanism of CD40-induced apoptosis of carcinoma cells involving TRAF3 and JNK/AP-1 activation. Cell Death Differ 13(10): 1789-1801.
- Hill, K. S., Errington, F., Steele, L. P., Merrick, A., Morgan, R., Selby, P. J., Georgopoulos, N. T., O'Donnell, D. M. and Melcher, A. A. (2008). OK432-activated human dendritic cells kill tumor cells via CD40/CD40 ligand interactions. J Immunol 181(5): 3108-3115.
- Young, L. S. and Eliopoulos, A. G. (2004). TNF receptors and their ligands: in sickness and in health, in life and death. Curr Opin Pharmacol 4(4): 311-313.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Ibraheem, K., Dunnill, C. J., Ioannou, M., Mohamed, A., Albarbar, B. and Georgopoulos, N. T. (2018). An in vitro Co-culture System for the Activation of CD40 by Membrane-presented CD40 Ligand versus Soluble Agonist. Bio-protocol 8(13): e2907. DOI: 10.21769/BioProtoc.2907.
Category
Cancer Biology > Cell death > Cell biology assays > Apoptosis
Cell Biology > Cell isolation and culture > Co-culture
Cell Biology > Cell signaling > Intracellular Signaling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.