- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Preserve Cultured Cell Cytonemes through a Modified Electron Microscopy Fixation

Published: Vol 8, Iss 13, Jul 5, 2018 DOI: 10.21769/BioProtoc.2898 Views: 6788
Reviewed by: Zinan ZhouTomas AparicioSilvia Caggia
Original research article
The authors used this protocol in:

Oct 2017


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Immunocytochemistry of cultured cells is a common and effective technique for determining compositions and localizations of proteins within cellular structures. However, traditional cultured cell fixation and staining protocols are not effective in preserving cultured cell cytonemes, long specialized filopodia that are dedicated to morphogen transport. As a result, limited mechanistic interrogation has been performed to assess their regulation. We developed a fixation protocol for cultured cells that preserves cytonemes, which allows for immunofluorescent analysis of endogenous and over-expressed proteins localizing to the delicate cellular structures.
Keywords: MEM-fixBackground
Cytonemes are classified as thin (~200 nm diameter) actin based filopodia, over 2 μm in length, which can transport morphogens (Ramírez-Weber and Kornberg, 1999). These signaling structures were first classified and described in detail in the developing Drosophila wing imaginal disc, and have subsequently been observed in mouse, chick and zebrafish model organisms (Ramírez-Weber and Kornberg, 1999; Sanders et al., 2013; Stanganello et al., 2015). In most of these cases, cytoneme detection was only possible with live imaging of over-expressed, fluorescently labeled proteins. Examination of cytonemes of cultured cells has been limited due to traditional fixation protocols failing to preserve these fragile filaments. These complications have been limiting factors in determining the cellular mechanisms driving cytoneme formation and function during development and tissue homeostasis, and determining whether these processes are corrupted in disease.
In order to overcome these limitations, we developed a modified electron microscopy fixative (MEM-fix)-based protocol that preserves cytonemes of cultured cells. Use of MEM-fix allows for the detection of endogenous and over-expressed proteins of interest in the filopodial structures via traditional immunofluorescent protocols (Bodeen et al., 2017). MEM-fix is generated by the addition of glutaraldehyde to a final working concentration of 0.5% to a standard 4% paraformaldehyde fixative solution. Glutaraldehyde, which is commonly used to fix cells for electron microscopy-based studies, was included because of its ability to effectively preserve subcellular structures. Although glutaraldehyde is not an optimal fixative for immunofluorescence microscopy due to its propensity to auto-fluoresce, we determined that addition of 26.4 mM sodium borohydride to the permeabilization buffer was sufficient to mitigate this undesirable side effect (Tagliaferro et al., 1997; Bacallao et al., 2006). Unfortunately, due to glutaraldehyde limiting antibody penetration into cells, MEM-fix is not conducive to staining of cytoplasmic or nuclear proteins, so should be limited to an examination of integral membrane or juxta-membrane proteins.
We recently used this technique to examine Hedgehog (Hh) morphogen transport through cytonemes of cultured Drosophila cells and mouse fibroblasts (Bodeen et al., 2017). Here, we provide an optimized protocol for imaging of cytonemes in NIH3T3 cells, and provide examples of its adaptability to additional cultured mammalian cell lines (Figure 1). MEM-fix protocol modifications to standard cell fixation methods allow for reproducible detection of cytonemes and immunofluorescence-based staining of trans-membrane and membrane-adjacent proteins in cultured cells. MEM-fix also preserves signal of fluorescently-labeled proteins, so is not solely dependent on immuno-detection based methods.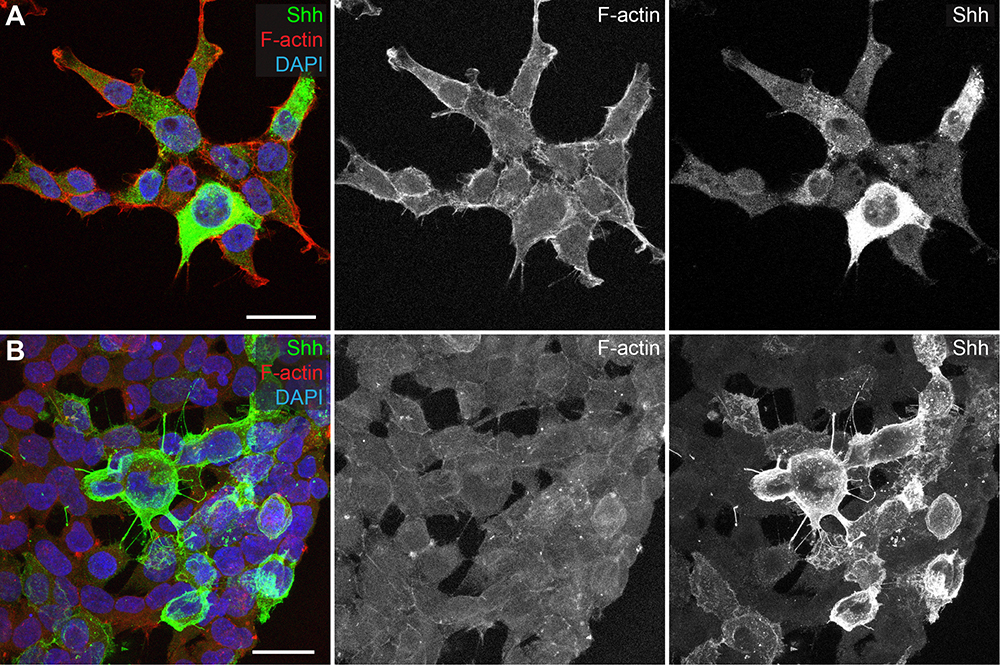
Figure 1. MEM-fix increases preservation of cytonemes in cultured cells compared to paraformaldehyde fixation. A and B. HEK293T cells transfected with Shh. A. Cells fixed with paraformaldehyde show some actin-based protrusions, but do not show Shh (green) positive filaments. B. Cells fixed with MEM-fix contain many cytoneme projections, marked by the presence of F-actin (red) and Shh. Scale bars = 25 µm.
Materials and Reagents
- SHARP® Precision Barrier Tips, For P-1000 and Eppendorf 1,000, 1,250 µl (Denville Scientific, catalog number: P1126 )
- SHARP® Precision Barrier Tips, For P-200, 200 µl (Denville Scientific, catalog number: P1122 )
- SHARP® Precision Barrier Tips, For P-20, 20µl (Denville Scientific, catalog number: P1121 )
- SHARP® Precision Barrier Tips, Extra Long for P-2 and P-10, 10 µl (Denville Scientific, catalog number: P1096-FR )
- Gold SealTM Rite-OnTM Micro Slides (Thermo Fisher Scientific, catalog number: 3050-002 )
- 12 mm Microscope Cover Glass-1.5 (Fisher Scientific, catalog number: 12-545-81 )
- TPP® centrifuge tubes, volume 50 ml, polypropylene (TPP Techno Plastic Products, catalog number: 91050 )
- TPP® centrifuge tubes, volume 15 ml, polypropylene (TPP Techno Plastic Products, catalog number: 91015 )
- StericupTM Sterile Vacuum Filter Units 500 ml (Merck, catalog number: SCGPU05RE )
- 24-well plate (Corning, Falcon®, catalog number: 353226 )
- 6-well plate NunclonTM Delta Surface (Thermo Fisher Scientific, catalog number: 140675 )
- Professional Kimtech ScienceTM KimwipesTM (KCWW, Kimberly-Clark, catalog number: 34155 )
- Premium Microcentrifuge Tubes: 1.5 ml (Fisher Scientific, catalog number: 05-408-129 )
- NIH3T3 (ATCC, catalog number: CRL-1658 )
- Ultrapure water
- DMEM (1x) 4.5 g/L D-glucose, [-] L-Glutamine, [-] HEPES, [-] Sodium Pyruvate (Thermo Fisher Scientific, GibcoTM, catalog number: 11960044 )
- Opti-MEMTM Reduced Serum Medium (Thermo Fisher Scientific, catalog number: 31985070 )
- Pen/Strep, 100x (Merck, catalog number: TMS-AB2-C )
- HyCloneTM Cosmic CalfTM Serum (BCS) (GE Healthcare, catalog number: SH30087.03 )
- MEM NEAA (100x) MEM Non-Essential Amino Acids (Thermo Fisher Scientific, GibcoTM, catalog number: 11140050 )
- L-Glutamine (100x) (100 ml) (Thermo Fisher Scientific, InvitrogenTM, catalog number: 25030081 )
- Sodium Pyruvate (100 mM) 100x (Thermo Fisher Scientific, GibcoTM, catalog number: 11360070 )
- Lipofectamine® 3000 transfection kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: L3000-015 )
- 70% (volume) ethanol diluted in water
- 0.05% Trypsin 0.53 nM EDTA, 1x [-] Sodium Bicarbonate (Corning, catalog number: 25-052-Cl )
- DPBS, 1x (Dulbecco's Phosphate-Buffered Saline) (Corning, CellgroTM, catalog number: 21-031-CM )
- Sodium phosphate dibasic (Sigma-Aldrich, catalog number: S0876 )
- Sodium phosphate monobasic (Sigma-Aldrich, catalog number: S5011 )
- Sodium borohydride (Sigma-Aldrich, catalog number: 213462-25G )
- Formaldehyde, 16%, methanol free, Ultra Pure EM Grade (Polysciences, catalog number: 18814-10 )
- Glutaraldehyde, 8% Aqueous Solution, EM Grade (Electron Microscopy Sciences, catalog number: 16019 )
- Normal Goat Serum (10 ml) (Jackson ImmunoResearch, catalog number: 005-000-121 )
- Triton X-100 (Sigma-Aldrich, catalog number: T9284 )
- Tween 20 (Acros Organics, catalog number: AC233360010 )
- Sonic hedgehog (Shh) antibody (H-160), rabbit polyclonal (Santa Cruz Biotechnology, catalog number: sc-9024 )
- Alexa FluorTM 633 Phalloidin (Thermo Fisher Scientific, catalog number: A22284 )
- Goat anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor 488 (Thermo Fisher Scientific, Invitrogen, catalog number: R37116 )
- DAPI Solution (1 mg/ml) (Thermo Fisher Scientific, catalog number: 62248 )
- Prolong® Diamond Antifade Mountant (Thermo Fisher Scientific, Invitrogen, catalog number: P36961 )
- NIH3T3 media (see Recipes)
- NIH3T3 serum/antibiotic-free media (see Recipes)
- 0.2 M Dibasic Sodium Phosphate solution (see Recipes)
- 0.2 M Monobasic Sodium Phosphate solution (see Recipes)
- Modified electron microscopy fixative (MEM-fix) (see Recipes)
- Permeabilization buffer (see Recipes)
- PBGT (see Recipes)
- Primary antibody solution (see Recipes)
- Secondary antibody solution (see Recipes)
Equipment
- Eppendorf Research Plus single channel pipette, 100-1,000 µl (Eppendorf, catalog number: 3123000063 )
- Eppendorf Research Plus single channel pipette, 20-200 µl (Eppendorf, catalog number: 3123000055 )
- Eppendorf Research Plus single channel pipette, 0.5-10 µl (Eppendorf, catalog number: 3123000020 )
- Eppendorf Research Plus single channel pipette, 0.1-2.5 µl (Eppendorf, catalog number: 3123000012 )
- Allegra X-12R centrifuge (Beckman Coulter, model: Allegra® X-12R , catalog number: 392302)
- Heracell VIOS 160i CO2 incubator (at 37 °C and 5% CO2), (Thermo Fisher Scientific, model: HeracellTM VIOS 160i, catalog number: 51030287 )
- AE50 analytical balance (Mettler-Toledo International, model: AE50 )
- Aspirator
- Precision dual-chamber water bath 288 (Thermo Fisher Scientific, catalog number: 2853 )
- Biological safety cabinet (The Baker Company, catalog number: B40-112 )
- Cell counting chamber (Hausser Scientific, catalog number: 3200 )
- FisherbrandTM Fine Point High Precision Forceps (Fisher Scientific, catalog number: 22-327379 )
- Confocal laser-scanning microscope (Leica Microsystems, model: Leica TCS SP8 )
Software
- Leica Application Suite X (used to generate Tiffs)
Procedure
- Day 1: Seeding cells (see Note 1)
Seed 0.5 x 106 NIH3T3 cells into 1.5 ml media per well of a 6-well cell culture dish. Use one well per experimental condition. If not performing transfection, begin at Step C1.
Note: All steps dealing with cells should be performed within a biological safety cabinet until ready to begin immunocytochemistry.
- Day 2: Transfections (see Note 2)
- Adherent cells should be at a confluence of 50-80% prior to transfection. Remove media and replace with 1 ml pre-warmed (37 °C) serum and antibiotic-free growth media to maximize transfection efficiency.
- Transfect each well with a total of 2 μg plasmid DNA encoding protein of interest according to transfection reagent protocol (we use Lipofectamine 3000 with P3000).
- Aspirate the media of transfected well(s) 6 h post transfection, and replace with 1.5 ml complete media.
- Adherent cells should be at a confluence of 50-80% prior to transfection. Remove media and replace with 1 ml pre-warmed (37 °C) serum and antibiotic-free growth media to maximize transfection efficiency.
- Day 3: Re-seeding cells (see Note 3)
- In the biological safety cabinet clean 12 mm cover slips in 70% ethanol. Place the clean coverslips in individual wells of a 24-well cell culture plate. Apply UV light to coverslips in the open 24-well plate for 15 min to sterilize prior to re-seeding cells (see Note 4).
- Aspirate the media from well(s) of 6-well plate and wash NIH3T3 cells with pre-warmed (37 °C) DPBS. Aspirate DPBS from well(s).
- Add 500 μl of pre-warmed 37 °C trypsin to each well, and place in an incubator for 3-5 min until cells detach. Add 2 ml media to neutralize trypsin, and count cells in counting chamber.
- Transfer cell suspension to a 15 ml centrifuge tube, and centrifuge at ~200 x g for 5 min at room temperature.
- While cells are in the centrifuge, add 500 μl media to each coverslip-containing well of the 24-well plate.
- Aspirate the cell supernatant and resuspend cell pellet in 5 ml pre-warmed media.
- Add an appropriate volume of cell suspension so that each well with a coverslip contains 0.1 x 106 cells.
- Allow 6-8 h for cells to fully attach to the coverslip and allow for cytoneme regrowth, or optimally wait overnight (> 10 h) for full attachment and outgrowth (see Note 5).
- In the biological safety cabinet clean 12 mm cover slips in 70% ethanol. Place the clean coverslips in individual wells of a 24-well cell culture plate. Apply UV light to coverslips in the open 24-well plate for 15 min to sterilize prior to re-seeding cells (see Note 4).
- Day 4: Immunocytochemistry
Crucial: From this point on, do not aspirate. Gently pipette when adding or removing any solution. Attempt to move the plate as little as possible, do not agitate plate during washes. This may fragment or break cytonemes. During incubations and washes, let solutions sit on cells without agitation.- Pipette off media and wash cells quickly 3 times with room temperature DPBS.
- Remove DPBS and add 400 μl of MEM-fix to cells, then incubate for 7 min at room temperature.
- Pipette off the MEM-fix and wash the cells three times in DPBS at room temperature, for 5 min per wash.
- While cells are being washed in DPBS, prepare permeabilization buffer. See Recipes for instructions.
- Remove last DPBS wash, and add 400 μl of permeabilization buffer to each well and incubate at room temperature for 1 h.
- While cells are permeabilizing/blocking, prepare primary antibody dilutions in PBGT. See recipes for instructions.
- Remove permeabilization buffer, and add 300 μl of primary antibody dilution to each well. Incubate the plate overnight at 4 °C.
- Pipette off media and wash cells quickly 3 times with room temperature DPBS.
- Day 5: Secondary antibodies and cover slip mounting
- Prepare 50 ml of PBGT. See Recipes for instructions.
- Remove primary antibody dilution, and wash the cells three times in 400 μl of PBGT at room temperature for 5 min per wash.
- While the cells are being washed, dilute secondary antibodies, phalloidin and DAPI in PBGT. See Recipes for instructions.
- Remove PBGT, and add 300 μl of secondary antibody dilutions to each well. Incubate plate at room temperature for 1 h in the dark or wrapped in aluminum foil (see Note 6).
- Remove secondary antibody dilution, and wash the cells three times in 400 μl of PBGT at room temperature for 5 min per wash. Keep the plate in the dark during washes.
- Remove final PBGT wash, and add 500 μl distilled water to each well to allow easier removal of coverslips and dilute salt content. Use forceps to gently remove coverslip from well. Remove any excess liquid from the edge of the coverslip by dabbing on a Kimwipe. Mount coverslips by gently placing them on a single drop of ProLong diamond applied to the slide. Avoid applying pressure or disturbing coverslip until ProLong diamond has cured.
- Let slide cure overnight in the dark at room temperature, and image on inverted confocal microscope within 2 weeks (see Notes 7 and 8).
- Prepare 50 ml of PBGT. See Recipes for instructions.
Data analysis
It is important to note the differences between cytonemes and other cellular projections when analyzing images, as this protocol also preserves stress fibers. We classify cytonemes in a cultured system as positive for signaling proteins, actin positive, and ~10 μm in length or greater, roughly 200 nm in diameter, and not originating from the basal surface of the cell that is in contact with the coverslip. This final criterion is important for the specific identification of cytonemes because cell adhesion sites including focal adhesions retain stress fibers along the basal surface of the cell. Stress fibers tend to be much more linear than cytonemes, and are typically observed to be attached to the surface of the coverslip (Vallenius, 2013). Cytonemes can form at any position along the cell and have dynamic projections in three dimensions (Figures 2 and 3). It is important to distinguish the differences between these two projections when quantifying cytonemes.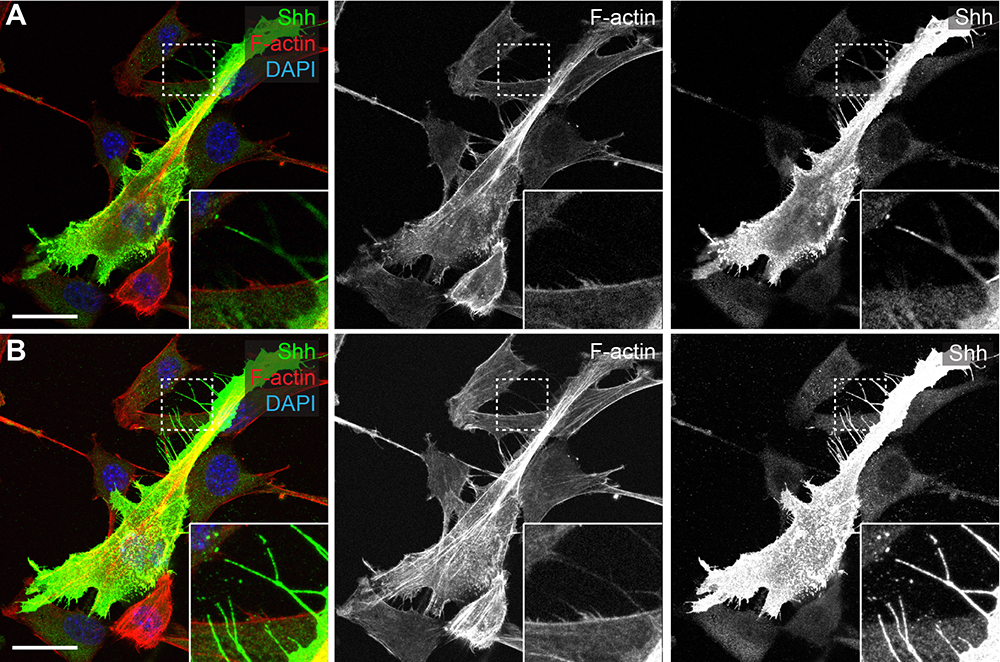
Figure 2. Maximum intensity projection (MIP) allows for accurate representation of cytoneme projections. A and B. NIH3T3 cell transfected with Shh. A. Single section NIH3T3 cell. Cytoneme position drifts in and out of focal plane and appears completely absent of F-actin (red). B. MIP encompassing the range of cytonemes in the same cell. Continuous cytoneme seen for both Shh (green) and F-actin channels. Scale bars = 25 µm.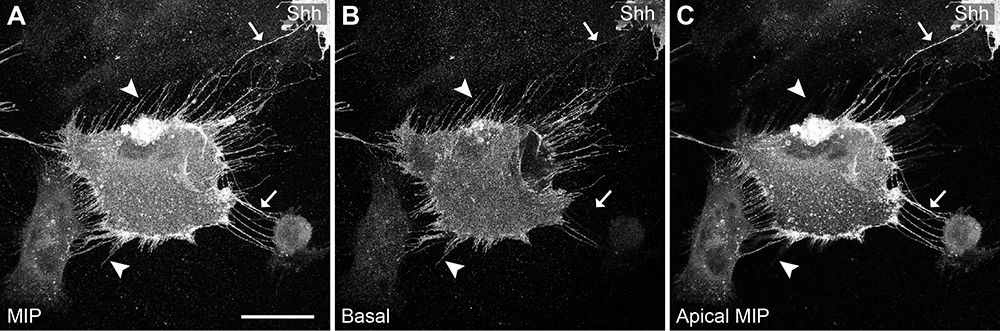
Figure 3. MEM-fix preserves stress fibers and cytonemes. A-C. C57BL/6 MEF cell transfected with Shh. A. MIP of entire cell shows many cellular projections, arrow heads show stress fibers, while arrows highlight cytonemes. B. Basal section of the cell shows stress fibers radiating out of cell body in all directions. Arrow head stress fibers still present, while arrow cytonemes absent. Some cytonemes will also be present in basal section. C. MIP of the cell excluding the basal section attached to the coverslip. Cytonemes are seen orienting to adjacent cells. Arrow head stress fibers absent, arrow cytonemes present. Scale bar = 25 µm.
Notes
- The procedure provided has been tailored for NIH3T3 cells, but can be applied to a wide range of different cell lines in cultured systems (as in Figure 1). We have applied this general protocol format to multiple cell lines with success in preserving and imaging cytonemes, even when altering culture methods for specific cell line requirements. This includes Mouse Embryonic Fibroblasts (MEF), Inner medullary collecting duct (IMCD3), HEK293T, IMR-32, and SK-N-SH cells.
- We transfected our cell with Lipofectamine 3000. To maximize transfection efficiency, plasmid DNA, P3000, and Lipofectamine were incubated in Opti-mem media, then and added to serum and antibiotic free NIH3T3 media. Cells can be transfected with any plasmid DNA and preferred transfection reagent under optimized conditions.
- It should be noted that cell re-seeding is not always necessary after transfection. However, in our experience it allows for much cleaner immunocytochemistry imaging, as it removes any dead cells and excess particles that may have accumulated on the coverslips from first seeding of the cells (Figure 4). This protocol has very mild washing steps and does not remove much debris. Trypsinizing transfected cells and re-seeding becomes more important when imaging cytonemes that end up attaching to the coverslip itself.
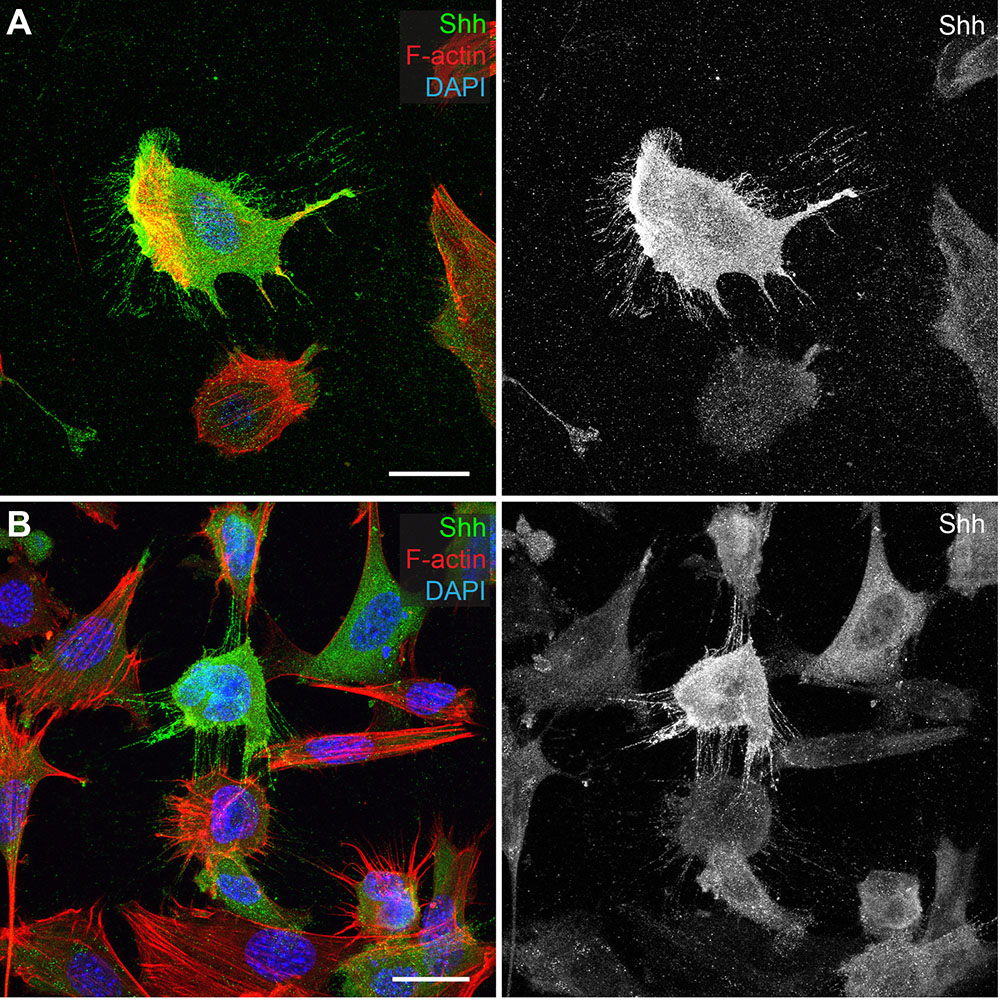
Figure 4. Re-seeding cells after transfections reduces nonspecific particle fluorescence and cellular debris. A and B. C57BL/6 MEF cells transfected with Shh. A. MEF cells seeded onto coverslip, transfected, and then stained with anti-Shh (green), phalloidin to mark F-actin (red) and DAPI (blue). B. MEF cells seeded, transfected, then trypsinized and re-seeded onto coverslip and stained with anti-Shh, phalloidin and DAPI. Scale bars = 25 µm. - Until comfortable with this protocol, it is recommended that you use a minimum of 2 coverslips per experimental condition.
- You may shorten the time period if working with suspension cell lines, or monitoring early cytoneme outgrowth.
- From this point on keep exposure of cells to direct light to a minimum.
- During image acquisition on the confocal microscope, it is recommended to take multiple Z-steps covering most of the cell. Cytonemes frequently project from various sections throughout the Z-axis of the cell, and may fall out of the focal plane of any single section. To ensure correct full cytoneme length, images representing cytonemes length and interactions with other cells should be shown as maximum intensity projections of the entire Z-range of the cytoneme.
- During image acquisition and identification of cytonemes, avoid imaging of projections that initiate along the base of the cell and remain in contact with the coverslip. This will prevent inclusion of stress fibers in images.
Recipes
- NIH3T3 media
DMEM (4.5 g/L glucose) is supplemented with 10% BCS, ~1% MEM NEAA, ~1% L-Glutamine, ~1% Pen/Strep, and ~1% sodium pyruvate
To make ~550 ml NIH3T3 media, add:
500 ml DMEM
50 ml HyCloneTM Cosmic CalfTM Serum (BCS)
5 ml MEM NEAA
5 ml L-Glutamine
5 ml Pen/Strep
5 ml Sodium Pyruvate to 500 ml StericupTM
Vacuum filter contents into receiver bottle and store at 4 °C for up to a month - NIH3T3 serum/antibiotic-free media
DMEM (4.5 g/L glucose) is supplemented with ~1% MEM NEAA, ~1% L-Glutamine, and ~1% sodium pyruvate
To make ~500 ml NIH3T3 media, add:
500 ml DMEM
5 ml MEM NEAA
5 ml L-Glutamine
5 ml Sodium Pyruvate to 500 ml StericupTM
Vacuum filter contents into receiver bottle and store at 4 °C for up to a month - 0.2 M Dibasic Sodium Phosphate solution
To make 1 L of 0.2 M Dibasic Sodium Phosphate solution, dissolve 28.392 g of Dibasic Sodium Phosphate in 1 L of ultrapure water - 0.2 M Monobasic Sodium Phosphate solution
To make 1 L of 0.2 M Monobasic Sodium Phosphate solution, dissolve 23.995 g of Monobasic Sodium Phosphate in 1 L of ultrapure water - Modified electron microscopy fixative (MEM-fix)
MEM-fix contains 4% formaldehyde, 0.5% glutaraldehyde and 0.1 M Phosphate buffer (pH 7.4)
To make 10 ml of MEM-fix:- Mix 8.1 ml 0.2 M Dibasic Sodium Phosphate solution and 1.9 ml 0.2 M Monobasic Sodium Phosphate solution in 15 ml centrifuge tube
- Remove 3.125 ml solution, so remaining volume is 6.875 ml
- Add 2.5 ml 16% formaldehyde and 625 μl 8% glutaraldehyde
- Invert tube several times to mix and use immediately prior to fixation
- Mix 8.1 ml 0.2 M Dibasic Sodium Phosphate solution and 1.9 ml 0.2 M Monobasic Sodium Phosphate solution in 15 ml centrifuge tube
- Permeabilization buffer
Permeabilization buffer consists of 5% Normal Goat or Donkey Serum (adjust as necessary to accommodate primary antibodies to be used), and 0.1% Triton X-100, in a 1 mg/ml Sodium Borohydride solution dissolved in distilled water
To make 10 ml of permeabilization buffer:- Dissolve 10 mg Sodium Borohydride in 10 ml distilled water in a 15 ml centrifuge tube. Sodium Borohydride dissolved in water releases gas, so make sure the lid is ajar to prevent excess gas build up and internal pressure in the centrifuge tube
- Remove 600 μl solution, so 9.4 ml Sodium Borohydride solution remains
- Add 500 μl Normal Goat Serum, and 100 μl 10% Triton X-100 (diluted in DPBS)
- Invert tube several times to mix. Use immediately and do not store with centrifuge tube lid sealed
- Dissolve 10 mg Sodium Borohydride in 10 ml distilled water in a 15 ml centrifuge tube. Sodium Borohydride dissolved in water releases gas, so make sure the lid is ajar to prevent excess gas build up and internal pressure in the centrifuge tube
- PBGT
PBGT consists of 5% Normal Goat Serum, 0.1% Tween-20 in DPBS
To make 50 ml of PBGT, add and mix:
47 ml of DPBS
2.5 ml Normal Goat Serum
0.5 ml 10% Tween-20 (diluted in DPBS) into a 50 ml centrifuge tube
Note: PBGT can be stored at 4 °C for several days until ready to use. - Primary antibody solution
Primary antibody dilution is 1:100 for anti-Shh
To make primary antibody solution for 2 wells of a 24 well plate, add 6 μl of anti-Shh to 600 μl of PBGT in a 1.5 ml microcentrifuge tube
Note: Prepare and use primary antibody solution immediately prior to use. - Secondary antibody solution
Dilute all secondary antibodies at 1:1,000, DAPI solution at 1:2,000, and phalloidin at 1:400, all diluted in PBGT
Note: Prepare and use secondary antibody solution immediately prior to use.
Acknowledgments
This work was supported by R01GM114049 and R35GM122546 to SKO, St. Jude Cancer Center support grant NCI P30 CA021765, and by ALSAC of St. Jude Children’s Research Hospital. This protocol was adapted from the MEM-fix protocol originally described in Bodeen et al. (2017).
Competing interests
The authors declare no conflicts of interest or competing interests.
References
- Bacallao, R., Sohrab, S., and Phillips, C. (2006). Guiding principles of specimen preservation for confocal fluorescence microscopy. In: Handbook of Biological Confocal Microscopy. Springer, 311-325.
- Bodeen, W. J., Marada, S., Truong, A. and Ogden, S. K. (2017). A fixation method to preserve cultured cell cytonemes facilitates mechanistic interrogation of morphogen transport. Development 144(19): 3612-3624.
- Ramírez-Weber, F. A. and Kornberg, T. B. (1999). Cytonemes: cellular processes that project to the principal signaling center in Drosophila imaginal discs. Cell 97(5): 599-607.
- Sanders, T. A., Llagostera, E. and Barna, M. (2013). Specialized filopodia direct long-range transport of SHH during vertebrate tissue patterning. Nature 497(7451): 628-632.
- Stanganello, E., Hagemann, A. I., Mattes, B., Sinner, C., Meyen, D., Weber, S., Schug, A., Raz, E. and Scholpp, S. (2015). Filopodia-based Wnt transport during vertebrate tissue patterning. Nat Commun 6: 5846.
- Tagliaferro, P., Tandler, C. J., Ramos, A. J., Pecci Saavedra, J. and Brusco, A. (1997). Immunofluorescence and glutaraldehyde fixation. A new procedure based on the Schiff-quenching method. J Neurosci Methods 77(2): 191-197.
- Vallenius, T. (2013). Actin stress fibre subtypes in mesenchymal-migrating cells. Open Biol 3(6): 130001.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hall, E. T. and Ogden, S. K. (2018). Preserve Cultured Cell Cytonemes through a Modified Electron Microscopy Fixation. Bio-protocol 8(13): e2898. DOI: 10.21769/BioProtoc.2898.
Category
Developmental Biology > Cell signaling > Fate determination
Developmental Biology > Morphogenesis > Organogenesis
Cell Biology > Cell imaging > Fixed-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.