- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Organotypic Explants of the Embryonic Rodent Hippocampus: An Accessible System for Transgenesis

(*contributed equally to this work) Published: Vol 8, Iss 6, Mar 20, 2018 DOI: 10.21769/BioProtoc.2764 Views: 7811
Reviewed by: Oneil G. BhalalaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2011
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
This protocol describes the technique of ex-vivo electroporation to target embryonic hippocampal progenitors in an organotypic slice preparation. This technique allows gene perturbation for examining developmental processes in the embryonic hippocampus while retaining the environment and connectivity of the cells. Gene perturbation can include Cre-mediated recombination, RNAi-mediated knockdown, gene overexpression, or a combination of any of these. Ex-vivo electroporation can be performed at a wide range of embryonic stages, giving temporal control to the experimenter. Spatial control can be achieved more easily by preparing the brain in a Petri dish to target particular regions of the hippocampus. The electroporated explant cultures provide a highly tractable system for the study of developmental processes that include progenitor proliferation, migration and cell fate acquisition.
Keywords: Mouse hippocampusBackground
The hippocampus presents a challenge in terms of accessibility due to its location in the caudomedial telencephalon. The embryonic hippocampus is even more inaccessible, requiring in utero surgical methods for experimental manipulation. Organotypic slice cultures circumvent this problem and at the same time retain many aspects of hippocampal field cytoarchitectonics, including molecular features and connectivity. While there are protocols that describe postnatal culturing of hippocampal explants from rodent brains (Stoppini et al., 1991; Opitz-Araya and Barria, 2011) these do not include genetic manipulation of the cells. The preparation of organotypic explants from the embryonic mouse hippocampus was first described in Tole et al. (1997). We extended this protocol by introducing ex-vivo electroporation of the embryonic brain prior to preparing the organotypic slices. Electroporation of the intact brain after introducing DNA into the telencephalic ventricle ensures that cells residing in and near the ventricular zone are targeted, and therefore provides an excellent means of inducing transgenesis in hippocampal progenitors. Data using this protocol were published in Subramanian et al. (2011). Here, we present detailed step-wise instructions including experimental ‘dos and don’ts’, and also illustrate key steps using photographs and movies, to aid new researchers in setting up this useful procedure.
Some advantages and applications of this protocol are:
1)Temporal control can be achieved by isolating the embryonic hippocampus at the desired stage to access early, mid, or late-gestation progenitors.
2)Spatial control can be achieved by orienting the electrodes to target the hippocampus or different areas of the cortex.
3)Cre-mediated recombination can be employed by electroporating Cre-GFP into embryos carrying the desired floxed alleles.
4)Overexpression constructs can be electroporated.
5)Embryonic lethal strains can be accessed by performing the procedure in the window of viability, and then further development can proceed in the organotypic explant.
Materials and Reagents
- Plastic Pasteur pipettes:
3 ml (P-3) and 2 ml (P-1) (Taurus Biomedical)
1.5 ml pipettes (RPI, catalog number: 147500 )
Note: Henceforth these will all be referred to as ‘pipettes’. Procuring the correct size of pipette and cutting the shaft to the correct aperture size (see Figure 1) is key to being able to manipulate brains, hemispheres and explants without damaging them.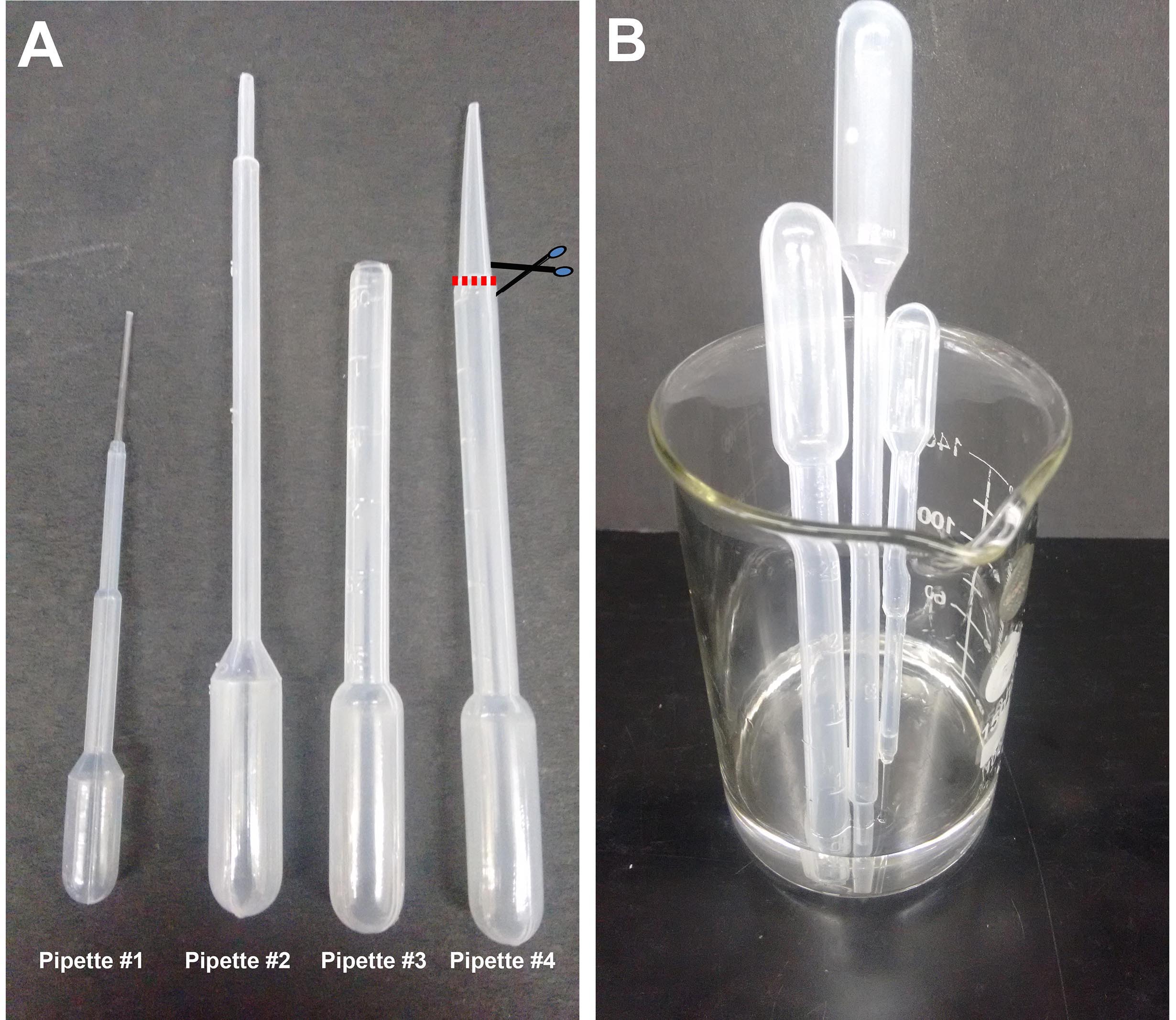
Figure 1. Plastic Pasteur pipettes. A. Different sizes of pipettes used in this procedure: #1 (1.5 ml pipette), #2 (2 ml pipette), #3 (3 ml pipette) prepared by cutting #4 as shown. The cut is made just at the point where the pipette shaft diameter begins to taper, approximately 2 cm from the tip. B. Beaker with pipettes being sterilized in alcohol. - 35 mm sterile Petri dishes (Laxbro, catalog number: PD-35 TC )
- Cell culture 6-well tissue culture plate (Thermo Fisher Scientific, catalog number: 140675 )
- Micropipette tip
- Glass Capillaries: Thin wall borosilicate tubing without filament (Sutter Instruments, catalog number: B100-75-10 )
- 100 mm Petri dishes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 150464 )
- Sterile blade
- Cell Culture Inserts: 0.4 µm (Millicell, Merck, catalog number: PICM03050 )
- Aspirator assembly (Sigma-Aldrich, catalog number: A5177-5EA )
- 50 ml syringe filter unit with 0.22 µm filter (Millex-GP, Merck, catalog number: SLGP033RS ) for sterilizing the culture medium
- 2 Squirt bottles (Tarsons, catalog number: 561100 ) (containing 70% and 100% ethanol)
- pCS2-EGFP plasmid (or any desired plasmid)
- Pregnant Swiss mice (SWR/J) (obtained from Tata Institute of Fundamental Research breeding facility)
- Absolute ethanol (Merck, catalog number: 107017 )
- Leibovitz’s L-15 medium (Thermo Fisher Scientific, GibcoTM, catalog number: 41300039 )
- Fast green dye (Sigma-Aldrich, catalog number: F7252 )
- Penicillin-streptomycin (P/S) (10,000 U/ml) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- Dulbecco’s modified Eagle medium (Thermo Fisher Scientific, GibcoTM,catalog number: 12800017 )
- B27TM supplement (50x), serum-free (Thermo Fisher Scientific, GibcoTM, catalog number: 17504044 )
- Working solution of DMEM medium (see Recipes)
Equipment
- Horizontal flow hood (Kirloskar, Envair Electrodyne, catalog number: KCH-B )
- Glass beaker (Borosil, catalog number: 1000D21 )
- Tissue chopper (McIlwain, model: MTC/2E )
- Stereo microscope (Olympus, catalog number: SZ61 ) placed in a horizontal flow tissue culture hood
- Cell culture incubator (Thermo Fisher Scientific, model: HeracellTM 150 )
- Fine tools for manipulating embryonic brains:
- Coarse forceps (Roboz Surgical Instrument, catalog number: RS-5040 ) for handling the chopper stage disc
- Fine scissors (Fine Science Tools, catalog number: 14060-11 ) and tissue forceps (Roboz Surgical Instrument, catalog number: RS-8166 ) for opening the dam and removing the uterus
- Electrodes (3 mm, BTX, Harvard Apparatus, catalog number: 450487 )
- Electroporator (BTX, Harvard Apparatus, model: ECM 830 )
- Dual-stage glass micropipette puller (NARISHIGE, model: PC-10 )
Procedure
Ethics statement: All procedures followed the Tata Institute of Fundamental Research Institutional Animal Ethics Committee guidelines.
- Set-up (pre-preparation)
Note: All steps after harvesting the embryos are performed in a horizontal flow hood.- Wipe the surface of the tissue culture hood with 70% ethanol.
- Pour 70% ethanol into a 200 ml glass beaker and sterilize pipettes #1, #2, and #3 by aspirating the ethanol all the way into the bulb and then ejecting it out. Allow the residual ethanol to drain.
- Wash the pipettes with L-15 medium to remove traces of ethanol and keep standing in a sterile empty beaker near the dissection microscope for easy access.
- Place dissection instruments in a plastic tray containing 70% ethanol for 10 min, and then dry before using. Place in sterile tray for easy access during the procedure.
- Keep approximately six 35 mm Petri dishes filled with L-15 on ice.
- Add 1.25 ml of DMEM medium in the required number of wells of a 6-well tissue culture plate. Using a sterile forceps place one sterile cell culture insert per well. Transfer the 6-well plate to a 37 °C incubator to prewarm the medium. Typically, 5-8 organotypic explants can be accommodated on one cell culture insert depending on the embryonic stage.
- Wipe the surface of the tissue culture hood with 70% ethanol.
- Preparation for injection
- Prepare 2 µg/µl of pCS2-EGFP plasmid (or any desired plasmid) in nuclease-free water. Lightly touch some fast green dye with the micropipette tip and introduce it into the DNA solution and mix. This enables visualization of the DNA solution during and after injection.
- For preparation of glass capillaries, set the capillary puller to one-step pull at a temperature of 62.5 °C.
- Pull capillaries using the puller and place them in a Petri dish on a capillary holder.
- Fit the capillary to a mouth aspirating rubber pipette and aspirate approximately 5-10 µl of the DNA solution containing fast green dye. This will last for approximately 2-3 ex-vivo injections.
- Prepare 2 µg/µl of pCS2-EGFP plasmid (or any desired plasmid) in nuclease-free water. Lightly touch some fast green dye with the micropipette tip and introduce it into the DNA solution and mix. This enables visualization of the DNA solution during and after injection.
- Harvesting embryos
- Sacrifice a pregnant dam of the desired stage (E13.5-18.5) using the procedures permitted by your Institute. The example shown below is E15.5 from a Swiss mouse. Nfia mutant embryos which were on the C57BL/6J background have also been used successfully in Subramanian et al., 2011.
- Wipe the abdominal skin with ethanol and make a longitudinal incision. Clean the incised area with 70% ethanol (squirt bottle) and using a new pair of sterile forceps and scissors make an incision in the abdominal wall.
- Lift out each arm of the uterus one by one taking care that it does not come in contact with the skin or hair of the dam. Cut the uterus at the lowest point and transfer to a dish containing sterile PBS. All procedures from this point onwards will take place inside the tissue culture hood.
- Using a dissection microscope, remove the embryos from the uterus carefully and place them in a sterile 100 mm Petri dish containing sterile 1x PBS.
- Decapitate the embryos and transfer the heads to a fresh sterile Petri dish containing L-15.
- Dissect out the brains and transfer using pipette #3 to a 35 mm dish containing L-15 kept on ice. After removing the brain, sever the hindbrain and midbrain, retaining only the forebrain including the olfactory bulbs. The presence of the olfactory bulb helps to position the telencephalic hemisphere in the correct orientation on the tissue chopper plate. The meninges should be retained and removed only after electroporation.
- Sacrifice a pregnant dam of the desired stage (E13.5-18.5) using the procedures permitted by your Institute. The example shown below is E15.5 from a Swiss mouse. Nfia mutant embryos which were on the C57BL/6J background have also been used successfully in Subramanian et al., 2011.
- Electroporation and organotypic slice preparation
A summary cartoon illustrating the overall procedure is shown in Figure 2, together with a sample organotypic explant at the end of the culture period.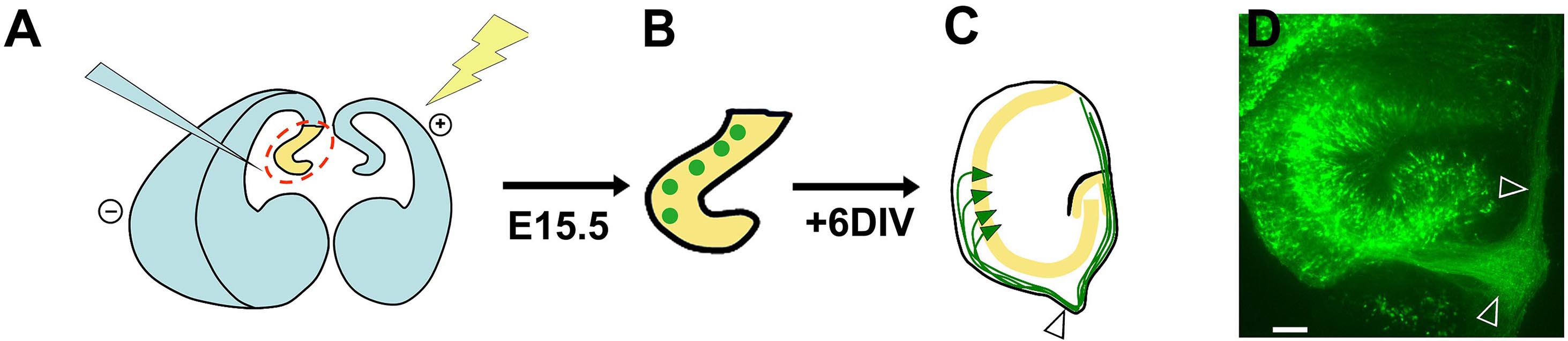
Figure 2. Summary schematic of ex-vivo electroporation and organotypic slice preparation. A. Injection of DNA and electroporation of the intact brain; B. Sections containing electroporated progenitors (green circles); C. After 6 d in vitro, the explants display GFP-expressing pyramidal neurons (green triangles) residing within the pyramidal cell layer (yellow). These neurons initially extend axons (green) which grow towards the developing fimbria (arrowhead) and then encircle the periphery of the explant, presumably because they cannot exit the fimbria onto the filter. D. An image of an organotypic explant displaying an axon bundle growing into the fimbria and then around the explant (arrowheads). Scale bar = 100 µm. This figure and legend are modified from Figure 5, Subramanian et al., PNAS, 2011.- Set the tissue chopper to 250 µm and insert a sterile blade onto the blade. Tighten the blade. Flush 100% ethanol over the blade using a squirt bottle and let the set-up dry.
- Wipe the paddle electrodes with ethanol and connect them to the electroporator.
- Set the parameters to the following: Voltage 50 V; Time 50 msec; Number of pulses 5; Pulse interval 999 msec.
- Inject the ventricle of the hemisphere to be electroporated with the desired DNA solution (mixed with Fast green), as shown in Figure 3A. The ventricles are hollow, and can be identified by their darker appearance, and the decrease in resistance when the micropipette punctures the neuroepithelium and enters the ventricle. It is recommended to perform unilateral injection and electroporation, however, the dye-DNA solution may sometimes diffuse into the connected contralateral ventricle (see Figure 3B). This is normal and does not affect the experimental setup. Fill the lateral ventricle with the solution until the dye reveals that the ventricular cavity is filled (see Figure 3B). Approximately 5-10 µl of solution can be injected into the ventricles depending on the age of the embryo used. Since the DNA is injected into the ventricles, the cells which have the best access to the DNA are the ventricular zone cells, which consist of dividing neuronal progenitors. This is the main population that is electroporated (Saito, 2006).
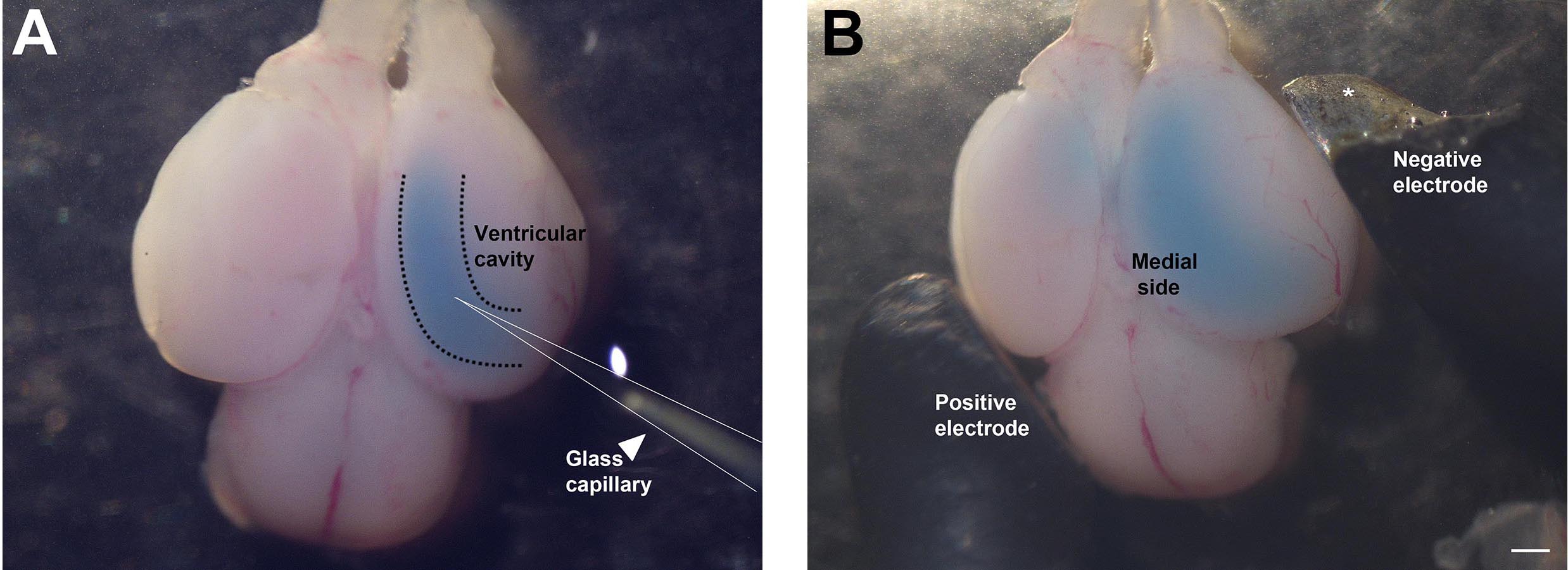
Figure 3. Injection of DNA solution into telencephalic hemisphere and electroporation. A. Glass capillary (arrowhead) injecting DNA into the ventricular cavity. B. Paddle electrodes (positive and negative) positioned at an angle suitable for electroporating the hippocampus. (*In Figure 3B, the insulation of the negative electrode is chipped at the tip, so a portion of the paddle appears silver. This does not affect the electroporation.) Scale bar = 500 µm. - Perform the electroporation immediately so that the DNA solution does not diffuse out of the ventricle. Place the electrodes at the angle shown in Figure 3B, such that the positive electrode is positioned on the contralateral side and the negative electrode is positioned on the ipsilateral side of the hemisphere to be electroporated. Since DNA is negatively charged, it will move towards the positive electrode and therefore display maximal electroporation on the medial side of the hemisphere.
- Initiate the pulse sequence on the electroporator. Successful passage of current is indicated by bubbles under the paddles around the region of electroporation. More bubbles are usually seen under the (-) paddle electrode.
- Transfer the electroporated brain to a 35 mm Petri dish containing ice-cold L-15 medium. Keep on ice for 5 min. In the meantime, electroporate other brains as per the experimental requirement. Transfer each brain to the ice-cold L-15.
- Once all the required brains are electroporated, separate the electroporated hemisphere and remove the meninges carefully.
- Place the hemisphere on the tissue chopper such that the medial face is in contact with the white disc on the stage, and the lateral (neocortex) faces up. Using pipette #1 gently remove excess medium. Excess medium will cause the hemisphere to wobble during the next step (chopping). While siphoning off the excess medium, orient the hemisphere’s rostro-caudal axis orthogonal to the blade so that coronal sections will be obtained (the olfactory bulb needs to point either right or left, not towards or away from you as in Figure 4).

Figure 4. Placement of the dissected electroporated hemisphere onto the stage of the tissue chopper. Note that the spring clips that hold the chopping stage disc in place should be oriented in the R-L direction to prevent them from interfering with the blade. The telencephalic hemisphere orientation should be aligned taking this into account (Olfactory bulb pointing R or L). - Initiate chopping (Videos 1 and 2). Immediately, remove the disk holding the chopped tissue using two pairs of strong forceps. Flush the tissue into a clean L-15 containing dish containing cold L-15 (Video 3).
 Video 1. Chopping of electroporated hemisphere (viewed from the front) Video 2. Chopping of electroporated hemisphere (viewed from the side)
Video 1. Chopping of electroporated hemisphere (viewed from the front) Video 2. Chopping of electroporated hemisphere (viewed from the side)
Video 3. Flushing chopped tissue into a Petri dish - The hemisphere will look like a loaf of sliced bread (Figure 5A). Turn the hemisphere over, so that the medial side faces up. Prod gently to release finger-like protrusions from the medial side that are individual slices of the hippocampus. Use the fine forceps to sever the hippocampus from the neocortex (dotted lines, Figure 5A). Collect hippocampal slices in a fresh 35 mm Petri dish containing ice cold L-15 medium.
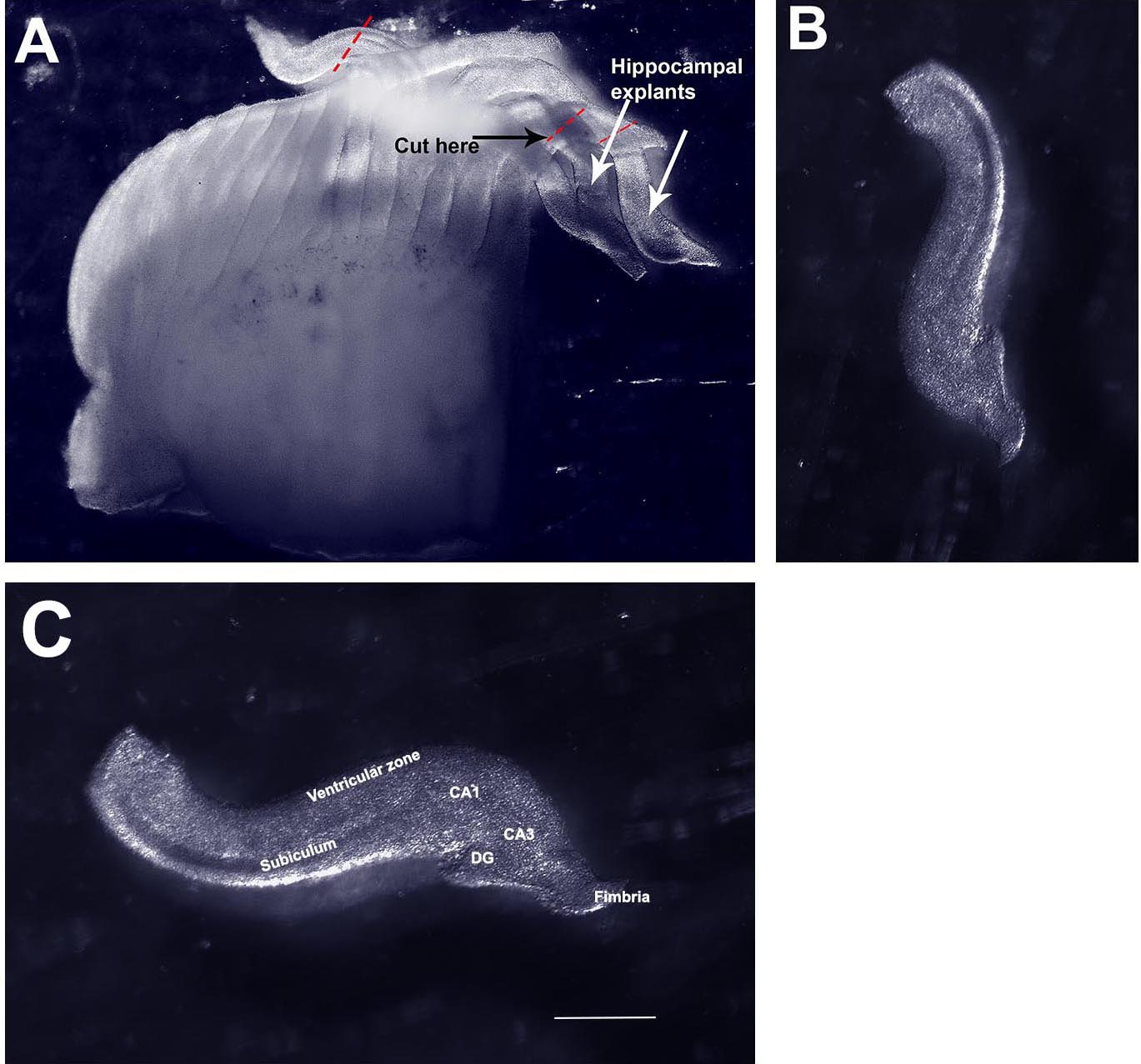
Figure 5. Isolating organotypic hippocampal slices from the chopped hemisphere. A. Image of a freshly chopped hemisphere displaying ‘sliced bread’ appearance. B. A single hippocampal explant isolated from the chopped hemisphere. C. The same explant in (B) re-oriented and labeled. CA1, CA3, hippocampal fields. DG, dentate gyrus. Scale bar = 500 µm. - Remove the 6-well dish containing the cell culture inserts from the incubator and place under the dissection microscope (Figure 6).

Figure 6. 6-well tissue culture dish with cell culture insert - Using pipette #2 take up 5-6 hippocampal explants with some L-15 medium. Place the explants onto the filter disc one by one by gently applying pressure on the bulb of the pipette so that only one explant is dispensed, each in its own droplet of L-15 medium (Figure 7A). Use pipette #1 to gently aspirate out excess medium and use the fluid flow generated by the aspiration to gently position the explants so they do not touch each other and also do not stick to the walls of the cell culture insert (Figure 7B). There should be enough medium to keep explants from drying out but excess medium should be avoided to prevent explants from folding or floating away (Figures 7C and 7D). The explants will not dry; they can access the DMEM medium in the well through the filter.
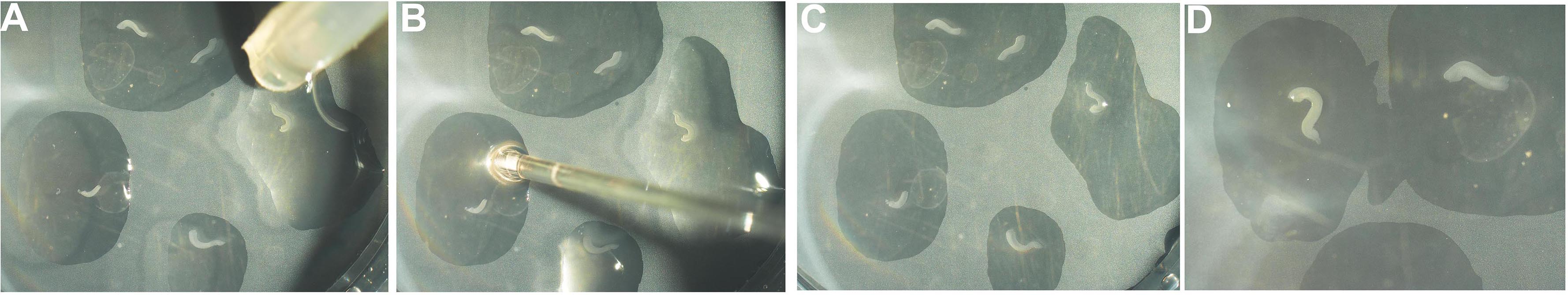
Figure 7. Placing hippocampal explants onto culture insert. A. The explants are placed in individual droplets of medium onto the cell culture insert using pipette #2. B. The excess fluid is removed using pipette #1. During this step, two explants that may have been deposited in the same droplet can be separated (C). D. Images of the explants when the procedure is complete. - Incubate the hippocampal explants at 37 °C, 5% CO2. Change the medium every alternate day.
- Within several hours, the electroporation can be visualized if a fluorescent reporter construct was used. Within the next 3-5 days, the labeled progenitors differentiate into pyramidal neurons, extending apical processes towards the center of the curled explant, and axons course around the explant to the region of the fimbria (Figure 8).
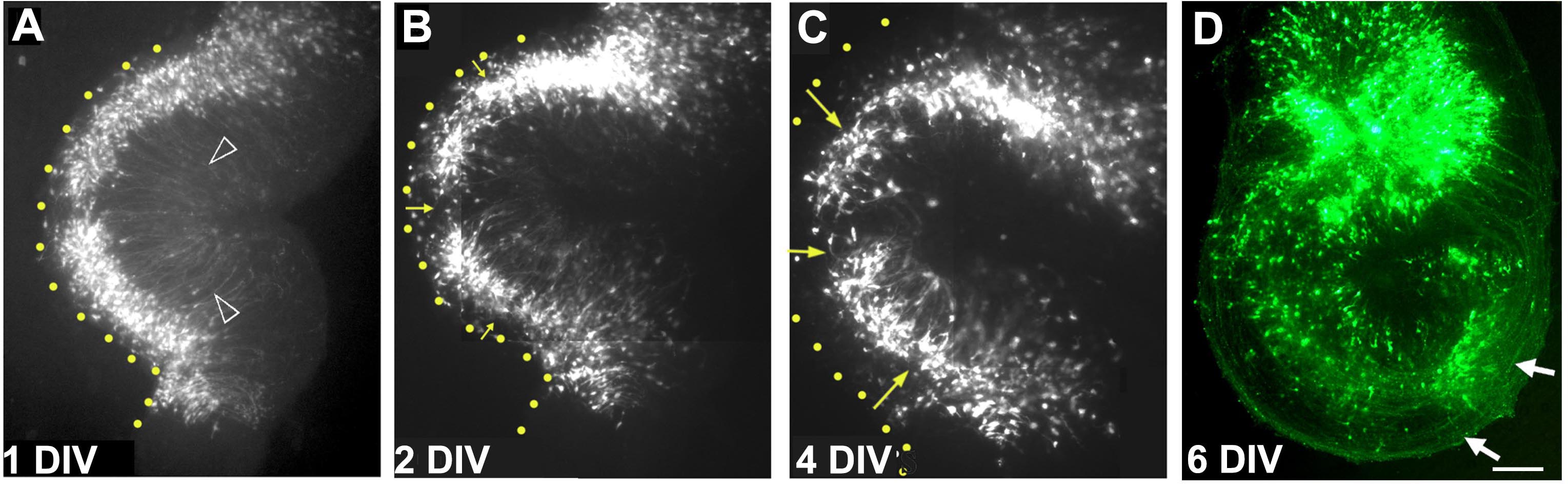
Figure 8. A single organotypic hippocampal explant imaged on consecutive days in vitro. A. After 1 day in vitro (DIV), electroporated cells are seen close to the ventricular zone (yellow dotted line). Apical processes are seen to extend toward the center of the explant arrowheads). (B) and (C) with additional days in vitro, cells begin to migrate away from the ventricular zone (yellow arrows) as they do in vivo, and form the pyramidal cell layer. D. By 6 days in vitro, the electroporated cells extend axons (white arrows) that can be seen encircling the explant. Images in (A-D) are composites consisting of multiple frames stitched together. Scale bar = 100 µm.
- Set the tissue chopper to 250 µm and insert a sterile blade onto the blade. Tighten the blade. Flush 100% ethanol over the blade using a squirt bottle and let the set-up dry.
Data analysis
This ex-vivo electroporated hippocampal organotypic preparation can be used to assess whether neurons or glia are generated from progenitors. The presence of GFP-expressing axons in the fimbria is indicative of neurogenesis. See Subramanian et al. (2011), Figure 5, for images of explants electroporated with neurogenic or gliogenic constructs. These explants can also be immunostained for cell type-specific markers and analyzed for the distribution of excitatory/inhibitory neurons or glia as required.
Notes
- Reproducibility and variability: The extent of electroporation is variable, but can be made more reproducible with practice. The key issues to pay attention to are the angle of the electrodes and how carefully one holds the brain i.e., the contact of the tissue with the paddle electrodes. The brain needs to be completely submerged in the medium during the electroporation.
- Technical tips and cautionary points:
- The speed of producing the explants is critical to explant health. Keeping the tissue on ice-cold L-15 when it is not under the microscope or chopping stage is critical.
- Being able to identify ‘good’ hippocampal finger-like protrusions in a chopped brain also comes with practice. Typically, easy-to-recognize hippocampal morphology is seen at the caudal end of the chopped brain, whereas explants prepared from more medial locations are recognizable only by a tiny fimbria. A good rule of thumb is that if it doesn’t have a fimbria, it’s probably not a hippocampal explant, but a curved piece of neocortex masquerading as a hippocampus.
- The cell culture inserts fit best in a 6-well plate. Do not try to use individual 35 mm dishes–they don’t have the requisite height. The dish will not close properly, so the medium will dry or become contaminated.
- The speed of producing the explants is critical to explant health. Keeping the tissue on ice-cold L-15 when it is not under the microscope or chopping stage is critical.
Recipes
- Working solution of DMEM medium (50 ml)
B27: 500 µl
Penicillin-streptomycin: 500 µl of a 100x stock
DMEM: 49 ml
Filter sterilize using a 0.22 µm filter and store at 4 °C. Use this solution up to 1 week as suggested in the document manual for B27
Acknowledgments
We thank Leora D’Souza for assistance with the videography, Dr. S. Suryavanshi and the TIFR animal breeding facility for excellent support. This protocol was developed in the Tole lab using support from a Wellcome Trust Senior Fellowship (056684/Z/99/Z), a Swarnajayanti Fellowship (4/3/2005-SF), grants from the Department of Biotechnology, Government of India and intramural funds from TIFR-DAE to ST; a Kanwal Rekhi Career Development Award from the TIFR Endowment Fund to LS. ST is a recipient of the Shanti Swarup Bhatnagar award (Council of Scientific and Industrial Research, Government of India) and the Infosys Prize for Life Sciences (Infosys Science Foundation).
This protocol was used in Subramanian et al. (2011) Proc Natl Acad Sci U S A 108(27): E265-274. (Current address: Eli & Edythe Broad Center of Regeneration Medicine & Stem Cell Research, University of California San Francisco, CA 94143, USA.)
The authors have no conflicts of interest or competing interests.
References
- Opitz-Araya, X. and Barria, A. (2011). Organotypic hippocampal slice cultures. J Vis Exp (48).
- Saito, T. (2006). In vivo electroporation in the embryonic mouse central nervous system. Nature Protoc 1(3): 1552–1558.
- Stoppini, L., Buchs, P. A. and Muller, D. (1991). A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37(2): 173-182.
- Subramanian, L., Sarkar, A., Shetty, A. S., Muralidharan, B., Padmanabhan, H., Piper, M., Monuki, E. S., Bach, I., Gronostajski, R. M., Richards, L. J. and Tole, S. (2011). Transcription factor Lhx2 is necessary and sufficient to suppress astrogliogenesis and promote neurogenesis in the developing hippocampus. Proc Natl Acad Sci U S A 108(27): E265-274.
- Tole, S., Christian, C. and Grove, E. A. (1997). Early specification and autonomous development of cortical fields in the mouse hippocampus. Development 124(24): 4959-4970.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Iyer, A., Subramanian, L. and Tole, S. (2018). Organotypic Explants of the Embryonic Rodent Hippocampus: An Accessible System for Transgenesis. Bio-protocol 8(6): e2764. DOI: 10.21769/BioProtoc.2764.
Category
Neuroscience > Development > Explant culture
Cell Biology > Tissue analysis > Electroporation
Cell Biology > Tissue analysis > Tissue isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.