- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Construction and Cloning of Minigenes for in vivo Analysis of Potential Splice Mutations
(*contributed equally to this work) Published: Vol 8, Iss 5, Mar 5, 2018 DOI: 10.21769/BioProtoc.2760 Views: 12318
Reviewed by: Nicoletta CordaniKate HannanPamela Maher
Original research article
The authors used this protocol in:

Jan 2016
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Disease-associated mutations influencing mRNA splicing are referred to as splice mutations. The majority of splice mutations are found on exon-intron boundaries defining canonical donor and acceptor splice sites. However, mutations in the coding region (exonic mutations) can also affect mRNA splicing. Exact knowledge of the disease mechanism of splice mutations is essential for developing optimal treatment strategies. Given the large number of disease-associated mutations thus far identified, there is an unmet need for methods to systematically analyze the effects of pathogenic mutations on mRNA splicing. As splicing can vary between cell types, splice mutations need to be tested under native conditions if possible. A commonly used tool for the analysis of mRNA splicing is the construction of minigenes carrying exonic and intronic sequences. Here, we describe a protocol for the design and cloning of minigenes into recombinant adeno-associated virus (rAAV) vectors for gene delivery and investigation of mRNA splicing in a native context. This protocol was developed for minigene-based analysis of mRNA splicing in retinal cells, however, in principle it is applicable to any cell type, which can be transduced with rAAV vectors.
Keywords: MinigeneBackground
A substantial portion of disease-associated mutations (at least 15%) are predicted to result in aberrant mRNA splicing (Cartegni et al., 2002; Singh and Cooper, 2012; Sterne-Weiler and Sanford, 2014). The ‘classical’ splice mutations are those affecting the canonical sequences defining the 5’- and 3’-splice sites (donor and acceptor splice sites, respectively). However, splice mutations can also occur in other non-coding and coding regions (Wang and Cooper, 2007; Scotti and Swanson, 2016). There is growing evidence that the frequency of splice mutations in coding regions (exonic mutations) has been underestimated (Julien et al., 2016; Soukarieh et al., 2016). Exonic splice mutations (i.e., point mutations, insertions or deletions) can induce exon skipping, intron retention or lead to the generation of novel donor or acceptor splice sites. Depending on the gene and exon composition, these mechanisms can have different impacts on the protein level ranging from haploinsufficiency to gain of function. Nevertheless, the exact knowledge of molecular mechanisms underpinning the disease-causing mutations is essential for developing optimal treatments.
mRNA splicing occurs in a highly cell type-specific manner, highlighting the need to analyze the impact of potential splice mutations in the tissue which is primarily affected by the mutation (Wang et al., 2008). Consequently, in the optimal case, mRNA splicing should be analyzed on the native gene and in the native tissue. This option, however, is rather demanding for several reasons:
1) It might require the elaborate generation of genetically modified human cell lines. This impedes a more systematic analysis of splice mutations for a single gene.
2) Many native cell types are highly specialized and their cultured counterparts (if available at all) do not reflect every morphological and molecular hallmark of the native cells including the composition and activity of the splicing machinery.
3) The generation of humanized animal models expressing the respective splice mutation in a given tissue is not only technically challenging, but also time-consuming and costly. Therefore, this approach also appears rather unsuitable for systematic testing of splice mutations for a given gene.
4) Often, native genes are too large to be cloned into classical expression vectors.
One alternative to circumvent a number of these obstacles is to use human minigenes designed for expression in appropriate animal models (e.g., mouse). We have evaluated this approach in recent studies addressing the effects of disease-associated mutations in different genes, e.g., PRPH2, on mRNA splicing (Becirovic et al., 2016b; Nguyen et al., 2016; Khan et al., 2017; Petersen-Jones et al., 2017). For stable and specific ectopic expression of the minigenes, we took advantage of rAAV vectors. These vectors are capable of transducing a variety of different cell types in vivo (Zincarelli et al., 2008; Lisowski et al., 2014). Furthermore, the design, cloning, production and purification of rAAV vectors can be completed in a few weeks and does not require elaborate technical equipment (Becirovic et al., 2016a).
Most native genes including PRPH2 exceed the limited packaging capacity of AAVs (approx. 4.7 kb) (Wu et al., 2010). Therefore, we designed PRPH2 minigenes lacking large intronic parts, which usually do not contain information required for correct mRNA splicing. For genes which do not contain large exon numbers or sizes, shortening of the intronic sections also allows for introducing the entire protein coding region into the rAAV vector-based minigenes.
This strategy (cf. Figure 1) was developed and evaluated to analyze the impact of known disease-linked mutations on mRNA splicing and protein expression in photoreceptor-specific genes, but should in principle also be transferable to other cell types.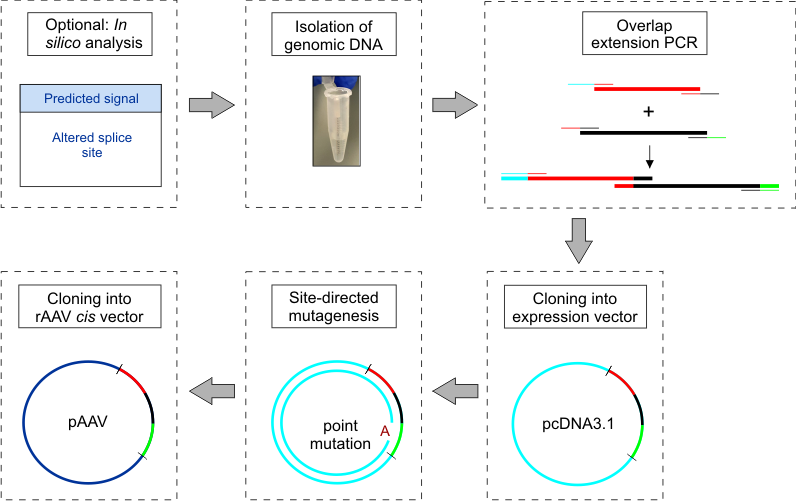
Figure 1. Workflow schematic for construction and cloning of minigenes
Materials and Reagents
- Isolation of human genomic DNA for cloning of minigenes
- 1.5 ml Eppendorf tubes (SARSTEDT, catalog number: 72.690.001 )
- Scalpel (Swann Morton, catalog number: 0510 )
- Gentra Puregene Buccal Cell Kit (QIAGEN, catalog number: 158845 )
- 2-Propanol (100%, ACS, ISO, Reag. Ph. Eur. grade)
- Ethanol (70%, ACS, ISO, Reag. Ph. Eur. grade)
- 1.5 ml Eppendorf tubes (SARSTEDT, catalog number: 72.690.001 )
- Overlap extension PCR for construction and cloning of minigenes
- PCR tubes (BRAND, catalog numbers: 781320 and 781334 )
- Expression vector of choice (e.g., pcDNA3.1 vector (+), Thermo Fisher Scientific, InvitrogenTM, catalog number: V79020 )
- Double distilled H2O (ddH2O)
- Herculase II Fusion DNA Polymerase (Agilent Technologies, catalog number: 600675 )
- dNTPs, 10 mM (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0192 )
- DNA Loading Dye (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0611 )
- GeneRuler 1 kb Plus DNA Ladder (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: SM1331 )
- EDTA (VWR, catalog number: 20302.293 )
- Tris-(hydroxymethyl) aminomethane (VWR, catalog number: 103156X )
- Boric acid (VWR, catalog number: 20185.360 )
- QIAquick Gel Extraction Kit (QIAGEN, catalog number: 28704 )
- 1x TBE buffer (see Recipes)
- PCR tubes (BRAND, catalog numbers: 781320 and 781334 )
- Site-directed mutagenesis to introduce point mutations of interest into minigene
- PCR tubes (BRAND, catalog numbers: 781320 and 781334 )
- QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, catalog number: 210518 )
- Double distilled H2O (ddH2O)
- Peptone (Applichem, catalog number: 403898.1210 )
- Yeast extract (Applichem, catalog number: A1552 )
- NaCl (VWR, catalog number: 27810.364 )
- D-(+)-Glucose (Sigma-Aldrich, catalog number: 49159 )
- Agar (Sigma-Aldrich, catalog number: A5054 )
- Appropriate antibiotic for the plasmid vector (e.g., Ampicillin, Carl Roth, catalog number: K029.2 )
- LB agar plates containing the appropriate antibiotic for the plasmid vector (see Recipes)
- LB medium (see Recipes)
- PCR tubes (BRAND, catalog numbers: 781320 and 781334 )
- Subcloning of minigenes into rAAV vector
- Appropriate rAAV cis vector plasmid (e.g., pAAV-MCS2, Addgene, catalog number: 46954 )
- Appropriate rAAV cis vector plasmid (e.g., pAAV-MCS2, Addgene, catalog number: 46954 )
Equipment
- Pipettes (e.g., Eppendorf)
- Vortexer (Heidolph Instruments, model: Reax top )
- Incubator (Thermo Fisher Scientific, Thermo ScientificTM, model: Heraeus B12 Function Line, catalog number: 50042307 )
- Microcentrifuge (Eppendorf, model: Centrifuge MiniSpin® , catalog number: 5452000018)
- Two water baths (Haake, catalog number: 003-2859 )
- Thermocycler (Thermo Fisher Scientific, model: ProFlexTM, catalog number: 4483636 )
- NanoDropTM (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: ND-2000C )
Software
- Human splicing finder software (e.g., http://www.umd.be/HSF3/)
- Tm Calculator Software (e.g., NEB Tm Calculator)
Procedure
- In silico splice analysis
In many cases, initial in silico prediction of mRNA splicing of the single mutations might be helpful. To predict the potential effects on splicing of your mutation of interest use a human splicing finder software (e.g., http://www.umd.be/HSF3/).- Go to http://www.umd.be/HSF3/ and select an analysis type. We recommend using the option ‘Analyze a sequence’.
- Choose a sequence. We recommend using the option ‘Pasting your own sequence’. Paste the wildtype version of your sequence of interest into the provided box and click on ‘Proceed to analysis’.
Note: Your sequence should contain at least 40 bp flanking the respective point mutation on both sides.
- Click on ‘Perform a quick mutation’, type in the position of your mutation of interest relative to the sequence you have entered before and select the new nucleotide. Then click on ‘Proceed to quick mutation analysis’.
- Human splice finder webserver output:
- Click on ‘Raw Data Tables’ to analyze the effect of the entered point mutation in detail.
- You are provided with five different data tables called ‘Potential splice sites’, ‘Potential Branch Points’, ‘Enhancer motifs’, ‘Silencer motifs’ and ‘Other splicing motifs’. Each of them shows the percental variation induced by the point mutation. Newly formed splice sites or motifs are indicated in green, a change in strength of an existing site or motif is indicated in light yellow.
- Click on ‘Raw Data Tables’ to analyze the effect of the entered point mutation in detail.
- Repeat the analysis for all point mutations of interest and select the ones predicted to affect splicing for further cloning.
- Go to http://www.umd.be/HSF3/ and select an analysis type. We recommend using the option ‘Analyze a sequence’.
- Isolation of human genomic DNA for cloning of minigenes
- Preheat one water bath to 65 °C and the other to 37 °C.
- To collect buccal cells, scrape the inside of the mouth 10 times with a Buccal Collection Brush (provided in the kit, Materials and Reagents A3).
Note: For best results, wait at least 1 h after eating or drinking to collect buccal cells.
- Dispense 300 µl Cell Lysis Solution (provided in the kit, Materials and Reagents A3) into a 1.5 ml Eppendorf tube. Remove the Collection brush from its handle using a sterile scalpel, and place the detached head in the tube.
- Incubate at 65 °C for 60 min to complete cell lysis.
- Remove the collection brush head from the Cell Lysis Solution, scraping it on the side of the tube to recover as much liquid as possible.
- Add 1.5 µl RNase A Solution (provided in the kit), and mix by inverting the tube 25 times.
- Incubate for 30 min at 37 °C. Incubate for 1 min on ice to cool the sample.
- Add 100 µl Protein Precipitation Solution (provided in the kit, Materials and Reagents A3), and vortex vigorously for 20 sec at high speed.
- Incubate for 5 min on ice.
- Centrifuge for 3 min at 13,000-16,000 x g to pellet the precipitated proteins.
- Pipet 300 µl isopropanol and 0.5 µl Glycogen Solution (provided in the kit, Materials and Reagents A3) into a clean 1.5 ml Eppendorf tube, and add the supernatant from the previous step by pouring or pipetting carefully. Make sure not to transfer the protein pellet.
- Mix by inverting the tube gently 50 times or by rotating it for 2.5 min at 20 rpm.
- Centrifuge at RT for 5 min at 13,000-16,000 x g.
- Carefully discard the supernatant, and drain the tube by inverting it on a clean piece of absorbent paper, taking care that the pellet remains in the tube.
- Add 300 µl of 70% ethanol and invert the tube several times to wash the DNA pellet.
- Centrifuge at RT for 1 min at 13,000-16,000 x g.
- Carefully discard the supernatant. Drain the tube on a clean piece of absorbent paper, taking care that the pellet remains in the tube. Allow to air dry for 5 min.
- Add 100 µl DNA Hydration Solution (provided in the kit, Materials and Reagents A3) and vortex for 5 sec at medium speed to mix.
- Incubate at 65 °C for 1 h to dissolve the DNA.
- Incubate at room temperature overnight with gentle shaking. Ensure tube cap is tightly closed to avoid leakage. Samples can then be centrifuged briefly and transferred to a storage tube.
- Preheat one water bath to 65 °C and the other to 37 °C.
- Overlap extension PCR for construction and cloning of minigenes
C1. Methodological description and design of overlap primers
To enable minigene-based analysis of mutations on mRNA splicing, large introns can be shortened. In our recent studies, we validated that approx. 200 bp of the intronic regions flanking the exons are sufficient for correct mRNA splicing (Becirovic et al., 2016b; Nguyen et al., 2016).
To generate the appropriate minigenes via standard cloning procedures, an overlap extension PCR can be used. For this purpose, as a first step the respective exons including ≥ 200 bp of the flanking introns must be amplified via standard PCR. The primers used for this PCRs must encompass defined overhangs of approx. 20 bp. These overhangs are complementary to the corresponding PCR fragment to be connected in the second overlap extension PCR. These create 40 bp overlap regions used for the amplification of the two fragments. We applied this strategy for creating minigenes consisting of three native exons including two shortened flanking introns (Figure 2). However, this method is also suitable for the generation of minigenes with a random number of exons and shortened introns.- Design PCR primers to amplify one exon plus ≥ 200 bp of the flanking intronic regions. Check the melting temperature (Tm) using a Tm Calculator Software (e.g., NEB Tm Calculator).
- To create the overlap regions, the primer must contain the approx. 20 bp long region complementary to the adjoining PCR amplicon (see Figure 2).
Example: When designing primers to amplify the second exon plus flanking intronic regions (amplicon #2), add the sequence of the final 20 bp in the exon 1 containing amplicon (amplicon #1) to the 5’ end of your forward primer. Next, add the reverse sequence of the first 20 bp of the exon 3 containing amplicon (amplicon #3) to the 5’ end of your reverse primer. These overlapping sequences will be needed for subsequent overlap extension PCRs.
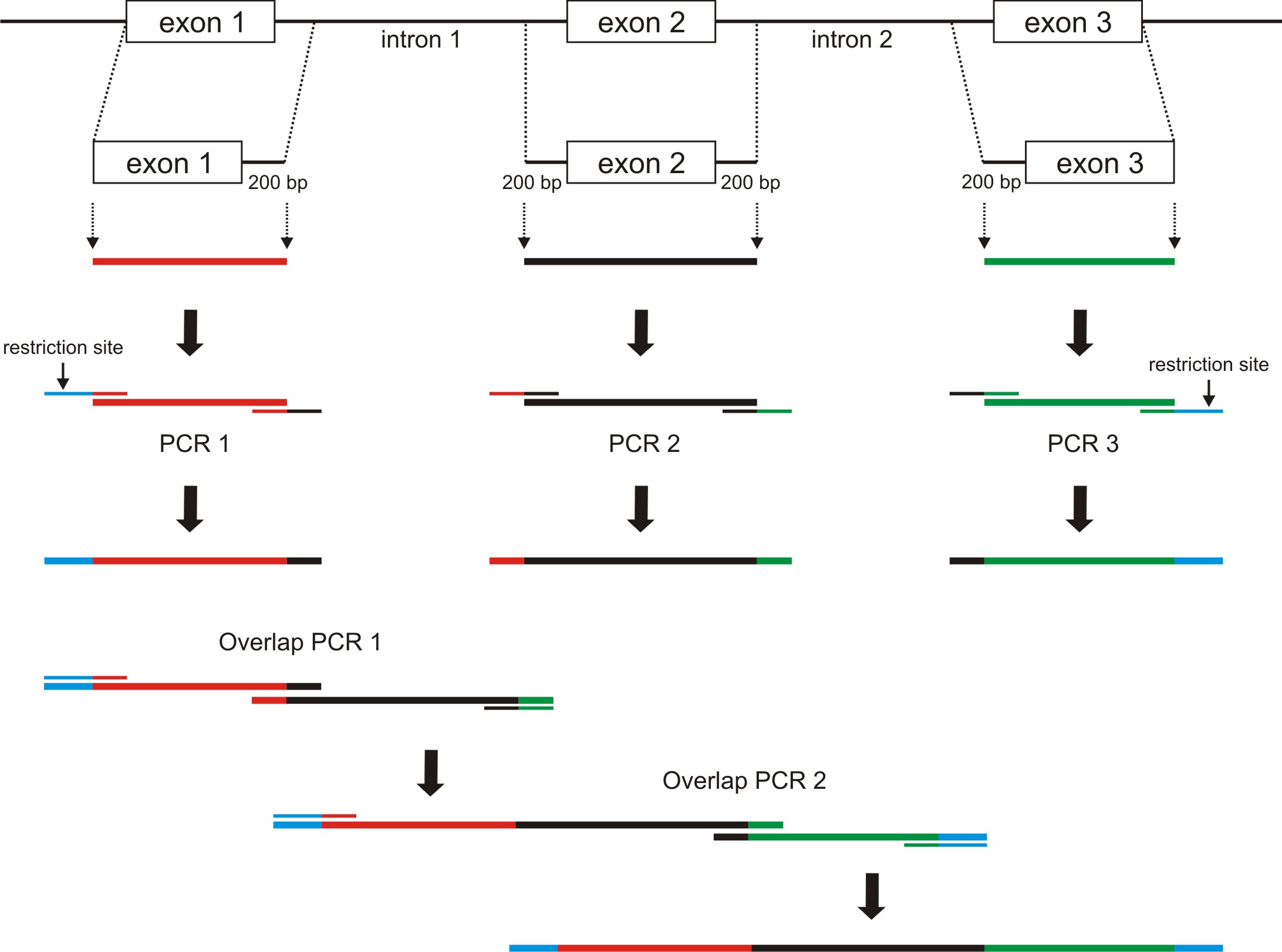
Figure 2. Schematic of overlap extension PCR for creating minigenes consisting of three exons and two flanking introns that were shortened. The single amplicons are depicted in different colors. Restriction sites are shown in blue. Primers are depicted bicolored according to their templates and matching overlaps. Two PCR amplicons from step 1 serve as templates for the overlap PCR 1 and can be combined together in a random manner (e.g., amplicons of PCR 1 + PCR 2). The final overlap PCR 2 can be performed with the remaining PCR fragment (in this example, the amplicon of PCR 3). For all overlap PCRs, only the 5’ primer of the first and the 3’ primer of the second PCR amplicon should be used.
- For the amplification of the first minigene exon: Add an appropriate restriction site to the 5’ end of your forward primer according to the restriction site of your expression vector (e.g., HindIII in pcDNA3.1). Consider adding an additional sequence (up to 4 bp) to the 5’ end of the restriction site for more efficient cleavage by the restriction enzymes.
For amplification of the last minigene exon: Add an appropriate restriction site to the 5’ end of your reverse primer according to the restriction site of your expression vector (e.g., NotI). Consider adding additional up to 4 bp to the 5’ end of the restriction site for more efficient cleavage by the restriction enzymes.
- Order PCR primers (approx. 0.05 µmol, at least HPSF purified) from commercial providers (e.g., Eurofins Genomics).
C2. PCR amplification of full-length exons and shortened introns- To amplify the single exon and flanking intronic regions, prepare the sample reaction as follows:
*Note: Provided in the kit (Materials and Reagents B4).X µl
Genomic DNA (100-300 ng)
5 µl
5x Herculase II Fusion buffer*
1 µl
dNTPs (10 mM)
1 µl
Forward primer (10 µM)
1 µl
Reverse primer (10 µM)
0.25 µl
Herculase II Fusion DNA Polymerase* Add to 25 µl
Double distilled H2O
- Mix all reagents in a PCR tube by pipetting up and down.
- For the PCR use the following cycling parameters:
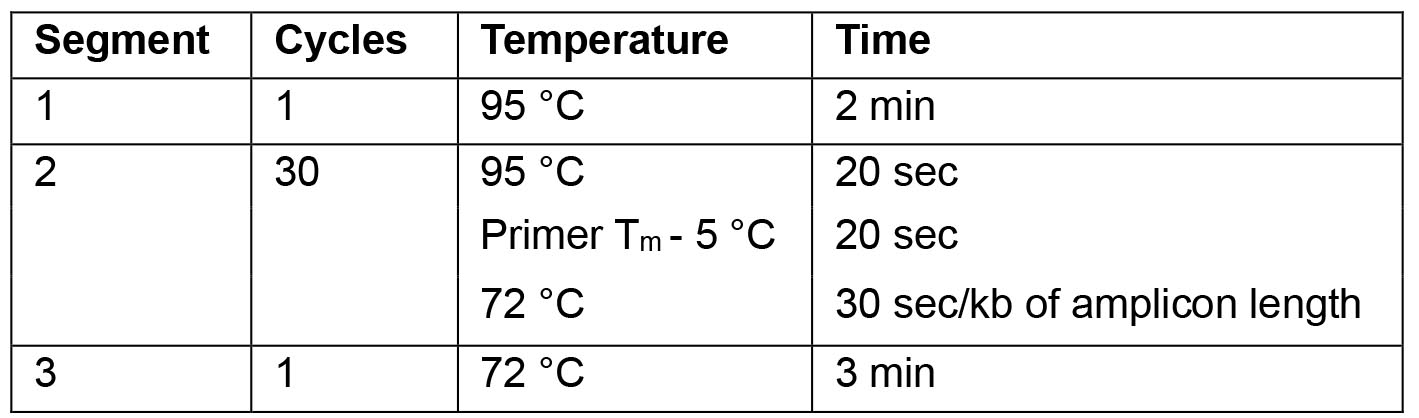
Note: For optimal results the annealing temperature can be adapted as required.
- After Amplification, add 5 µl 6x DNA Loading Dye to your PCR reaction, load it on a 0.7% agarose gel together with a DNA standard of your choice (e.g., GeneRuler 1 kb Plus DNA Ladder) and run it in 1x TBE buffer. Check for correct amplification.
- If amplification was successful, i.e., if you see a band of correct size, excise it using a scalpel and put the gel slice into an Eppendorf tube. Purify DNA using the gel extraction kit according to the manufacturer’s instructions.
- Measure the DNA concentration using NanoDrop.
- Repeat Step C2 for all exons.
C3. Overlap extension PCR- To join the first two amplicons containing exon 1 and 2, prepare the sample reaction as follows:
X µl
Amplicon #1 (100-150 ng)
X µl
Amplicon #2 (use molar ratio 1:1)
5 µl
5x Herculase II Fusion buffer
1 µl
dNTPs (10 mM)
1 µl
Forward primer of Amplicon #1 (10 µM)
1 µl
Reverse primer of Amplicon #2 (10 µM)
0.25 µl
Herculase II Fusion DNA Polymerase
Add to 25 µl
Double distilled H2O
- Mix all reagents in a PCR tube by pipetting up and down.
- For the PCR use the following cycling parameters:
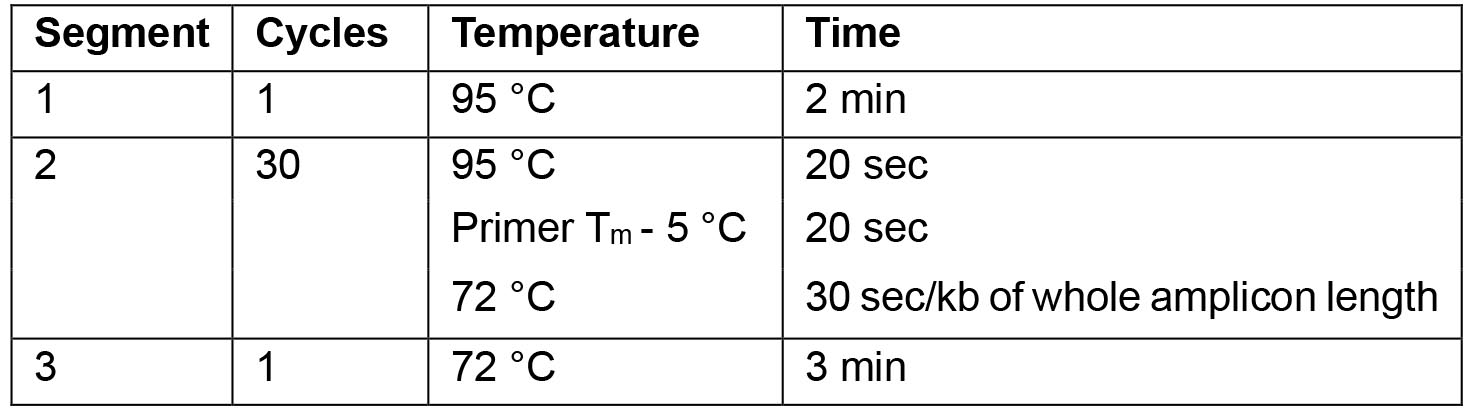
Note: For optimal results the annealing temperature can be adapted as necessary.
- Load all of your PCR reaction on an agarose gel and check for correct amplification.
- If amplification was successful, i.e., if you see a band of correct size, excise the band and purify DNA using the gel extraction kit according to the manufacturer’s instructions.
- Measure the DNA concentration using NanoDrop.
- Repeat Overlap extension PCR to connect the new amplicon with the subsequent amplicon. For this, use the purified DNA of both amplicons as template; and the forward primer of amplicon #1 and the reverse primer of amplicon #3 as Overlap extension PCR primers.
- Depending on the number of amplicons to be joined together, Steps 1-7 can be repeated.
- Use the chosen restriction sites to clone your minigene into the respective expression vector using standard cloning techniques.
- Check for the correct sequence of the minigene by sequencing (e.g., Eurofins Genomics).
Notes:- Consider using an expression vector carrying a reporter gene (e.g., GFP) at the 5’ or 3’ end of your minigene. In case minigenes will also be used for protein analysis, this will enable a convenient visualization of protein expression. Check for the correct reading frame and the presence of a start codon and stop codon at appropriate positions by sequencing prior to proceeding with the next steps.
- This vector is required for initial in vitro testing of the minigene expression and validation of mRNA splicing. Do not use rAAV vectors for cloning at this stage, as the site-directed mutagenesis (see the following section) will be prevented by the secondary structure of the rAAV vector ITRs.
- Consider using an expression vector carrying a reporter gene (e.g., GFP) at the 5’ or 3’ end of your minigene. In case minigenes will also be used for protein analysis, this will enable a convenient visualization of protein expression. Check for the correct reading frame and the presence of a start codon and stop codon at appropriate positions by sequencing prior to proceeding with the next steps.
- Design PCR primers to amplify one exon plus ≥ 200 bp of the flanking intronic regions. Check the melting temperature (Tm) using a Tm Calculator Software (e.g., NEB Tm Calculator).
- Site-directed mutagenesis to introduce point mutations of interest into minigenes
D1. Design of mutagenic oligonucleotide primers- Use the QuikChange primer design program and select the QuikChange kit that you require (e.g., QuikChange Lightning).
- Paste the DNA sequence of your gene of interest in the box, then click on ‘Upload now’.
- To generate a point mutation select the nucleotide to be changed by ticking the corresponding box. Subsequently, choose the substituting nucleotide in the box of ‘Site 1’.
- Click on ‘Design Primers’.
- Obtain the sequence of the forward and reverse primer and additional information (e.g., length, Tm).
- Order primers from commercial providers (e.g., Eurofins Genomics).
Notes:- The desired mutation should lie within the middle of both primers with flanking 10 to 15 bp of correct sequence on each side.
- Their length should be between 25 and 45 bp.
- Tm ≥ 78 °C (verify by Tm calculator, e.g., NEB)
- The desired mutation should lie within the middle of both primers with flanking 10 to 15 bp of correct sequence on each side.
D2. Mutant strand synthesis reaction- Prepare the sample reaction as follows:
Notes:5 µl
10x QuikChange Lightning Buffer*
X µl
10-100 ng of dsDNA template
X µl
125 ng of forward primer
X µl
125 ng of reverse primer
1 µl
dNTP mix*
1.5 µl
QuickSolution reagent*
1 µl
QuickChange Lightning Enzyme*
Add to 50 µl
Double distilled H2O
- *Provided in the kit (Materials and Reagents C2).
- We recommend 10-30 ng of dsDNA template.
- *Provided in the kit (Materials and Reagents C2).
- Mix all reagents in a PCR tube by pipetting up and down.
- For the PCR use following cycling parameters:
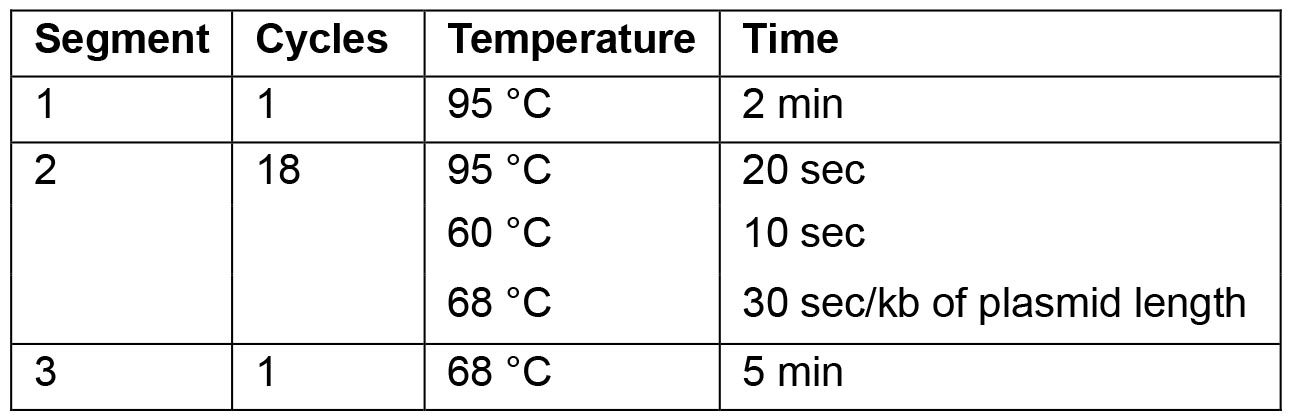
Note: For optimal results the annealing temperature can be adapted as necessary.
- Check for successful amplification by loading 3-5 µl of the PCR product on an agarose gel.
D3. DpnI digestion of template- Add 2 µl of the provided DpnI restriction enzyme to the amplification reaction and mix the solution by pipetting up and down.
- Incubate the mixture at 37 °C for 1-2 h to digest the non-mutated, methylated template DNA.
D4. Transformation- Thaw an aliquot (45 µl) of the provided XL10-Gold ultracompetent cells (or an alternative strain of competent cells) on ice.
- Transfer 2-4 µl of the DpnI-treated DNA to the cells and swirl gently to mix the components.
- Incubate the transformation reaction on ice for 30 min.
- Heat-pulse the tube at 42 °C for 30 sec.
- Incubate the tube on ice for 2 min.
- Plate the whole transformation reaction on an agar plate containing the appropriate antibiotic for the plasmid vector.
- Incubate agar plate at 37 °C for >16 h.
D5. Picking colonies, plasmid isolation, digestion- Pick 24 well-separated colonies from the agar plate and inoculate culture tubes containing 5 ml LB medium and the appropriate concentration of antibiotic. Shake at 250 rpm and 37 °C for > 7 h.
- Isolate plasmid DNA using alkaline lysis or a plasmid miniprep kit according to the manufacturer’s instructions.
- Digest the isolated DNA with appropriate restriction enzymes to verify the presence of your minigene in the plasmid.
Note: Check whether site-directed mutagenesis creates new or deletes existing restriction sites. If this is the case, the digestion with appropriate restriction enzymes directly allows determining the success of the QuikChange PCR.
- Sequence to confirm the correct plasmids prior to use.
- Use the QuikChange primer design program and select the QuikChange kit that you require (e.g., QuikChange Lightning).
- Subcloning of minigenes into rAAV cis vector plasmid
Use appropriate restriction sites to clone your mutant minigenes into a rAAV vector using standard cloning techniques.
Note: Check for ITR integrity prior to rAAV production. The AAV ITRs contain SmaI (or XmaI) sites, which can be used as diagnostics for ITR integrity.
Notes
- Not all genes are suitable for an in vivo splice analysis using rAAV-mediated gene expression. If the size of the expression cassette containing a promoter, the minigene and a poly-A signal exceeds 4.7 kb, lentiviral-derived vector systems might be used.
- Instead of PCR-based cloning, the minigenes can also be ordered via gene synthesis from commercial providers.
- In the case of using minigenes for mRNA splicing only, the exon to be analyzed should be flanked by at least two additional native exons including the respective full-length or shortened intronic sequences. In contrast to the middle exon, the remaining two exons do not necessarily need to be flanked with the corresponding intronic sequence on both sides.
- As deep intronic regions might also contain splice regulatory elements, shortening of the native introns should be done only if necessary.
- In case no PCR products are obtained for some overlap PCRs, consider varying the length of the overlap region. Alternatively, change the melting temperature and/or the order of the template PCR amplicons used for initial overlap PCRs in Step C1.2 (cf. Figure 2).
Recipes
- 1x TBE buffer

- LB-medium/LB agar plates
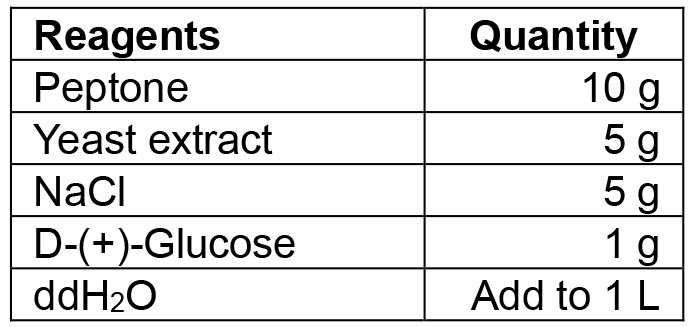
Mix all reagents. If making agar plates, also add 15 g agar to the solution. Autoclave the solution at 121 °C for 10 min. If making selective media or agar plates, add the desired antibiotic in the appropriate concentration once the medium has cooled down to 50 °C. For agar plates, pour the medium into Petri dishes
Acknowledgments
We thank Berit Noack for the valuable technical support. This work was funded by the Deutsche Forschungsgemeinschaft, grant number BE 4830/1-1. The authors declare that they have no competing interests.
References
- Becirovic, E., Bohm, S., Nguyen, O. N., Riedmayr, L. M., Hammelmann, V., Schon, C., Butz, E. S., Wahl-Schott, C., Biel, M. and Michalakis, S. (2016a). AAV vectors for FRET-based analysis of protein-protein interactions in photoreceptor outer segments. Front Neurosci 10: 356.
- Becirovic, E., Bohm, S., Nguyen, O. N., Riedmayr, L. M., Koch, M. A., Schulze, E., Kohl, S., Borsch, O., Santos-Ferreira, T., Ader, M., Michalakis, S. and Biel, M. (2016b). In vivo analysis of disease-associated point mutations unveils profound differences in mRNA splicing of peripherin-2 in rod and cone photoreceptors. PLoS Genet 12(1): e1005811.
- Cartegni, L., Chew, S. L. and Krainer, A. R. (2002). Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 3(4): 285-298.
- Julien, P., Minana, B., Baeza-Centurion, P., Valcarcel, J. and Lehner, B. (2016). The complete local genotype-phenotype landscape for the alternative splicing of a human exon. Nat Commun 7: 11558.
- Khan, A. O., Becirovic, E., Betz, C., Neuhaus, C., Altmuller, J., Maria Riedmayr, L., Motameny, S., Nurnberg, G., Nurnberg, P. and Bolz, H. J. (2017). A deep intronic CLRN1 (USH3A) founder mutation generates an aberrant exon and underlies severe Usher syndrome on the Arabian Peninsula. Sci Rep 7(1): 1411.
- Lisowski, L., Dane, A. P., Chu, K., Zhang, Y., Cunningham, S. C., Wilson, E. M., Nygaard, S., Grompe, M., Alexander, I. E. and Kay, M. A. (2014). Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature 506(7488): 382-386.
- Nguyen, O. N., Bohm, S., Giessl, A., Butz, E. S., Wolfrum, U., Brandstatter, J. H., Wahl-Schott, C., Biel, M. and Becirovic, E. (2016). Peripherin-2 differentially interacts with cone opsins in outer segments of cone photoreceptors. Hum Mol Genet 25(12): 2367-2377.
- Petersen-Jones, S. M., Occelli, L. M., Winkler, P. A., Lee, W., Sparrow, J. R., Tsukikawa, M., Boye, S. L., Chiodo, V., Capasso, J. E., Becirovic, E., Schön, C., Seeliger, M. W., Levin, A. V., Michalakis, S., Hauswirth, W. W. and Tsang, S. H. (2017). Patients and animal models of CNGβ1-deficient retinitis pigmentosa support gene augmentation approach. JCI 128(1).
- Scotti, M. M. and Swanson, M. S. (2016). RNA mis-splicing in disease. Nat Rev Genet 17(1): 19-32.
- Singh, R. K. and Cooper, T. A. (2012). Pre-mRNA splicing in disease and therapeutics. Trends Mol Med 18(8): 472-482.
- Soukarieh, O., Gaildrat, P., Hamieh, M., Drouet, A., Baert-Desurmont, S., Frebourg, T., Tosi, M. and Martins, A. (2016). Exonic splicing mutations are more prevalent than currently estimated and can be predicted by using in silico tools. PLoS Genet 12(1): e1005756.
- Sterne-Weiler, T. and Sanford, J. R. (2014). Exon identity crisis: disease-causing mutations that disrupt the splicing code. Genome Biol 15(1): 201.
- Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., Kingsmore, S. F., Schroth, G. P. and Burge, C. B. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456(7221): 470-476.
- Wang, G. S. and Cooper, T. A. (2007). Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet 8(10): 749-761.
- Wu, Z., Yang, H. and Colosi, P. (2010). Effect of genome size on AAV vector packaging. Mol Ther 18(1): 80-86.
- Zincarelli, C., Soltys, S., Rengo, G. and Rabinowitz, J. E. (2008). Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther 16(6): 1073-1080.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Riedmayr, L. M., Böhm, S., Michalakis, S. and Becirovic, E. (2018). Construction and Cloning of Minigenes for in vivo Analysis of Potential Splice Mutations. Bio-protocol 8(5): e2760. DOI: 10.21769/BioProtoc.2760.
Category
Neuroscience > Sensory and motor systems > Retina
Developmental Biology > Morphogenesis > Cell structure
Molecular Biology > RNA > RNA splicing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.