- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Terminal Deoxynucleotidyl Transferase Mediated Production of Labeled Probes for Single-molecule FISH or RNA Capture



Published: Vol 8, Iss 5, Mar 5, 2018 DOI: 10.21769/BioProtoc.2750 Views: 13960
Reviewed by: Gal HaimovichJoshua S TitlowKarthik Krishnamurthy
Original research article
The authors used this protocol in:

Oct 2017
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Arrays of short, singly-labeled ssDNA oligonucleotides enable in situ hybridization with single molecule sensitivity and efficient transcript specific RNA capture. Here, we describe a simple, enzymatic protocol that can be carried out using basic laboratory equipment to convert arrays of PCR oligos into smFISH and RAP probesets in a quantitative, cost-efficient and flexible way.
Keywords: Terminal deoxynucleotidyl transferaseBackground
The use of multiple, singly-labeled, short oligonucleotides of synthetic origin has vastly improved the detection of specific transcripts with high specificity and single molecule sensitivity (Femino et al., 1998; Raj et al., 2008). Such probe molecules have improved penetration and require milder hybridization conditions than the classically used long nucleic acid probes, resulting in better preservation of the structure of the specimen (e.g., Little et al., 2015, Gaspar et al., 2017a). Since in this design multiple oligonucleotides–typically 24-96–target different portions of the same transcript, there occurs an accumulation of signal on the specific target molecules over the aspecific background, as opposed to the equal signal produced by long multiply labeled probes (Raj et al., 2008). Moreover, as the labeling of the individual short probes is quantitative–as opposed to the stochastic labelling of the long probes–the signal intensity directly and linearly correlates with the transcript copy number at a given spot, allowing precise recording/counting of the target RNA molecules (Raj et al., 2008, Little et al., 2015). Until now, the production of smFISH probe arrays has depended on chemical synthesis and labeling that rendered such single molecule FISH application inflexible and costly. Here, we describe an effective and cost-efficient enzymatic three-pot probe production (3P3) assay that makes use of terminal deoxynucleotidyl transferase (TdT) and custom labeled terminator nucleotides to convert any custom-assembled array of cheap PCR oligos into smFISH probes bearing fluorescent or non-fluorescent labels of the experimenter’s choice (Gaspar et al., 2017b). These enzymatically produced 3P3 probes are chemically nearly identical to smFISH probes from other sources. Thus the same protocols–optimized for a given specimen under study–can be used to perform single molecule FISH (reviewed in Gaspar and Ephrussi, 2015) and RNA capture analyses(see e.g., Gaspar et al., 2017a and Khong et al., 2017).
Materials and Reagents
- 1.5 ml Eppendorf tube (e.g., Sigma-Aldrich, catalog number: Z336769 )
- 0.2 ml thin-walled PCR tube (e.g., Corning, catalog number: 6571 )
- 2 cm thick adhesive tape (Tesa)
- Glass slides (e.g., VWR, catalog number: 631-0411 ) and coverslips (e.g., 22 x 22 x 0.17 mm, Marienfeld-Superior, catalog number: 0107052 ) for sample preparation
- 15 ml tubes (e.g., Corning, Falcon®, catalog number: 352097 )
- 0.22 μm filter (e.g., Corning, catalog number: 431227 )
- 3 cm wide foldback paperclips (e.g., Staples, catalog number: WW-9130156 )
- Amine reactive labels (tested and working):
- BDP-FL-NHS (Lumiprobe, catalog number: 11420 )
- Atto-tec Atto488-NHS (Atto-tec, catalog number: AD 488-31 ), Atto532-NHS (Atto-tec, catalog number: AD 532-31 ), Atto565-NHS (Atto-tec, catalog number: AD 565-31 ) and Atto633-NHS (Atto-tec, catalog number: AD 633-31 )
- AlexaFluor488-NHS (Thermo Fisher Scientific, InvitrogenTM, catalog number: A20000 )
- Abberior STAR 470SXP-NHS (Abberior, catalog number: 1-0101-008-3 ) and Abberior STAR RED-NHS (Abberior, catalog number: 1-0101-011-3 )
- biotin-NHS (Sigma-Aldrich, catalog number: H1759 )
- BDP-FL-NHS (Lumiprobe, catalog number: 11420 )
- Anhydrous DMSO (e.g., Sigma-Aldrich, catalog number: 276855 )
- Optional: silica gel (e.g., Merck, catalog number: 1.01969.1000 ) (see Note 1)
- Amino-11-ddUTP (Lumiprobe, catalog number: 15040 ) or 5-propargylamino-ddUTP (Jena Biosciences, catalog number: NU-1619 )
- 1 M NaHCO3, pH 8.4 (e.g., Sigma-Aldrich, catalog number: S5761 )
- A custom designed target specific array of non-overlapping ssDNA oligonucleotides (desalting purification is sufficient, see Software section for the design)
- 20 U/μl Terminal deoxynucleotidyl transferase (TdT) with 5x TdT buffer (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EP0161 )
- 1-3 M Na-acetate, pH 5.5 (e.g., Sigma-Aldrich, catalog number: S2889 )
- 5 mg/ml linear acrylamide (e.g., Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9520 )
- Ethanol (e.g., Merck, EMD Millipore, catalog number: 1.00983 )
- 100% ethanol, -20 °C
- 80% ethanol, 4 °C
- 70% ethanol, RT
- 100% ethanol, -20 °C
- Nuclease free ddH2O (e.g., New England Biolabs, catalog number: B1500S )
- 40% Acrylamide/Bis solution, 29:1 (e.g., Bio-Rad Laboratories, catalog number: 1610146 )
- Urea (e.g., Sigma-Aldrich, catalog number: U5378 )
- N,N,N’,N’-Tetramethylethylenediamine (TEMED) (e.g., Sigma-Aldrich, catalog number: T9281 )
- 10% (w/v) ammonium persulfate (APS) (e.g., Sigma-Aldrich, catalog number: A3678 )
- 6x gel loading dye (e.g., New England Biolabs, catalog number: B7021S )
- SYBR-GOLD (e.g., Thermo Fisher Scientific, InvitrogenTM, catalog number: S11494 )
- Optional: colorimetric Biotin Assay Kit (e.g., Sigma-Aldrich, catalog number: MAK171 ) (see Note 8)
- 20 mg/ml Proteinase-K (e.g., Thermo Fisher Scientific, InvitrogenTM, catalog number: AM2546 )
- Mounting medium
- VectaShield (Vector Laboratories, catalog number: H-1000 )
- 80% TDE (see Recipes)
- VectaShield (Vector Laboratories, catalog number: H-1000 )
- Pierce® Avidin agarose (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 20219 )
- Dynabeads® MyOneTM C1 (Thermo Fisher Scientific, InvitrogenTM, catalog number: 65001 )
- Quick-RNATM MicroPrep Kit (Zymo Research, catalog number: R1050 )
- Tris-HCl pH 7.0 (e.g., Sigma-Aldrich, Roche Diagnostics, catalog number: 10812846001 )
- Colorimetric Biotin Assay Kit (Sigma-Aldrich, catalog number: MAK171 )
- Tris base (e.g., Sigma-Aldrich, catalog number: T1503 )
- Ethylenediaminetetraacetic acid (EDTA) (e.g., Sigma-Aldrich, catalog number: E5391 )
- Sodium chloride (NaCl) (e.g., Merck, catalog number: 106404 )
- Potassium chloride (KCl) (e.g., Merck, catalog number: 104936 )
- Potassium dihydrogen phosphate dihydrate (KH2PO4·2H2O) (e.g., Merck, catalog number: 104873 )
- Sodium phosphate dibasic (Na2HPO4) (e.g., Merck, catalog number: 106342 )
- EM-grade paraformaldehyde (e.g., Electron Microscopy Sciences, catalog number: 15710 )
- Triton X-100 (e.g., Sigma-Aldrich, catalog number: X100 )
- Boric acid (e.g., Merck, catalog number: 100165 )
- Ethylene carbonate (e.g., Sigma-Aldrich, catalog number: E26258 )
- 50 mg/ml heparin (e.g., Sigma-Aldrich, catalog number: H3393 )
- 10 mg/ml salmon sperm DNA (e.g., Sigma-Aldrich, catalog number: D7656 )
- 2,2’-Thiodiethanol (Sigma-Aldrich, catalog number: 166782 )
- 20% (v/v) SDS (e.g., Sigma-Aldrich, catalog number: 05030 )
- PMSF (e.g., Sigma-Aldrich, catalog number: P7626 )
- cOmplete® mini EDTA-free protease inhibitor (Roche Diagnostics, catalog number: 11836170001 )
- RiboLock RNase Inhibitor (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EO0381 )
- Sodium citrate (e.g., Sigma-Aldrich, catalog number: S1804 )
- TE buffer (see Recipes)
- 1x PBS (see Recipes)
- Fixative (see Recipes)
- PBT (see Recipes)
- 1.5x PAGE loading buffer (see Recipes)
- 1x and 10x TBE (see Recipes)
- 15% PA - 8 M Urea stock (see Recipes)
- 20x SSC buffer (see Recipes)
- 2x full-HYBEC (see Recipes)
- 2x wash-HYBEC (see Recipes)
- Lysis buffer (see Recipes)
- Capturing hybridization buffer (see Recipes)
- Low salt wash buffer (see Recipes)
- High salt wash buffer (see Recipes)
- Elution buffer (see Recipes)
Equipment
- Optional: inert gas (e.g., Argon) glove-box (e.g., Inert Technology, model: PureLab HE 2GB ) (see Note 1)
- PCR machine with programmable hot-lid (e.g., Bio-Rad Laboratories, catalog number: 1851148 )
- -20 °C freezer
- Refrigerated table-top centrifuge (e.g., Eppendorf, catalog number: 5426000018 )
- Erlenmeyer flask
- Handcast PAGE system including a 1 mm spacer plate (e.g., Bio-Rad Laboratories, catalog number: 1653311 ), a short plate (e.g., Bio-Rad Laboratories, catalog number: 1653308 ) and a 15-well comb (e.g., Bio-Rad Laboratories, catalog number: 4560016 )
- Vertical Electrophoresis Cell (e.g., Bio-Rad Laboratories, catalog number: 1658005 )
- Electrophoresis power supply (e.g., Bio-Rad Laboratories, catalog number: 1645050 )
- Gel documentation system with filters to image fluorescence of SYBR-GOLD and the fluorescent dye used for labeling (e.g., Bio-Rad Laboratories, catalog number: 17001402 )
- P2, P200 and P1000 pipettes
- Rocking thermoblock (e.g., Eppendorf, model: ThermoMixer® C , catalog number: 5382000015)
- Microscope for imaging (we use a Leica SP8 (Leica, model: Leica TCS SP8 ) equipped with a 63x NA=1.4 oil immersion objective and two HyD detectors)
- Tissue grinder (e.g., DWK Life Sciences, Kimble, catalog numbers: 8853000015 or 8853000040 )
- Rotator (e.g., Cole-Parmer, Stuart, model: Rotator SB3 )
- Magnetic rack (e.g., New England Biolabs, catalog number: S1507S )
- Moisture free chamber (see Note 1)
- UV/VIS spectrophotometer (e.g., Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM 8000 , catalog number: ND-8000-GL)
- Nutator (e.g., Labnet International, model: S0500 )
Software
- A probe designer algorithm, e.g., the StellarisTM Probe Designer
(https://www.biosearchtech.com/support/tools/design-software/stellaris-probe-designer, registration required) or the provided smFISHprobe_finder.R script (Supplementary File 1, see Notes 2 and 17) - MS Office Excel to run the interactive probe_calculator.xls sheet (Supplementary File 2)
- ImageJ/FIJI (https://imagej.nih.gov/ij/) with the xsPT plugin (https://github.com/Xaft/xs/blob/master/_xs.jar)
- Optional: deconvolution software, e.g., Huygens Essentials (https://svi.nl/Huygens-Essential) or DeconvolutionLab2 (Sage et al., 2017; http://bigwww.epfl.ch/deconvolution/deconvolutionlab2/)
- R (preferentially with RStudio) for data analysis
- smFISH_analysis.R to analyze the sensitivity and specificity of smFISH (Supplementary File 3)
Procedure
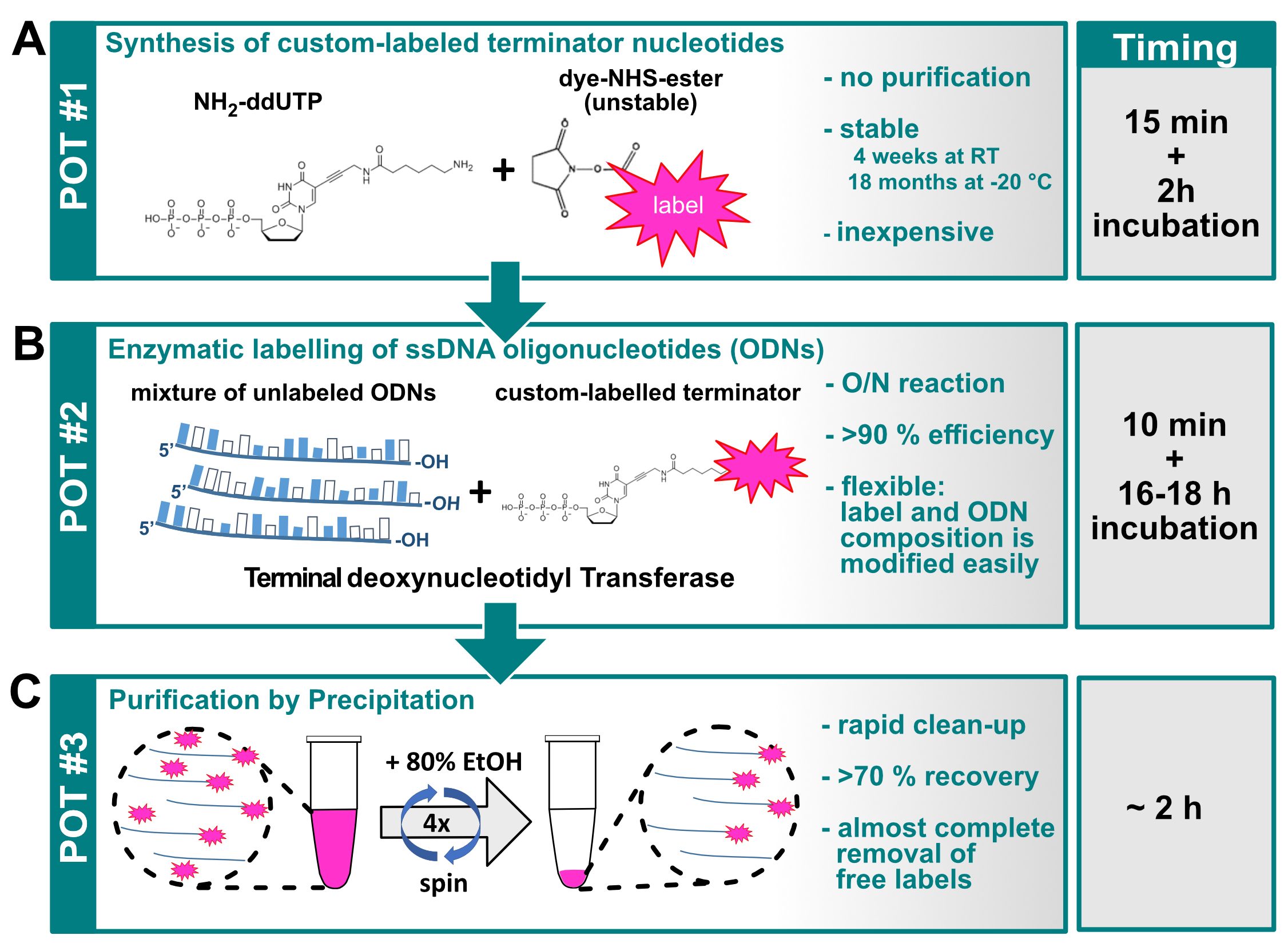
Figure 1. Graphical overview of the three-pot probe production assay. A. Conjugation of the label to the NH2-ddUTP terminator nucleotide; B. TdT mediated labeling of ssDNA oligonucleotides; C. Purification of labeled ssDNA molecules (probes).
- Preparing the dye conjugated ddUTP (First pot, Figure 1A)
- Reconstitute the dye-NHS ester to 40 mM final concentration in anhydrous DMSO in a moisture free environment. (see Note 1)
- In a clean 1.5 ml Eppendorf tube, aliquot X μl of 20 mM Amino-11-ddUTP or 5-propargylamino-ddUTP. (see Note 3)
- Add 0.2X μl of 1 M NaHCO3 (pH = 8.4).
- Finally, add X μl of 40 mM dye-NHS ester (see Note 4). Mix well and incubate at RT for 2 h sealed from light.
- After the incubation, add 1.8X μl dH2O to expand the reaction volume to 4X μl. This results in a 5 mM stock of dye conjugated ddUTP (see Note 5). The resulting labeled nucleotide is stable for over a year when stored at -20 °C.
- Reconstitute the dye-NHS ester to 40 mM final concentration in anhydrous DMSO in a moisture free environment. (see Note 1)
- Production of labeled ssDNA oligonucleotides (second pot, Figure 1B)
- Prepare an equimolar mixture of all different ssDNA oligos that should be labeled together, i.e., that target the same transcript. The total concentration of oligos should not be lower than 100 μM. (see Note 6)
- To label 1 nmol of oligonucleotide mixture, prepare the following labeling mixture in a 0.2 ml thin-walled PCR tube:
Please use the interactive probe_calculator.xls Excel sheet (Supplementary File 2) to get the reaction composition adjusted to the dye-ddUTP used for labeling.Amount Final concentration 1 nmol oligo-mixture (e.g., 4 μl of 250 μM stock) 66.67 μM 3 μl 5x TdT reaction buffer (with Co2+) 1x 3-5 nmol dye-ddUTP (e.g., 0.6-1.0 μl of 5 mM stock) 200-330 nM 1x standard amount of TdT 0.4-0.8 U/μl dH2O to 15 μl - Incubate at 37 °C O/N (16-18 h) in a PCR machine with the hot-lid set to 37 °C.
- Prepare an equimolar mixture of all different ssDNA oligos that should be labeled together, i.e., that target the same transcript. The total concentration of oligos should not be lower than 100 μM. (see Note 6)
- Purification and spectroscopic analysis of the labeled oligonucleotide mixture (third pot, Figure 1C)
- After the O/N incubation, add 60 μl of 1 M Na-acetate (pH = 5.5), 125 μl of dH2O and 1.5 μg linear acrylamide to the reaction mixture.
- Transfer the entire 200 μl into a clean 1.5 ml Eppendorf tube. Add 800 μl 100% ethanol prechilled at -20 °C. Invert the tube a couple of times and place it into the -20 °C freezer for about 15-20 min.
- In the meantime, cool the table-top centrifuge to 4 °C.
- After the -20 °C incubation, spin the oligonucleotide mixture at 16,000 x g for 20 min at 4 °C.
- Remove the supernatant and add 1 ml 80 % ethanol prechilled at 4 °C. Vortex until the pellet dissociates from the bottom of the tube.
- Spin at 16,000 x g for 5 min at 4 °C.
- Wash away the pellet from the wall of the tube with 1 ml 80% ethanol and transfer the entire volume including the floating pellet into a clean 1.5 ml Eppendorf tube. Repeat Steps C5 and C6 two more times.
- Remove the supernatant from the last wash and let the pellet dry on air. (see Note 7)
- Resuspend the dried pellet in 15-50 μl nuclease-free dH2O.
- Measure the absorbance of the labeled oligonucleotide mix at 260 nm and at the dye absorption maximum (e.g., 570 nm for Atto565).
- In order to calculate the concentration and the degree-of-labeling (DOL), measure the absorbance of the unlabeled, undiluted oligonucleotide mix at 260 nm. (see Note 8)
- Calculate the molar extinction coefficient (𝜖oligo) of the oligonucleotide mixture by dividing the measured OD260 nm value with the concentration of the mixture (in M). Increase this value by 9,000 mol-1 cm-1 to correct for the UTP added to the 3’ of the ssDNA molecules.
- Calculate the concentration of the labeled oligo by dividing the dye-corrected OD260 nm absorption by 𝜖oligo:coligo = (OD260 nm - cf260 nm x ODdye)/𝜖oligo (see Note 9).
- Calculate the concentration of the dye as follows: cdye = ODdye/𝜖dye (provided by the dye manufacturer).
- Typically, only a small fraction of the dye molecules is present as contaminants (i.e., free from ssDNA), therefore the DOL is estimated as follows: DOL = cdye/coligo.
- The fraction of recovered oligonucleotides is assessed by taking the ratio of the recovered and initial amounts of the oligo (recovery% = coligo x Vresuspension/ninitial).
- The measured OD values can be entered into the interactive probe_calculator.xls Excel sheet (Supplementary File 2) to obtain the concentration and the DOL of the labeled oligonucleotide mixture.
- Store the labeled probes at -20 °C.
- After the O/N incubation, add 60 μl of 1 M Na-acetate (pH = 5.5), 125 μl of dH2O and 1.5 μg linear acrylamide to the reaction mixture.
- Trouble-shooting of 3P3 probe production
Labeled oligo mixtures with 0.9 < DOL ≤ 1.0 are considered good quality products that can be used in smFISH applications.- The most typical cause of DOLs lower than 0.9 is the composition of the oligonucleotide mixture. We currently lack an understanding of which property(s) of the mixture–e.g., formation of intra- and intermolecular hybrids in the mixture–influence the labeling efficiency. However, if DOL < 0.9 is obtained, we recommend ‘splitting’ the oligo mixture into two-three non-overlapping fractions, i.e., preparing two-three mixtures of the ssDNA oligos present in the original mixture. Individual labeling of these mixtures can help in identifying any molecules that behave extraordinarily in the labeling reaction. In most cases, we found that this ‘splitting’ almost completely alleviates the low labeling efficiency problem, i.e., the DOL of each of the split mixtures increases above 0.9.
- Another solution we found was to re-label the already labeled oligonucleotide mixture, i.e., start over the labeling protocol from Step B2 using the labeled ssDNA molecules as input. (REF)
- The second most frequent reason for low DOL is the non-accurate (lower than actual) measurement of the molar extinction coefficient of the unlabeled oligo mixture. This can be noticed that when calculating the fraction of recovered oligonucleotides (recovery %) one obtains a value higher than one. In such cases, we recommend re-measuring the OD260 nm of the unlabeled oligo mixture at different concentrations (e.g., at 50, 100 and 200 μM) and estimating the 𝜖oligo by taking the slope of the line fitted to the data points.
- We found that terminal transferase activity of TdT varies from batch to batch. This may result in improper labeling (DOL < 0.9) when using 1x standard amount of the enzyme. We recommend to redefine the standard amount when starting a new aliquot of TdT, by setting up several labeling reactions of a single ssDNA oligo with Atto633-ddUTP (Procedures B and C) using increasing concentrations of TdT–e.g., 1x, 2x and 3x of the standard amount found in the recipe generator. Use the smallest concentration of the enzyme that yields DOL > 0.9 as the new standard amount.
- DOL > 1.0 may also indicate an imprecisely determined 𝜖oligo. This would appear as a huge loss of the oligos (recovery < 50%). Try the same solution as in Step D3.
- If the 𝜖oligo is appropriate, DOL > 1.0 indicates free dye contamination. In such cases it is advised to measure the DOL by PAGE (Procedure E). If the DOL determined by densitometry is ~1.0 and the difference of the two DOLs is small (max. 0.05-0.1), the free dye contamination is considered harmless in the downstream applications. If it is higher, re-purification of the oligonucleotide mixture by another method (e.g., by size exclusion chromatography) is recommended. We observed such high free dye contamination when using Abberior470SX-, Atto488- and AlexaFluor488-ddUTP (Gaspar et al., 2017b).
- The most typical cause of DOLs lower than 0.9 is the composition of the oligonucleotide mixture. We currently lack an understanding of which property(s) of the mixture–e.g., formation of intra- and intermolecular hybrids in the mixture–influence the labeling efficiency. However, if DOL < 0.9 is obtained, we recommend ‘splitting’ the oligo mixture into two-three non-overlapping fractions, i.e., preparing two-three mixtures of the ssDNA oligos present in the original mixture. Individual labeling of these mixtures can help in identifying any molecules that behave extraordinarily in the labeling reaction. In most cases, we found that this ‘splitting’ almost completely alleviates the low labeling efficiency problem, i.e., the DOL of each of the split mixtures increases above 0.9.
- PAGE analysis of the labeled oligonucleotides
Gel electrophoresis provides a simple means to confirm the DOL estimated from spectroscopy data and to calculate DOL in case of non-fluorescent modifications (e.g., biotinylation). PAGE analysis also allows the quality control of most of the labeled ddUTP analogs, i.e., to determine whether there are unconjugated ddUTP molecules in the dye conjugated ddUTP stock (produced in Procedure A) that will result in reduced DOL of oligonucleotides.
IMPORTANT: Only oligo-mixtures containing ssDNA molecules of identical length (e.g., only 20 mers) should be analyzed by PAGE.- Wipe the glass plates for gel casting clean with 70% ethanol, assemble the cassette and seal with 20 mm wide transparent tape, leaving the top (where the comb will be inserted) open. Put two fold-back clamps on the two sides of the cassette such that the clamps clamp above the spacer between the two glass plates.
- To cast a 10 x 8 cm acrylamide gel, add 8 ml of 15% PA - 8 M urea stock (see Recipes), 40 μl 10% APS and 5 μl TEMED into an Erlenmeyer flask. Mix well by swirling the flask and pour the mixture between the two plates of the assembled cassette. Fill to the top.
- Insert the comb, place the cassette horizontally and wait until the gel polymerizes (15-20 min).
- Remove the clamps, the tape and the comb. Rinse the outside of the cassette with dH2O to remove gel pieces polymerized on the outer surface and assemble the PAGE chamber.
- Fill with 1x TBE buffer (see Recipes) and pre-run the gel for 30 min with 2.5 mA/cm current (20 mA for an 8 cm long gel).
- In the meantime, prepare 5 μl of each sample by mixing 3 μl 1.5x PAGE loading buffer (see Recipes) and 2 μl oligonucleotide mixture containing 15-60 pmoles of labeled oligo. As size marker use a dilution row of the unlabeled oligonucleotide mixture (e.g., 1.5, 3 and 6 pmoles), no boiling is necessary.
- After the pre-run (E5), rinse all wells on the gel with 1x TBE using a P200 pipette to remove the accumulated urea that would prevent the loading.
- Load the samples and run the gel until the xylene cyanol (blue) and the bromophenol blue (purple) markers (from the 6x gel loading dye) reach about the one-third and two-third of the gel length.
- Image the fluorescently labeled pool of molecules on a gel-imager with appropriate filter sets to excite and detect the incorporated fluorescent dyes.
- Incubate the gel with SYBR-GOLD (or similar RNA/ssDNA dye) diluted 1:10,000 in 1x TBE for 10-15 min.
- Re-image the gel to detect both the non-modified and modified pools of ssDNA.
- Due to the addition of a bulky terminator nucleotide, labeled oligonucleotides run slower and thus they are well separated from their non-modified peers during PAGE. The amount of non-modified oligos can be measured by comparing the corresponding SYBR-GOLD fluorescence intensity to that of the dilution row of the unlabeled oligonucleotide mixture used as loading control (Figures 2A and 2A’).
- The presence of unconjugated ddUTP will result in production of unlabeled, ddUTP-terminated oligonucleotides. On gel, they appear as an intermediate band migrating between the unlabeled and labeled, fluorescent pool of oligos.
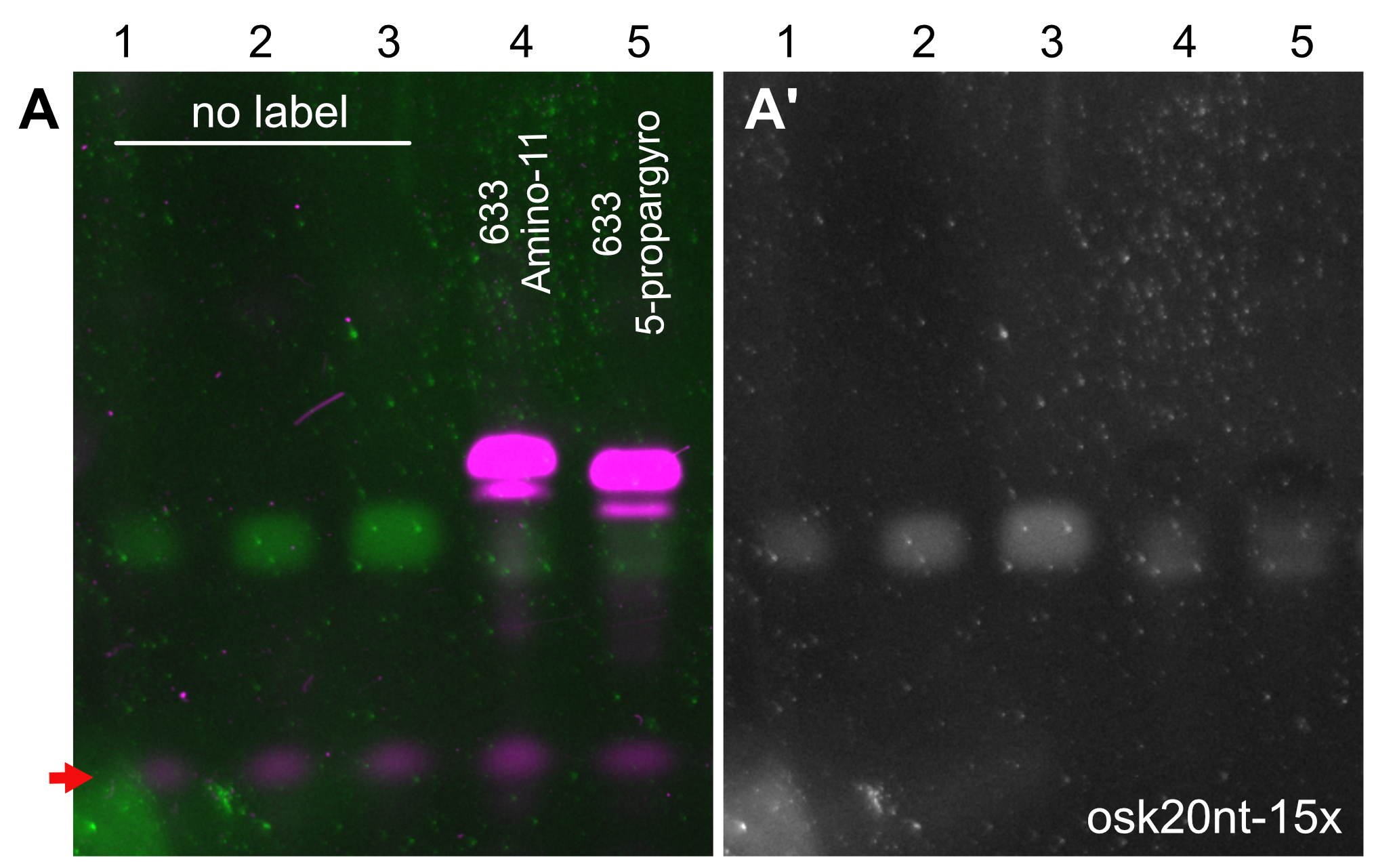
Figure 2. PAGE analysis of the labeled oligonucleotide mixtures. Labelling of the osk20nt-15x (Gaspar et al., 2017b) probe mixture with Atto633-amino-11-ddUTP (lane 4) or Atto633-5-propargylamino-ddUTP (lane 5). The fluorescently labeled ssDNA species (magenta, A) migrate slower in the gel than the unlabeled oligonucleotides (green, A, gray, A’). Note that the probe molecules labeled with far-red fluorescence are not visible in the SYBR-GOLD channel, possibly because of a very efficient energy transfer form SYBR GOLD to Atto633 that quenches the SYBR GOLD fluorescence. While this is the case with other far-red dyes also (e.g., Abberior-RED), when there is no quenching dye present (e.g., biotinylated probes) or when the FRET fluorescence can be detected by the SYBR GOLD imaging setup (e.g., in case of Atto565), the labeled probes appear in the SYBR GOLD channel (Gaspar et al., 2017b). 1.5, 3, 6, 20 and 20 pmol ssDNA mixture was loaded to lanes 1-5, respectively. After a labeling reaction with either of the terminator nucleotides, only low amounts (< 1.5 pmol) of unlabeled oligonucleotides were left in the mixtures (lanes 4 and 5A and 5A’), indicating near-quantitative labeling with both terminator nucleotides. The red arrow indicates fluorescence of the bromophenol blue dye.
- Wipe the glass plates for gel casting clean with 70% ethanol, assemble the cassette and seal with 20 mm wide transparent tape, leaving the top (where the comb will be inserted) open. Put two fold-back clamps on the two sides of the cassette such that the clamps clamp above the spacer between the two glass plates.
- Single molecule FISH in Drosophila ovaries
- Dissect ovaries into a 1.5 ml Eppendorf tube containing 300-500 μl fixative (for ovary dissection, please refer to Gaspar and Ephrussi, 2017).
- Fix for 20 min by nutating the dissected material.
- Remove the fixative and rinse the ovaries with 1 ml PBT (see Recipes).
- Wash ovaries in 1 ml fresh PBT for 10 min while nutating.
- Replace PBT and add Proteinase-K to 2 μg/ml final concentration. Nutate for 5 min at room temperature (RT). (see Note 10)
- Preheat 0.5 ml 0.05 v/v % SDS in PBS (see Recipes) to 95 °C.
- After the 5 min Proteinase-K digestion, remove the PBT and immediately apply the preheated SDS/PBS to the ovaries. Incubate them for 5 min at 95 °C.
- Add 1 ml RT PBS to the ovaries to cool the solution.
- Replace the wash solution with 200 μl 2x full-HYBEC (see Recipes).
- Transfer the tube to a rocking thermoblock set to 37-42 °C and shake at 1,000 RPM for 10 min.
- In the meantime, prepare the probe solution (5-12.5 nM/individual probe) in 50 μl of 2x full-HYBEC. Transfer the probe solution to the thermoblock and allow it to warm up (2-3 min). (see Note 11)
- After the 10 min incubation of the ovaries, apply the probe solution by mixing it into the 200 μl 2x full-HYBEC already on the specimen.
- Incubate for 1.5-3 h at 37-42 °C while rocking at 1,000 RPM.
- 10-15 min before the end of the incubation, prewarm two times 1 ml 2x wash-HYBEC (see Recipes) to the hybridization temperature.
- Remove the hybridization solution–it may be kept at 4 °C for another hybridization–and wash with 1 ml prewarmed 2x wash-HYBEC for 2 x 15 min.
- Wash once with 1 ml RT PBT for another 15 min.
- Thoroughly remove the PBT and apply mounting medium, e.g., 80% TDE (see Recipes) or VectaShield. (see Note 12)
- After allowing the mounting medium to soak for at least one hour, mount the ovaries onto glass slides and image them using a high NA (> 1.1 NA) objective (Figure 3A).
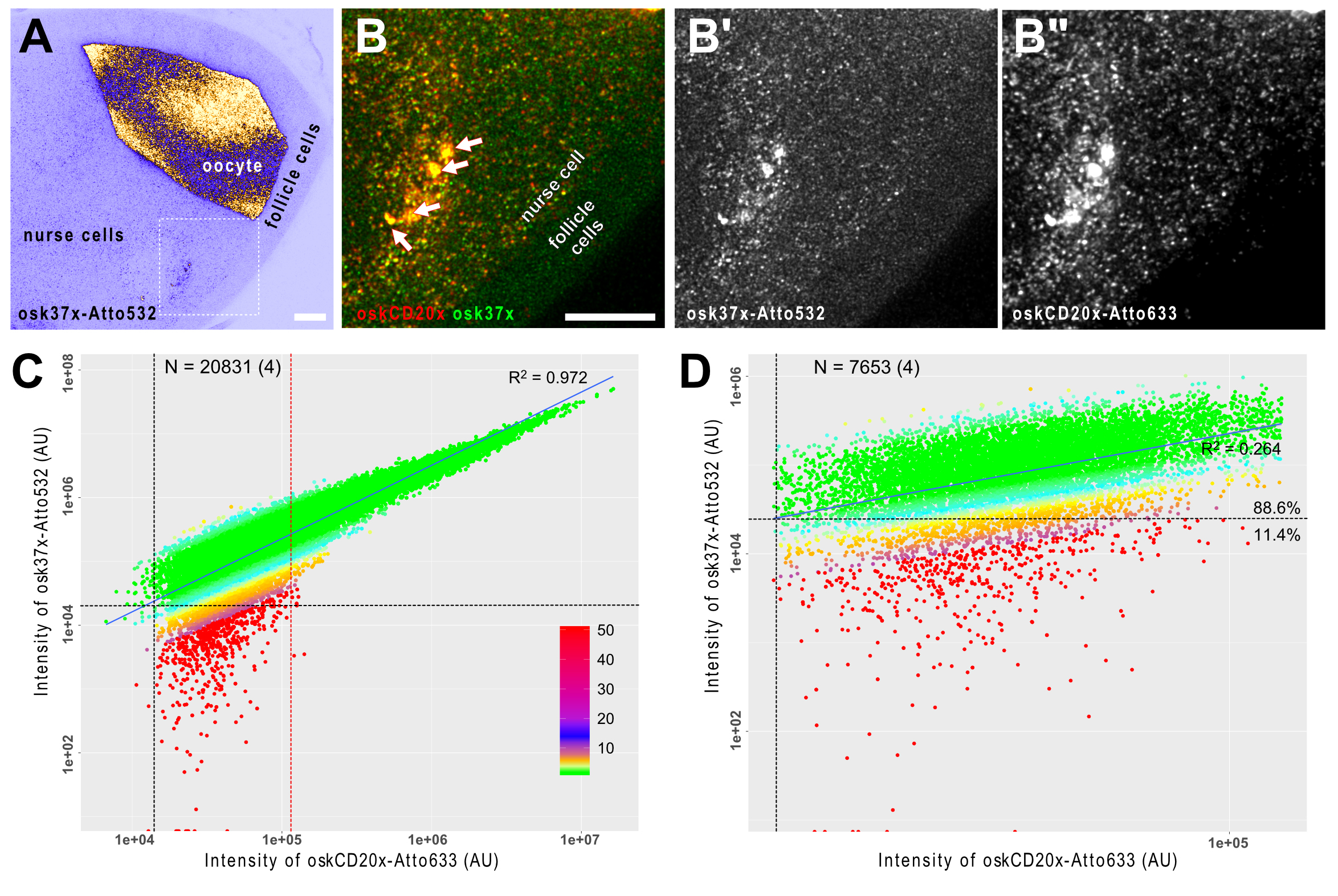
Figure 3. smFISH analysis of oskar mRNA in developing Drosophila egg-chambers. A. oskar mRNA is produced in the transcriptionally active nurse cells (left) and it is transported into the oocyte, where it localizes eventually at the posterior pole (on the right). The female germ-line cells (nurse cells + oocyte) are encapsulated by a layer of somatic epithelium, the follicle cells that do not express oskar mRNA. B. Close-up of the boxed region in A. oskar mRNA was detected by two different probe sets osk37x-Atto532 (green, osk3UTR#1-15 + oskCD#1-22) and oskCD20x-633 (red, oskCD#23-42, Gaspar et al., 2017b). There is a high degree of co-localization of the two puncta-like signal in the nurse cell compartment (B) and no such strong accumulations of the probes is detected in the follicle cells (B-B”). Arrows indicate multiple transcriptional loci inside the polyploid nucleus of the nurse cell (B). Scale bars represent 10 μm. C and D. Correlation of the signal intensities of the two channels. C. When all objects across the egg chamber are considered, a strong linear correlation (R2 = 0.972) is established, indicating that the RNA content of the detected objects is variable. By fitting multiple Gaussian functions to the signal intensity distribution (Little et al., 2015; Gaspar et al., 2017b), it is possible to filter the smFISH objects containing a single copy of oskar RNA. D. In this regime, the correlation of the signal intensities is low to moderate (R2 = 0.264), however, vast majority of the objects can be detected in both channels–e.g., 88.6% of the objects analyzed in the Atto633 channel are also detectable in the Atto532 channel also. Vertical and horizontal black dashed lines represent the detection thresholds for the Atto633 and Atto532 channels, respectively. Red dashed line shows the boundary between objects with single and multiple copies of RNA (C). Colours represent the relative intensity difference between the two channels (see panel C for key). The number of smFISH objects (number of experiments) is indicated in C and D.
- Dissect ovaries into a 1.5 ml Eppendorf tube containing 300-500 μl fixative (for ovary dissection, please refer to Gaspar and Ephrussi, 2017).
- RNA capture from ovarian lysate
- Isolate ovaries from well-fed 2-3 day old Drosophila melanogaster in PBS (see Note 13).
- Remove PBS and resuspend with 3 volumes lysis buffer.
- Mechanically homogenize using a tissue grinder.
- Transfer to fresh 15 ml tubes and clear the lysate by centrifugation (5 min at 140 x g).
- Transfer the supernatant to a fresh tube and dilute with 2 volumes capturing hybridization buffer (see Recipes).
- Preclear by adding 1:50 v/v Pierce® Avidin Agarose prewashed with a 1:2 mix of lysis buffer and capturing hybridization buffer.
- Keep on a rotator for 30 min at room temperature.
- Remove Avidin agarose by centrifugation (5 min at 140 x g).
- Split the precleared lysate (keep 0.1% as input sample) and supplement it with 0.25 µg (see Note 14) of targeting and non-targeting control biotinylated DNA probes per ml of ovaries and incubate at 37 °C for 2 h on a rotator.
- Pre-wash 3.75 µl magnetic Streptavidin beads per ml lysate with a 1:2 mix of lysis buffer and capturing hybridization buffer.
- Add magnetic Streptavidin beads to lysate and keep rotating for 1 h at 37 °C.
- Collect the beads using a magnetic rack.
- Wash beads three times for 5 min at 37 °C with low salt wash buffer (see Recipes).
- Wash beads two times for 5 min at 37 °C with high salt wash buffer (see Recipes).
- Wash beads three times for 5 min at 37 °C with low salt wash buffer.
- Elute the RNA from the beads by adding elution buffer (see Recipes) and boil at 95 °C for 5 min. Use approx. 0.75 µl TE buffer per µl Streptavidin bead slurry used in Step H10.
- Extract the RNA using an RNA extraction kit and determine the amount of captured RNA of targeting and non-targeting probes by qRT-PCR or Northern blot.
- Isolate ovaries from well-fed 2-3 day old Drosophila melanogaster in PBS (see Note 13).
Data analysis
- Determining specificity and sensitivity of smFISH
- The rationale behind using an array of singly labeled probes to detect target transcripts is the local increase of signal on the specific RNA molecule–due to specific hybridization of the entire array–relative to the background resulting from e.g., random aspecific binding of individual probe molecules (Femino et al., 1998; Raj et al., 2008). Consequently, most of the aspecificity appears as background that can be filtered out (e.g., by the xsPT plugin during Step 7 of this section). In extreme cases, however, the probes may accumulate in specific subcellular structures/organelles, mainly in the nucleolus. If this is observed, we recommend increasing the stringency of the hybridization and the subsequent two wash steps (Steps 10-15 of Procedure F) by increasing the temperature and/or decreasing the salt concentration in the corresponding buffers. Decreasing the probe concentration and increasing the relative amount of the organic solvent (e.g., ethylene carbonate) could be also tried out, in this order (see Notes 16 and 18).
- To test whether the given smFISH setup–including the probe set and the applied protocol–is detecting all of the target molecules and only the target molecules, a two color smFISH reaction against the transcript of interest can be performed (Figures 3B-3B” and Video 1).
 Video 1. A short tutorial (~20 min) on how to use ImageJ and R to establish single molecule sensitivity of an smFISH experiment. A further tutorial on using the xsPT plugi-in is available in Gaspar et al., 2017.
Video 1. A short tutorial (~20 min) on how to use ImageJ and R to establish single molecule sensitivity of an smFISH experiment. A further tutorial on using the xsPT plugi-in is available in Gaspar et al., 2017. - Split the unlabeled ssDNA oligonucleotides into two non-overlapping arrays and label them with two spectrally distinguishable fluorophores–e.g., Atto532 and Atto633–as described in Procedures B and C. (see Note 6)
- Carry out an smFISH experiment–e.g., as described in Procedure F–using the two sets of probes simultaneously.
- Acquire images of both fluorophores while avoiding cross-talk of the two channels–e.g., by performing sequential scan during confocal scanning microscopy.
- Optional: perform deconvolution on the resulting images to enhance signal to noise ratio and facilitate downstream analysis.
- Analyze the overlap of the two signals e.g., by using the xsPT plugin of ImageJ (Gaspar and Ephrussi, 2017).
- xsPT segments one of the channels (for more details, see Gaspar and Ephrussi, 2017) and performs tracking through an image sequence, e.g., a Z-stack allowing to recognize objects in 3D.
- Set the parameters of the plugin to detect all–or at least most of–the objects in the reference channel (for details, please refer Video S4 and S5 of Gaspar and Ephrussi, 2017). In the tracking part, set a maximum displacement of 1 px (the center of an object is not expected to shift much between the slices), set ‘Prefer short steps’ and uncheck ‘Manual tracking’ (i.e., perform automated tracking). Let the plugin run.
- RNPs–and other structures smaller than the diffraction limit–are expected to be observed in at least three consecutive slices when the image is four-fold over-sampled according to the modified Nyquist criterion (z-step size is typically between 180-300 nm). Remove trajectories that are shorter than three frames (‘Min. trajectory = 3’ and press ‘Filter trajectories’). You may further filter the recognized objects by selecting a region of interest in the image and pressing ‘Filter trajectories’.
- Save the filtered trajectories. The integrated signal intensities of both the reference and any other channels will be stored in the resulting .csv file. Four of such .csv files (Supplementary Files 4-7) are provided as examples. These–and any user generated files–can be analyzed using the smFISH_analysis.R script (Supplementary File 3).
- Determine a detection threshold for the reference channel (e.g., the minimum or the lowest 0.1th percentile of the integrated signal intensity).
- Repeat Steps A9-A12 using the other channel as the reference. This analysis results in independent measurements of the detection threshold for both channels.
- Test what fraction of the objects of the non-reference channel has higher signal intensity than the corresponding detection threshold, which was determined in the parallel analysis. If this fraction is over 80% for both channels, the smFISH analysis is considered to be at single molecule sensitivity (low false negative detection) and highly specific (low false positive detection rate) (Figure 3D).
- Lower than 80% co-detection of the same transcripts with the two probe sets–in extreme cases no overlap of the two signals or no signal at all–indicate insufficient sensitivity that is usually due to too high stringency during the hybridization and the two subsequent washes (Steps 10-15 of Procedure F). To overcome this problem, we recommend decreasing the temperature, increasing the salt concentration in the corresponding buffers as well as the probe concentration and decreasing the relative amount of the organic solvent during hybridization (try implementing these changes sequentially, in the listed order) (see Notes 16 and 18).
- Plotting the signal intensities of the reference and the non-reference channel can give insight into the RNA copy number of the objects (Figure 3C). In case of single RNA molecules, the two signals are not expected to correlate well as they are primarily governed by stochastic events (e.g., the hybridization efficiency of the probes, Figure 3D). If there are more than one copy of target transcripts in the objects, these stochastic events tend to average out yielding an improved linear relationship between the signals of the two channels which get stronger and stronger with the increase of the signal intensity.
- The rationale behind using an array of singly labeled probes to detect target transcripts is the local increase of signal on the specific RNA molecule–due to specific hybridization of the entire array–relative to the background resulting from e.g., random aspecific binding of individual probe molecules (Femino et al., 1998; Raj et al., 2008). Consequently, most of the aspecificity appears as background that can be filtered out (e.g., by the xsPT plugin during Step 7 of this section). In extreme cases, however, the probes may accumulate in specific subcellular structures/organelles, mainly in the nucleolus. If this is observed, we recommend increasing the stringency of the hybridization and the subsequent two wash steps (Steps 10-15 of Procedure F) by increasing the temperature and/or decreasing the salt concentration in the corresponding buffers. Decreasing the probe concentration and increasing the relative amount of the organic solvent (e.g., ethylene carbonate) could be also tried out, in this order (see Notes 16 and 18).
- Determining the efficiency and specificity of RNA capture
- After RNA extraction, set up reverse transcription of the 0.1% input sample and eluted sample from the non-targeting and specific capture (see Note 15).
- From the cDNA, make a dilution series of the ‘input’ (e.g., 0.1%, 0.01%, 0.001% and 0.0001%) and dilute ‘captured’ sample to at least ten-fold. Set up triplicates of qRT-PCR reactions to amplify the target transcript and other, abundant non-target RNAs (e.g., the 18S ribosomal RNA) from the ‘input’ dilutions and the eluted ‘capture’ samples using appropriate PCR primer pairs.
- Calculate the amount of ‘captured’ RNA by comparing it to the regression of the diluted ‘input’ sample. For more details, see Gaspar et al., 2017b.
- After RNA extraction, set up reverse transcription of the 0.1% input sample and eluted sample from the non-targeting and specific capture (see Note 15).
Notes
- You may use an argon-filled chamber. Another, simple means to obtain moisture free environment is to fill 50-100 g of silica gel into an about 20 x 20 x 20 cm polystyrene or plastic box with a lid.
- The smFISHprobe_finder.R script designs target specific antisense ssDNA oligonucleotides such that the terminal uracil nucleotide added during the labeling will become part of the hybrid. Any large gaps (> the double of minLength) between such probes are further scanned for potential probes that do not satisfy this terminal U rule but satisfy the other search criteria. Alternatively, the probe finder script can be used without the terminal U rule (lines 93-96 of smFISHprobe_finder.R script). Of note, we have found no adversary effect of a non-complimentary uracil nucleotide during hybridization at the 3’ end of the produced probes (data not shown).
- We found no practical differences between Amino-11-ddUTP or 5-propargylamino-ddUTP regarding dye conjugation and enzymatic incorporation of the resulting terminator nucleotide (Figure 2).
- We found that BDP-FL-NHS ester is quite hydrophobic and requires higher organic solvent concentration. When using this dye, add 1.8x DMSO before slowly titrating in the dye in Step A4 and skip Step A5. If precipitation occurs add more DMSO and calculate the final concentration accordingly.
- Originally, we added Tris-HCl, pH 7.5 to a 10 mM final concentration to quench the remaining NHS ester activity. However, we found no adverse effects of omitting this quenching step on the downstream enzymatic reaction.
- Although all oligonucleotides targeting the same transcript could be combined in one master-mix, we recommend preparing at least two non-overlapping mixtures, e.g., by combining every other oligonucleotides, by splitting them into 5’ and 3’ halves or grouping them by oligonucleotide length. Label these mixtures separately–this will facilitate troubleshooting, i.e., when the low degree of labeling is observed (Procedure E) and also allows benchmarking the smFISH performance (Procedure G). We typically order the oligos resuspended in H2O to 250 μM concentration.
- To facilitate drying, make sure that all residual ethanol is removed from the walls of the tube by a quick pulse of centrifugation after removing the bulk of the supernatant.
- To determine the degree of biotinylation, Colorimetric Biotin Assay Kit based on color loss of 2-(4-hydroxyphenylazo)-benzoic acid could be used. However, we found that incorporation of biotin-ddUTP is always 100% (see Figure 1 of Gaspar et al., 2017b) and therefore we do not routinely determine the degree of biotinylation.
- The organic dyes usually have also some absorption at 260 nm. The correction factor at 260 nm (cf260 nm) provided by the dye manufacturer specifies this absorbance as the fraction of the maximal absorption of the dye. This cf260 nm and the absorption of the dye at maximum allows for the correction of the OD260 nm value when calculating the oligonucleotide concentration.
- Steps 5-8 of Procedure F (smFISH) will result in a limited proteolysis and denaturation of proteins and RNA secondary structures and thus they may facilitate target recognition of the probes. However, if the fluorescence of fluorescent proteins (e.g., Emerald-GFP, as in Gaspar et al., 2017a) is to be preserved these steps should be omitted. In case of oskar mRNA, we found a minuscule effect of omitting these steps on the hybridization results.
- The final probe concentration we use is between 1-2.5 nM/probe. We find that lower concentrations may impair the sensitivity of the reaction while higher concentrations–although rarely they may cause staining artifacts–do not affect the quality of the hybridization but result in unnecessary waste of material.
- We found that 80% TDE boosts the brightness of GFP and red fluorescent proteins without affecting the signal of Atto565 and Atto633. However, the green and yellow fluorescent dye are substantially dimmer in this mounting medium and thus we recommend using e.g., VectaShield.
- If high amounts of starting material are required due to low abundance of target RNA, we use a method for mass-isolation of ovaries (similar to Jambor et al., 2015).
- The probe concentration should be adjusted depending on the amount of RNA present in your sample. We estimated the amount of the desired target RNA by qRT-PCR or Northern blot together with known standards of target RNA.
- Make sure not to oversaturate the RT reaction. The maximum volume is determined by the concentration of the ‘input’ sample.
- We recommend changing only one parameter at a time. It is a good practice to include the original value and two-three modified values of the parameter in these optimization steps. We find that once the listed parameters are optimized for the specimen, they are an excellent starting point for all smFISH reactions performed with yet uncharacterized probe sets, and in 95% of the cases those parameters provide good quality results.
- However, ‘aspecificity’ that arises from (partial) complementarity of a large fraction of the probe array to another transcript(s) may result in signal indistinguishable from that produced by the specific hybrids. This phenomenon can be only tested in control samples that lack the RNA-to-be-detected (RNA null mutants or efficient RNAi knock-downs). Since these control samples are not readily available in most cases, we recommend BLAST-ing the probe sequences against the host transcriptome during the design phase (before starting Procedure B) to rule out such cryptic targets.
- If an already tested and proven-to-work probe set results in such low sensitivity hybridization, the cause is likely nuclease contamination in one of the buffers. In such a case, it is recommended to replace all buffers used for the hybridization.
Recipes
- TE buffer
10 mM Tris base
1 mM EDTA
Adjust pH to 8.0 - 1x PBS
137 mM NaCl
2.7 mM KCl
10 mM Na2HPO4
1.8 mM KH2PO4
Adjust pH to 7.4
Store at RT - Fixative
10 ml 16% (v/v) EM-grade paraformaldehyde
30 ml 1x PBS
Sterile filter with 0.22 μm filter
Store at 4 °C - PBT
0.1% Triton X-100 in 1x PBS
Store at RT - 1.5x PAGE loading buffer
¾ 8 M urea in 1x PBS
¼ 6x gel loading dye
Store at RT - 10x TBE
1 M Tris base
1 M boric acid
20 mM EDTA
Store at RT - 15% PA - 8 M urea stock (200 ml)
75 ml 40% (v/v) Acrylamide/Bis solution
20 ml 10x TBE
96 g urea
Top up to 200 ml with dH2O
Stir and heat (max 100 °C) until the urea completely dissolves
Store at RT isolated from light - 20x SSC
3 M NaCl
300 mM Na-citrate
pH 7.0
Store at RT - 2x wash-HYBEC
2x SSC
15% (v/v) ethylene carbonate
1 mM EDTA
0.1% Triton X-100
Store at RT - 2x full-HYBEC
2x SSC
15% (v/v) ethylene carbonate
1 mM EDTA
50 µg/ml heparin
100 µg/ml salmon sperm ssDNA
0.1% Triton X-100
Store at RT - 80% TDE
80% (v/v) 2,2’-thiodiethanol (Sigma-Aldrich)
20% (v/v) 1x PBS
Mix well and store at RT - Lysis buffer
50 mM Tris-HCl pH 7.0
10 mM EDTA
1% (v/v) SDS
Fresh 1 mM PMSF
Fresh cOmplete® mini EDTA-free protease inhibitor*
1:2,000 fresh RiboLock RNAse Inhibitor - Capturing hybridization buffer
50 mM Tris-HCl pH 7.0
750 mM NaCl
1 mM EDTA
1% (v/v) SDS
15% (v/v) ethylene carbonate
Fresh 1 mM PMSF
Fresh cOmplete® mini EDTA-free protease inhibitor*
1:2,000 fresh RiboLock RNAse Inhibitor - Low salt wash buffer
2x SSC
0.5% (v/v) SDS
Fresh 1 mM PMSF
Fresh cOmplete® mini EDTA-free protease inhibitor* - High salt wash buffer
750 mM NaCl
30 mM sodium citrate pH 7.0
0.5% (v/v) SDS
Fresh 1 mM PMSF
Fresh cOmplete® mini EDTA-free protease inhibitor* - Elution buffer
10 mM Tris-HCl pH 7.0
1 mM EDTA
Store at RT
*Note: Add according the manufacturer’s instructions.
Acknowledgments
This work was funded by the EMBL. This protocol is adapted from Gaspar et al., 2017b. The authors declare no conflicts of competing interests.
References
- Femino, A. M., Fay, F. S., Fogarty, K. and Singer, R. H. (1998). Visualization of single RNA transcripts in situ. Science 280(5363): 585-590.
- Gaspar, I. and Ephrussi, A. (2015). Strength in numbers: quantitative single-molecule RNA detection assays. Wiley Interdiscip Rev Dev Biol 4(2): 135-150.
- Gaspar, I. and Ephrussi, A. (2017). Ex vivo ooplasmic extract from developing Drosophila oocytes for quantitative TIRF microscopy analysis. Bio-protocol 7(13).
- Gaspar, I., Sysoev, V., Komissarov, A. and Ephrussi, A. (2017a). An RNA-binding atypical tropomyosin recruits kinesin-1 dynamically to oskar mRNPs. EMBO J 36(3): 319-333.
- Gaspar, I., Wippich, F. and Ephrussi, A. (2017b). Enzymatic production of single-molecule FISH and RNA capture probes. RNA 23(10): 1582-1591.
- Jambor, H., Surendranath, V., Kalinka, A. T., Mejstrik, P., Saalfeld, S. and Tomancak, P. (2015). Systematic imaging reveals features and changing localization of mRNAs in Drosophila development. eLife 4: e05003
- Khong, A., Matheny, T., Jain, S., Mitchell, S. F., Wheeler, J. R. and Parker, R. (2017). The stress granule transcriptome reveals principles of mRNA accumulation in stress granules. Mol Cell 68(4): 808-820 e805.
- Little, S. C., Sinsimer, K. S., Lee, J. J., Wieschaus, E. F. and Gavis, E. R. (2015). Independent and coordinate trafficking of single Drosophila germ plasm mRNAs. Nat Cell Biol 17(5): 558-568.
- Raj, A., van den Bogaard, P., Rifkin, S. A., van Oudenaarden, A. and Tyagi, S. (2008). Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5(10): 877-879.
- Sage, D., Donati, L., Soulez, F., Fortun, D., Schmit, G., Seitz, A., Guiet, R., Vonesch, C. and Unser, M. (2017). DeconvolutionLab2: An open-source software for deconvolution microscopy methods-image processing for biologists. Methods 115: 28-41.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Gáspár, I., Wippich, F. and Ephrussi, A. (2018). Terminal Deoxynucleotidyl Transferase Mediated Production of Labeled Probes for Single-molecule FISH or RNA Capture. Bio-protocol 8(5): e2750. DOI: 10.21769/BioProtoc.2750.
Category
Developmental Biology > Cell growth and fate > Oocyte
Cell Biology > Cell imaging > Fluorescence
Molecular Biology > RNA > RNA detection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.