- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Conditional Knockdown of Proteins Using Auxin-inducible Degron (AID) Fusions in Toxoplasma gondii
Published: Vol 8, Iss 4, Feb 20, 2018 DOI: 10.21769/BioProtoc.2728 Views: 18440
Reviewed by: David CisnerosNoelia LanderAnonymous reviewer(s)
Original research article
The authors used this protocol in:
May 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Toxoplasma gondii is a member of the deadly phylum of protozoan parasites called Apicomplexa. As a model apicomplexan, there is a great wealth of information regarding T. gondii’s 8,000+ protein coding genes including sequence variation, expression, and relative contribution to parasite fitness. However, new tools are needed to functionally investigate hundreds of putative essential protein coding genes. Accordingly, we recently implemented the auxin-inducible degron (AID) system for studying essential proteins in T. gondii. Here we provide a step-by-step protocol for examining protein function in T. gondii using the AID system in a tissue culture setting.
Keywords: AuxinBackground
Auxins are a class of phytohormones that signal by targeting certain proteins for proteasomal degradation in plants (Teale et al., 2006). Kohei Nishimura et al. had the clever idea of transferring components of this plant-specific signaling system to other eukaryotes for conditional regulation of proteins of interest (POIs), creating the auxin-inducible degron (AID) system (Nishimura et al., 2009). This system has since been adapted successfully in several eukaryotes, including the apicomplexan parasite Plasmodium (Kreidenweiss et al., 2013; Philip and Waters, 2015). Just two transgenic components are needed to implement this system, a plant auxin receptor called transport inhibitor response 1 (TIR1) and a POI tagged with an AID. Treatment with an auxin (e.g., 3-indolacetic acid/IAA) activates the SCFTIR1 ubiquitin ligase complex which exclusively targets AID-tagged proteins for ubiquitin-dependent proteasomal degradation (Figure 1). We recently engineered an RHΔhxgprtΔku80 line of T. gondii to stably express TIR1 from Oryza sativa (RH TIR1-3FLAG) (Brown et al., 2017; Long et al., 2017a). In this background, we were able to use CRISPR/Cas9 genome editing (Shen et al., 2014; Sidik et al., 2014; Shen et al., 2017) to tag essential T. gondii genes of interest with AID-3HA or mini-AID(mAID)-3HA, regulate their expression with auxin, and identify phenotypes associated with their loss (Brown et al., 2017; Long et al., 2017a and 2017b).
There are several advantages for using this system for conditional knockdowns in T. gondii. First, POI-AID fusions are expressed from their endogenous promoters, maintaining normal expression timing and levels. Second, auxin is non-toxic to parasite and host cell cultures at 1 mM but can function as low as ~50 µM. Third, auxin is added only when knockdown is desired and is commercially available for less than $5 USD per gram. Last and most importantly, POI-AID fusions are fully degraded in as little as 15 min following auxin treatment. For these reasons, we were compelled to elaborate on our published methods in this detailed protocol to facilitate the establishment of this system in other apicomplexan laboratories. 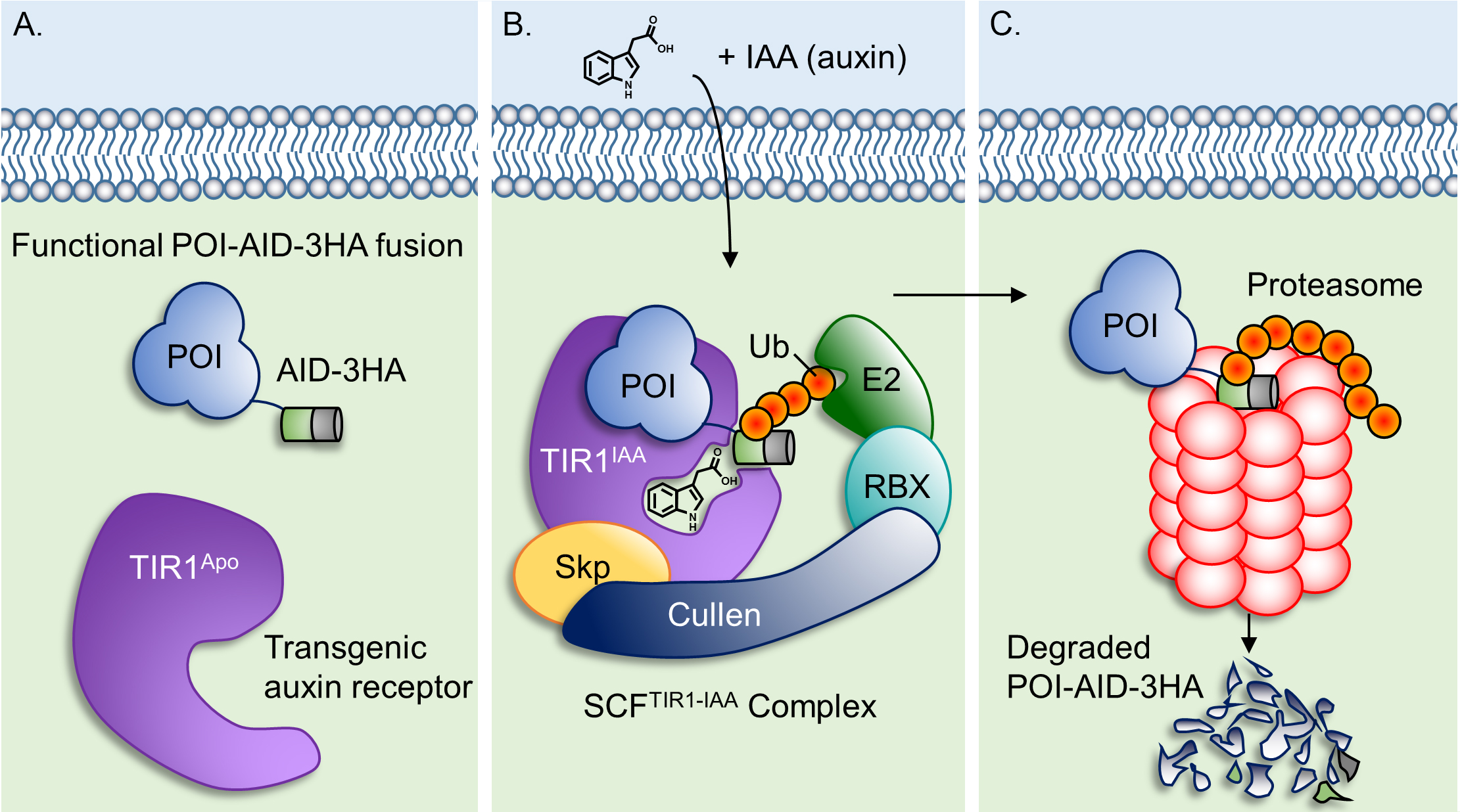
Figure 1. Model of the auxin inducible-degron system. A. In the absence of auxin, the plant auxin receptor TIR1 is in its inactive ‘Apo’ state, allowing the protein of interest (POI)-AID-3HA fusion to express and function normally. B. Auxin-bound TIR1 assembles into an active Skp-Cullen-F Box (SCFTIR1) ubiquitin ligase complex where it recognizes and polyubiquitinates AID. C. The polyubiquitin modification targets POI-AID-3HA for proteasomal degradation.
Materials and Reagents
- Microcentrifuge tubes (1.7 ml)
- PCR tubes (0.2 ml)
- T-25 and T-175 culture flasks (Corning, catalog numbers: 430639 , 431080 )
- 96-well tissue culture plates (TPP, catalog number: 92696 )
- 24-well plates (TPP, catalog number: 92024 )
- 22 G blunt needles (CML Supply, catalog number: 901-22-100M )
- 3.0 µm pore size 47 mm filter membrane (GE Healthcare, Whatman, catalog number: 111112 )
- 10 ml syringes (BD, catalog number: 309695 )
- 50 ml polystyrene conical vials (Fisher Scientific, catalog number: 05-539-10 )
- 1 L Stericup Filter Units (Merck, catalog number: SCVPU11RE )
- 13 mm cell scrapers (TPP, catalog number: 99002 )
- 47 mm polycarbonate syringe filter holder (GE Healthcare, Whatman, catalog number: 420400 )
- Filter paper for Western blot wet transfer (GE Healthcare, Whatman, catalog number: 3030-917 )
- Nitrocellulose membrane (GE Healthcare, Amersham, catalog number: 10600003 )
- Petri dishes (Sigma-Aldrich, catalog number: P5606 )
- Pipette tips for Gilson pipettes (Gilson, catalog numbers: F171101 , F171301 , F171501 )
- Sterile serological pipettes (5 ml, 10 ml, 25 ml)
- Electroporation cuvettes 4 mm gap (BTX, catalog number: 45-0126 )
- pSAG1::Cas9-U6::sgUPRT plasmid (Addgene, catalog number: 54467 ) (Shen et al., 2014)
- pYFP-AID-3HA, Floxed HXGPRT plasmid (Addgene, catalog number: 87260 ) (Long et al., 2017a)
- pYFP-mAID-3HA, Floxed HXGPRT plasmid (Addgene, catalog number: 87259 ) (Brown et al., 2017)
- T. gondii line RH TIR1-3FLAG (genotype: RHΔhxgprtΔku80; TUB1:TIR1-3FLAG, SAG1:CAT) (Brown et al., 2017; Long et al., 2017a)
- NEB5α chemically-competent E. coli with SOC medium (New England Biolabs, catalog number: C2987I )
- Human foreskin fibroblasts (HFF) (ATCC, catalog number: SCRC-1041 )
- Q5 Site-Directed Mutagenesis Kit with chemically competent E. coli (New England Biolabs, catalog number: E0554S )
- Mutagenesis primers for reprogramming pSAG1::Cas9-U6::sgUPRT (IDT, 25 nmole, standard desalting)
- 2x SDS-PAGE sample buffer (Sigma-Aldrich, catalog number: S3401 )
- 1 kb DNA ladder (New England Biolabs, catalog number: N3232 )
- Agarose (Fisher Scientific, catalog number: BP160 )
- SDS-PAGE 4-15% gradient Tris-glycine polyacrylamide gels (Bio-Rad Laboratories, catalog number: 4561086 )
- 6x Gel Loading Dye (New England Biolabs, catalog number: B7025 )
- LB broth (BD, catalog number: 244610 )
- Ampicillin (Sigma-Aldrich, catalog number: A9518 )
- Plasmid miniprep kit (Macherey-Nagel, catalog number: 740588 )
- M13 Reverse universal primer (5’-ACAGGAAACAGCTATGAC) (Genewiz)
- Q5 DNA Polymerase (New England Biolabs, catalog number: M0491 )
- dNTPs 10 mM each (New England Biolabs, catalog number: N0447 )
- Gene of interest tagging primers for amplifying (m)AID-3HA, Floxed HXGPRT tagging cassette with short homology flanks (IDT, 25 nmole, standard desalting)
- Gene of interest diagnostic tagging primers (IDT, 25 nmole)
- Agarose Gel and PCR Cleanup Kit (Macherey-Nagel, catalog number: 740609 )
- Trypsin-EDTA (Sigma-Aldrich, catalog number: T3924 )
- ATP (Sigma-Aldrich, catalog number: A6419 )
- Glutathione (Sigma-Aldrich, catalog number: G6013 )
- Mycophenolic acid (Sigma-Aldrich, catalog number: M3536 )
- Xanthine (Sigma-Aldrich, catalog number: X4002 )
- Proteinase K (Sigma-Aldrich, catalog number: P2308 )
- Taq polymerase (New England Biolabs, catalog number: M0273 )
- Ethanol (EtOH) (Pharmco-AAPER, catalog number: 11100020 )
- GelRed nucleic acid stain (Biotium, catalog number: 41001 )
- Licor anti-mouse 800CW secondary antibody (LI-COR, catalog number: 925-32210 )
- Licor anti-rabbit 680RD secondary antibody (LI-COR, catalog number: 925-68071 )
- Mouse anti-HA monoclonal antibody (BioLegend, catalog number: 901501 )
- Non-fat powdered milk (Nestle Carnation)
- Rabbit anti-Aldolase (T. gondii) (Starnes et al., 2009) or other T. gondii loading control antibody
- Tris base (Sigma-Aldrich, catalog number: T6066 )
- Glacial acetic acid (Fisher Scientific, catalog number: A38-500 )
- 0.5 M EDTA pH 8.0 (Merck, catalog number: 324504 )
- Boric acid (Sigma-Aldrich, catalog number: B6768 )
- Glycine (Sigma-Aldrich, catalog number: G7128 )
Note: This product has been discontinued. - Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: L5750 )
- Methanol (Fisher Scientific, catalog number: A412P )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P5405 )
- Sodium chloride (NaCl) (Fisher Scientific, catalog number: S271 )
- Sodium phosphate dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: S3264 )
- Potassium phosphate dibasic (K2HPO4) (Sigma-Aldrich, catalog number: P8281 )
- Tween-20 (Sigma-Aldrich, catalog number: P2287 )
- Dulbecco’s Modified Eagle’s Medium (DMEM) (Thermo Fisher Scientific, GibcoTM, catalog number: 12100046 )
- Sodium bicarbonate (Sigma-Aldrich, catalog number: S5761 )
- 200 mM L-glutamine (Thermo Fisher Scientific, GibcoTM, catalog number: 25030149 )
- 10 mg/ml gentamicin (Thermo Fisher Scientific, GibcoTM, catalog number: 15710072 )
- Characterized fetal bovine serum (FBS) (GE Healthcare, catalog number: SH30071.01HI )
- Hanks’ balanced salt solution (Sigma-Aldrich, catalog number: H9269 )
- HEPES (Sigma-Aldrich, catalog number: H3375 )
- EGTA (Merck, catalog number: 324626 )
- EDTA (Merck, catalog number: 324504 )
- Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: P5655 )
- Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M4880 )
- Calcium chloride dihydrate (CaCl2·2H2O) (Sigma-Aldrich, catalog number: C5080 )
- 3-indoleacetic acid (IAA/auxin) (Sigma-Aldrich, catalog number: I2886 )
- Agar (Fisher Scientific, catalog number: BP1423 )
- 10x Tris-Acetate-EDTA (TAE) buffer (see Recipes)
- 10x Tris-Borate-EDTA (TBE) buffer (see Recipes)
- 10x SDS-PAGE running buffer (see Recipes)
- 10x protein transfer buffer (see Recipes)
- 1x protein transfer buffer (see Recipes)
- 10x phosphate buffered saline (PBS) (see Recipes)
- Phosphate buffered saline + Tween-20 (PBST) (see Recipes)
- PCR lysis buffer (see Recipes)
- D10 medium (see Recipes)
- Hank’s balanced salt solution with HEPES and EGTA (HHE) (see Recipes)
- 0.1 M KPO4 buffer pH 7.6 (for Cytomix buffer) (see Recipes)
- Cytomix electroporation buffer pH 7.6 (Soldati and Boothroyd, 1993) (see Recipes)
- 500 mM 3-indoleacetic acid (IAA/auxin) (1,000x Stock) (see Recipes)
Equipment
- Autoclave (Steris, model: SG-120 )
- Benchtop centrifuge with 15 ml and 50 ml conical vial holders (Eppendorf, model: 5810 R )
- Biological safety cabinet (Baker, model: SterilGuard® II )
- CO2 incubator (Thermo Fisher Scientific, Thermo ScientificTM, model: FormaTM 310 )
- Detergent free pyrex glassware and stir bars for media preparation
- Electroporator (BTX, model: ECM 830 )
- Gel documentation system (Bio-Rad Laboratories, model: Gel DocTM XR+ )
- Incubating orbital shaker (VWR, model: Model 3500I )
- Inverted phase contrast microscope (Nikon Instruments, model: Eclipse TS100 )
- Hemacytometer (Sigma-Aldrich, model: Bright-LineTM )
- Licor Odyssey imaging system (LI-COR, model: Odyssey® CLx )
- Microcentrifuge (Eppendorf, model: 5417 R )
- Microvolume spectrophotometer (Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM One )
- Microwave (VWR, catalog number: 75856-526)
Manufacturer: Argos Technologies, catalog number: 111092 . - Pipet aid (Thermo Fisher Scientific, Thermo ScientificTM, model: S1 )
- Pipettes (Pipetman, Gilson, models: P2 , P20 , P200 , P1000 )
- Protein wet transfer blotting apparatus (Bio-Rad Laboratories, model: Mini Trans-Blot® )
- Refrigerator Freezer (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 10ECEETSA )
- SDS-PAGE electrophoresis apparatus (Bio-Rad Laboratories, model: Mini-Protean Tetra Cell )
- Standard orbital shaker (VWR, model: Model 1000 )
- Thermal cycler (Thermo Fisher Scientific, Applied BiosystemsTM, model: VeritiTM 96-well )
Software
- Google Chrome (Google) or other web browsing software
- Snapgene (Snapgene) or other plasmid viewing software
- Image studio lite (LI-COR) or other gel analysis software
- Axiovision (Carl Zeiss Microscopy) or other cellular imaging software
- ImageJ (Developed by Wayne Rasband) or other image quantification software
- GraphPad Prism (GraphPad Software Inc.) or other graphing and statistical analysis software
Procedure
- Generating gene-specific Cas9/sgRNA plasmids for C-terminal tagging
- Genomic sequence retrieval
- From www.toxodb.org, navigate to the gene of interest (GOI)’s gene page (Figure 2A). In general GT1 sequences are used when working with the RH TIR1-3FLAG line. Click the ‘Download Gene’ link above the Gene ID (Figure 2A).
- From the ‘Download Gene’ page on ToxoDB.org, download the 3’ tagging region by selecting the following options: Choose a Report: FASTA; Type of sequence: Genomic; Region: Translation stop -80, Translation stop +200; Download type: Show in browser. Select ‘Get Sequences’ link (Figure 2B).
- From the sequence retrieval page, select the genomic sequence without the FASTA identifiers and copy to clipboard (Figure 2C).
- From www.toxodb.org, navigate to the gene of interest (GOI)’s gene page (Figure 2A). In general GT1 sequences are used when working with the RH TIR1-3FLAG line. Click the ‘Download Gene’ link above the Gene ID (Figure 2A).
- Protospacer selection
- From the Eukaryotic Pathogen CRISPR guide RNA/DNA Design Tool webpage (EuPaGDT, http://grna.ctegd.uga.edu/), begin by entering a ‘Job Name’. Use the default settings recommended for SpCas9: ’20 nt gRNA NGG PAM’. Under the genome selection tab, select the T. gondii lineage and database (e.g., T. gondii GT1 ToxoDB-32). Paste the copied genomic sequence from ToxoDB.org and select ‘Get guide RNA’ (Figure 2D).
- From the gRNA output page on http://grna.ctegd.uga.edu/, select a 20 nt gRNA protospacer sequence with an NGG PAM downstream of the translation stop codon (e.g., STOP-N…N<100GATCGACTTAGTAACGCATCCGG) (Figure 2E). Optional: Add a ‘G’ to the 5’ end of the protospacer sequence if it does not begin with one to enhance sgRNA transcription (Doench et al., 2014).
- From the Eukaryotic Pathogen CRISPR guide RNA/DNA Design Tool webpage (EuPaGDT, http://grna.ctegd.uga.edu/), begin by entering a ‘Job Name’. Use the default settings recommended for SpCas9: ’20 nt gRNA NGG PAM’. Under the genome selection tab, select the T. gondii lineage and database (e.g., T. gondii GT1 ToxoDB-32). Paste the copied genomic sequence from ToxoDB.org and select ‘Get guide RNA’ (Figure 2D).
- Mutagenesis of pSAG1::Cas9-U6::sgUPRT (Addgene #54467) (Shen et al., 2014)
- The plasmid pSAG1::Cas9-U6::sgUPRT transiently expresses SpCas9-HA-GFP nuclease and a single guide RNA (sgRNA) that binds Cas9 and directs it to perform a double stranded break in the TgUPRT gene. The gene target specificity of the Cas9/sgRNA ribozyme is based on a 20 nt protospacer sequence at the 5’-most end of the sgRNA. To switch the Cas9 target from TgUPRT to a different gene, the 20 nt protospacer sequence will need to be changed to the one designed in Step A2 Protospacer Selection. Use Q5 Site-Directed Mutagenesis (New England Biolabs) to mutate pSAG1::Cas9-U6::sgUPRT, changing the 20 nt TgUPRT protospacer to the 20 nt protospacer selected for the gene of interest (Figure 2E). A standard 25 µl reaction consists of:
1 µl (1 ng) pSAG1::Cas9-U6::sgUPRT
1.25 µl (10 µM) reverse primer: (5’-AACTTGACATCCCCATTTAC)
1.25 µl (10 µM) forward primer:
(5’-(G)NNNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAGAAATAGC)
9 µl deionized H2O
12.5 µl 2x Q5 Hot-Start PCR mix - Q5 Mutagenesis thermal cycling
Step 1. Denature at 98 °C for 30 sec
Step 2. Denature at 98 °C 10 sec
Step 3. Anneal at 55 °C 30 sec
Step 4. Extend at 72 °C for 5 min
Step 5. Repeat Steps 2-4 24 times
Step 6. Extend at 72 °C for 2 min
Step 7. Hold at 4 °C until further use. - Resolve 1 µl of PCR reaction on a 0.8% agarose gel (TAE or TBE) with a 1 kb DNA ladder to check for amplification. The expected amplicon size is 9.68 kb.
- Kinase, Ligase, Digest (KLD) reaction (included in the Q5 Site-Directed Mutagenesis Kit)
1 µl PCR product
3 µl deionized H2O
5 µl 2x KLD buffer
1 µl 10x KLD enzyme mix
Mix and incubate at room temperature for 5 min. - Transform NEB5α chemically-competent E. coli with 1 µl of the KLD reaction. Incubate on ice for 30 min. Heat chemically-transformed cells for 30 sec at 42 °C. Add 950 µl SOC medium (New England Biolabs) to transformed cells and incubate at 37 °C with mixing for 1 h. Spread 100 µl of transformed E. coli onto LB agar plates with 100 µg/ml ampicillin. Incubate plates at 37 °C overnight. The next day, pick 2-3 colonies from each LB agar plate to grow up in 5 ml LB broth + 100 µg/ml ampicillin overnight cultures for plasmid extraction.
- Pellet overnight bacterial cultures at 3,000 x g with a benchtop centrifuge for 10 min.
- Extract plasmid DNA from bacterial pellets using a plasmid extraction kit (Macherey-Nagel). Pre-warm sterile-filtered Buffer EB to 70 °C. Elute DNA into a sterile 1.7 ml microfuge tube with 50 µl pre-warmed sterile-filtered Buffer EB.
- Measure DNA concentration and purity by spectrophotometry (absorbance at 260 nm/280 nm, NanoDrop (Thermo Fisher)). The expected yield is approximately 500-1,000 ng/µl.
- Confirm mutated protospacer sequence of pSAG1::Cas9-U6::sg”GOI” by Sanger sequencing using the M13 reverse universal primer (5’-ACAGGAAACAGCTATGAC) (Genewiz).
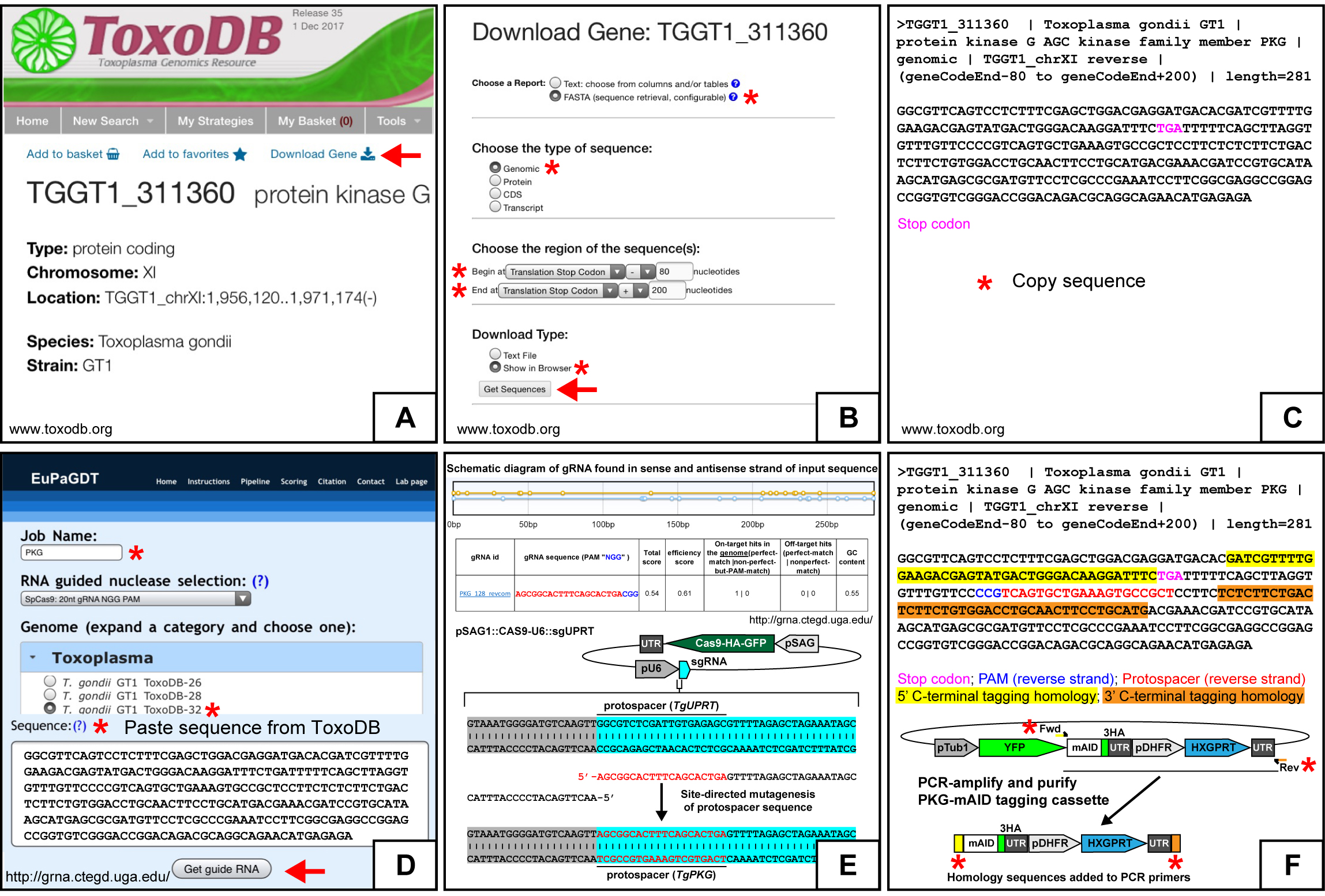
Figure 2. Generation of constructs for C-terminal mAID tagging. A-E. Generating gene-specific Cas9/sgRNA plasmids. The gene page of TgPKG (TGGT1_311360; example gene for C-terminal tagging) from ToxoDB.org. The red arrow points to the download gene link for acquiring the TgPKG genomic sequence needed. B. The ‘Download Gene’ page for TgPKG from ToxoDB.org. The red asterisks highlight fields that need to be entered as shown. The red arrow points to the get sequences link. C. The FASTA genomic sequence of TgPKG (-80 to +200 from the translation stop codon) from ToxoDB.org needed for gRNA and donor homology design for C-terminal tagging. D. Guide RNA design tool (cropped to fit) from EuPaGDT (http://grna.ctegd.uga.edu/). The red asterisks indicate the basic fields that need to be completed. The red arrow points to the ‘Get guide RNA’ link. E. Output of gRNA protospacers for TgPKG genomic sequence (-80 to +200 from the translation stop codon) on each strand from EuPaGDT (upper schematic). Selected gRNA protospacer and protospacer adjacent motif (PAM) sequence with quality scores from EuPaGDT (table). Swapping the TgUPRT protospacer with the designed TgPKG protospacer in pSAG1::Cas9-U6::sgUPRT by site-directed mutagenesis (lower schematic). F. Generation of a TgPKG-specific C-terminal mAID-3HA, HXGPRT tagging cassette. The FASTA genomic sequence of TgPKG (-80 to +200 from the translation stop codon) from ToxoDB.org with the indicated highlighted features. PCR amplification of mAID-3HA, HXGPRT tagging cassette from pYFP-mAID-3HA, Floxed HXGPRT with 5’ and 3’ regions of donor homology (40 bp) to TgPKG (flanking the Cas9 break-site) added to the forward and reverse primers, respectively (asterisks).
- The plasmid pSAG1::Cas9-U6::sgUPRT transiently expresses SpCas9-HA-GFP nuclease and a single guide RNA (sgRNA) that binds Cas9 and directs it to perform a double stranded break in the TgUPRT gene. The gene target specificity of the Cas9/sgRNA ribozyme is based on a 20 nt protospacer sequence at the 5’-most end of the sgRNA. To switch the Cas9 target from TgUPRT to a different gene, the 20 nt protospacer sequence will need to be changed to the one designed in Step A2 Protospacer Selection. Use Q5 Site-Directed Mutagenesis (New England Biolabs) to mutate pSAG1::Cas9-U6::sgUPRT, changing the 20 nt TgUPRT protospacer to the 20 nt protospacer selected for the gene of interest (Figure 2E). A standard 25 µl reaction consists of:
- Genomic sequence retrieval
- Generation of a gene-specific AID tagging cassette
- Microhomology flank selection and primer design
- The 5’ microhomology flank is the last 40 nt of the gene of interest that immediately precedes the translation stop codon. The 3’ microhomology flank will be the first 40 nt that immediately follows the selected protospacer/PAM site. These flanks will be appended to the (m)AID tagging cassette by PCR using ~60 nucleotide DNA oligo primers (Figure 2F).
- Order tagging primers. The tagging forward primer will be the 40 nt 5’ homology flank + GCTAGCAAGGGCTCGGGC. The tagging reverse primer will be the 40 nt 3’ homology flank (reverse complement) + ATAGGGCGAATTGGAGCTCC. We purchase 25 nmole DNA oligo primers with ‘standard desalting’ from Integrated DNA Technologies (www.idtdna.com).
- The 5’ microhomology flank is the last 40 nt of the gene of interest that immediately precedes the translation stop codon. The 3’ microhomology flank will be the first 40 nt that immediately follows the selected protospacer/PAM site. These flanks will be appended to the (m)AID tagging cassette by PCR using ~60 nucleotide DNA oligo primers (Figure 2F).
- PCR amplification and clean-up of an AID tagging amplicon
- Q5 PCR set up. Set up the Q5 PCR reaction using pYFP-mAID-3HA, Floxed HXGPRT (Addgene #87259) (Brown et al., 2017) or pYFP-AID-3HA, Floxed HXGPRT (Addgene #87260) (Long et al., 2017b) plasmids as the template (Figure 2F). A standard 500 µl reaction consists of:
100 µl 5x Q5 Buffer
10 µl dNTPs
5 µl (50 µM) tagging forward primer (Figure 2F)
5 µl (50 µM) tagging reverse primer (Figure 2F)
5 µl (5 ng) plasmid template
5 μl Q5 DNA polymerase
370 µl deionized H2O
Split the PCR mix into 5 PCR tubes, 100 µl each - Q5 PCR thermal cycling.
Step 1. Denature at 98 °C for 30 sec
Step 2. Denature at 98 °C for 10 sec
Step 3. Anneal at 60 °C for 30 sec
Step 4. Extend at 72 °C for 2.5 min
Step 5. Repeat Steps 2-4 29 times
Step 6. Extend at 72 °C for 2.5 min
Step 7. Hold at 4 °C until further use - Checking PCR quality. Resolve 1 µl of PCR reactions on 0.8% agarose gel (TAE or TBE) with a 1 kb DNA ladder to check for amplification. The tagging amplicon will consist of a 5’ homology flank-linker-(m)AID-3HA, Floxed HXGPRT-3’ homology flank (expected amplicon sizes: 2.9 kb for mAID, 3.4 kb for AID).
- PCR cleanup with a PCR Cleanup Kit (Macherey-Nagel). Pool the 5 PCR reactions and add 1,000 µl Buffer NTI. Load 750 µl mix into DNA column. Centrifuge at 11,000 x g with a microfuge for 1 min. Discard flow through. Load remaining 750 µl mix into the column. Centrifuge at 11,000 x g for 1 min. Discard flow through. Wash column with 700 µl Buffer A3. Centrifuge at 11,000 x g for 1 min. Discard flow through. Repeat wash step and discard flow through. Centrifuge additional 3 min at 11,000 x g. Pre-warm sterile-filtered Buffer EB to 70 °C. Elute DNA into a sterile 1.7 ml microfuge tube with 30 µl pre-warmed sterile-filtered Buffer EB.
- Measure DNA concentration and purity by spectrophotometry (Absorbance at 260 nm/280 nm, NanoDrop (Thermo Fisher). Aim for having at least 10 µg tagging cassette for parasite transfection. Repeat PCR and cleanup steps if more DNA is needed.
- Q5 PCR set up. Set up the Q5 PCR reaction using pYFP-mAID-3HA, Floxed HXGPRT (Addgene #87259) (Brown et al., 2017) or pYFP-AID-3HA, Floxed HXGPRT (Addgene #87260) (Long et al., 2017b) plasmids as the template (Figure 2F). A standard 500 µl reaction consists of:
- Microhomology flank selection and primer design
- Cell culture
- Human foreskin fibroblasts (HFF) culture
- Starter cultures. Grow starter HFF cells in 35 ml D10 medium (see Recipes) in sterile T-175 flasks in a 37 °C 5% CO2 incubator.
- HFF passage. Once confluent, remove D10 medium and wash with 35 ml sterile 1x PBS and discard wash. Add 5 ml trypsin, replace cap, and incubate cells at 37 °C for 1-5 min until cells detach from one another (as detected using 10x magnification). Firmly tap the side of the flask a few times forcefully to detach cells from the plastic. Add 175 ml fresh D10 medium to trypsinized cells and mix to generate a 1:5 cell suspension. Passage 35 ml into new T-175. Passage 5 ml into T-25s to make HFF feeder flasks to support Toxoplasma gondii culture. Passage 200 µl per well of 96-well plates and 1 ml per well of 24-well plates for parasite cloning following transfection.
- Starter cultures. Grow starter HFF cells in 35 ml D10 medium (see Recipes) in sterile T-175 flasks in a 37 °C 5% CO2 incubator.
- Parasite culture
- Parasite information. The T. gondii line RH TIR1-3FLAG (genotype: RHΔhxgprtΔku80; TUB1:TIR1-3FLAG, SAG1:CAT) is needed as it provides three key elements for the AID system: Δku80 restricts non-homologous end-joining, reducing random integration of the AID tagging cassette; Δhxgprt provides the proper genetic background for positive selection of the AID tagging cassette based on HXGPRT-mediated resistance to Mycophenolic acid; TIR1-3FLAG is the plant auxin receptor required for in auxin induced SCFTIR1-targeting of AID-tagged proteins for proteasomal degradation.
- Parasite passage. Inoculate confluent HFF T-25 feeder flasks with RH TIR1-3FLAG parasites using 350 µl of freshly egressed parasites every two days or immediately following natural egress as needed. Culture parasites in 37 °C 5% CO2 in D10 medium.
- Parasite information. The T. gondii line RH TIR1-3FLAG (genotype: RHΔhxgprtΔku80; TUB1:TIR1-3FLAG, SAG1:CAT) is needed as it provides three key elements for the AID system: Δku80 restricts non-homologous end-joining, reducing random integration of the AID tagging cassette; Δhxgprt provides the proper genetic background for positive selection of the AID tagging cassette based on HXGPRT-mediated resistance to Mycophenolic acid; TIR1-3FLAG is the plant auxin receptor required for in auxin induced SCFTIR1-targeting of AID-tagged proteins for proteasomal degradation.
- Human foreskin fibroblasts (HFF) culture
- (m)AID tagging using CRISPR genome editing (e.g., TgPKG-mAID-3HA; Figure 3)
- Parasite transfection
- Culture parasites for transfection. Infect confluent host cells grown in T-25 flasks with sufficient RH TIR1-3FLAG parasites (95 µl of freshly egressed parasites from T-25) to achieve around 75% host cell lysis 2 days post-infection. Scrape the monolayers and pass the cell suspension through a 22 G blunt-end needle 3-5 times to disrupt the host cells and release all parasites. Avoid needle sticks and carefully dispose of used needles into a biohazard sharps container. Pre-wet 3.0 μm polycarbonate membranes by passing 5 ml HHE through the filter with a 10 ml syringe into a waste container. Pass the syringe-lysed parasites through the filter with a 10 ml syringe into a 50 ml polystyrene conical vial. Pass an additional 5 ml HHE through the filter into the 50 ml conical to collect additional parasites from the filter. Centrifuge the filtered culture at 400 x g with a benchtop centrifuge for 10 min at 18 °C, resuspend the pellets in 10 ml HHE, count the cell density with a hemacytometer, and centrifuge again under the same conditions. Subsequently, resuspend the parasite cell pellet in Cytomix buffer (see Recipes) to achieve a parasite density of 4 x 107 ml-1.
- Prepare the transfection mix in an electroporation cuvette
250 µl parasites in Cytomix buffer
10 µg GOI-specific CRISPR/Cas9 plasmid (Figures 2A-2E)
10 µg GOI-specific AID-tagging PCR amplicon (Figure 2F)
3 µl ATP (0.2 M, 100x)
3 µl glutathione (0.5 M, 100x)
Adjust to 300 µl with Cytomix buffer. - Electroporation. Electroporate parasites according to electroporator manufacturer’s instructions. We use the BTX ECM-830 electroporator with 4 mm gap cuvettes, with the following parameters:
1,700 V
176 μsec of pulse length
2 pulses
100 msec interval between pulses
Immediately after electroporation, transfer the parasites into fresh T-25 flasks with confluent host cells and grow them at 37 °C, 5% CO2.
- Culture parasites for transfection. Infect confluent host cells grown in T-25 flasks with sufficient RH TIR1-3FLAG parasites (95 µl of freshly egressed parasites from T-25) to achieve around 75% host cell lysis 2 days post-infection. Scrape the monolayers and pass the cell suspension through a 22 G blunt-end needle 3-5 times to disrupt the host cells and release all parasites. Avoid needle sticks and carefully dispose of used needles into a biohazard sharps container. Pre-wet 3.0 μm polycarbonate membranes by passing 5 ml HHE through the filter with a 10 ml syringe into a waste container. Pass the syringe-lysed parasites through the filter with a 10 ml syringe into a 50 ml polystyrene conical vial. Pass an additional 5 ml HHE through the filter into the 50 ml conical to collect additional parasites from the filter. Centrifuge the filtered culture at 400 x g with a benchtop centrifuge for 10 min at 18 °C, resuspend the pellets in 10 ml HHE, count the cell density with a hemacytometer, and centrifuge again under the same conditions. Subsequently, resuspend the parasite cell pellet in Cytomix buffer (see Recipes) to achieve a parasite density of 4 x 107 ml-1.
- Drug selection and parasite cloning
- Drug selection. At 24 h after electroporation and recovery, begin drug selection for the (m)AID-3HA, Floxed HXGPRT construct using mycophenolic acid (25 µg/ml) supplemented with xanthine (50 µg/ml). Repeat passages until the drug-resistant pool becomes stable (usually 2-3 passages until the culture stabilizes (i.e., lyses the monolayer every two days with a 350 µl inoculum of freshly egressed parasites).
- Parasite cloning by limiting dilution. Following natural egress of the drug-resistant population, purify and count the parasites as described above. Dilute the purified parasites with D10 medium (without drug) to achieve 3 parasites/150 μl. Subsequently, add 150 μl/well of this diluted culture into 96-well plates with confluent host cells and let the parasites grow at 37 °C, 5% CO2 for 7 days without movement to allow plaques (macroscopic focal destruction of the host cell monolayer by lytic parasite replication) to develop.
- Drug selection. At 24 h after electroporation and recovery, begin drug selection for the (m)AID-3HA, Floxed HXGPRT construct using mycophenolic acid (25 µg/ml) supplemented with xanthine (50 µg/ml). Repeat passages until the drug-resistant pool becomes stable (usually 2-3 passages until the culture stabilizes (i.e., lyses the monolayer every two days with a 350 µl inoculum of freshly egressed parasites).
- Screening clones for genetic recombination
- Identifying clones. After plaque development, visually check each well of the 96-well plates under an inverted-phase contrast microscope (such as a Nikon TS100) and look for wells that contain only one plaque (Refer to Ufermann et al., 2017 for high resolution examples of plaque morphology). Transfer the parasites of positive wells into 24-well plates containing confluent host cell monolayers and grow them until they naturally egress.
- Making lysates for diagnostic PCR. Inoculate a small aliquot (e.g., 10 μl) from each well into a new 24-well plate containing a confluent monolayer of host cells to continue culturing the parasites. The rest of the lysed out culture will be used for diagnostic PCR. Add 1 ml egressed parasite clones into 1.7 ml microfuge tubes. For a negative control, add 1 ml of egressed wild-type parasites (the parental line) to a separate 1.7 ml microfuge tube. Pellet the parasite suspensions at 400 x g with a microfuge for 10 min at 18 °C and remove supernatant from parasite pellet. To each pellet, add 100 μl Proteinase K (PK) (0.2 mg/ml) in 1x PBS and mix. Heat at 37 °C for 10 min, 50 °C 10 min, and 98 °C for 10 min. Freeze samples or chill on ice and proceed with diagnostic PCR.
- Diagnostic PCR set up. Two simple PCRs should be performed to determine the tagging success. PCR1 should exclusively amplify the unmodified gene locus, while PCR2 should exclusively amplify the tagged gene locus (Figure 3A). Each 25 μl diagnostic PCR reaction consists of:
2.5 μl 10x Taq buffer
0.5 μl dNTPs
0.25 μl (50 μM) forward primer (same for PCR1 and PCR2)
0.25 μl (50 μM) reverse primer (unique for PCR1 and PCR2)
1 μl PK-treated parasite lysate
0.25 μl Taq DNA polymerase (New England Biolabs)
20.25 μl deionized H2O
The forward primer for PCR1 and PCR2 should prime ~350 bp upstream (5’ side) of the translation stop codon. The PCR1 reverse primer should prime in the 3’ UTR, ~300 bp downstream of the translation stop codon. The PCR2 reverse primer should prime in the mAID or AID sequence ~+50 from start of linker. - Diagnostic PCR thermal cycling
Step 1. Denature at 98 °C for 30 sec
Step 2. Denature at 98 °C 10 sec
Step 3. Anneal at 60 °C 30 sec
Step 4. Extend at 68 °C for 1 min
Step 5. Repeat Steps 2-4 29 times
Step 6. Extend at 68 °C for 1 min
Step 7. Hold at 4 °C until further use - Resolving diagnostic PCR amplicons. Resolve 10 µl of PCR reactions on 1% agarose gel (TAE or TBE) with a 1 kb DNA ladder to check for positive amplification (Figure 3C). PCR1 should yield an amplicon of ~650 bp for the wild-type, unmodified locus. PCR2 should yield an amplicon of ~400 bp for successfully tagged clones. PCR positive clones can be further examined by other means such as immunofluorescence microscopy (Figure 3D) or Western blotting (Figure 3E) for the (m)AID-3HA epitope tag using anti-HA antibodies.
- Subcloning tagged clones. Once positive clones are identified and confirmed, they should be transferred from 24-well plates into T-25 flasks and cryopreserved for future use.
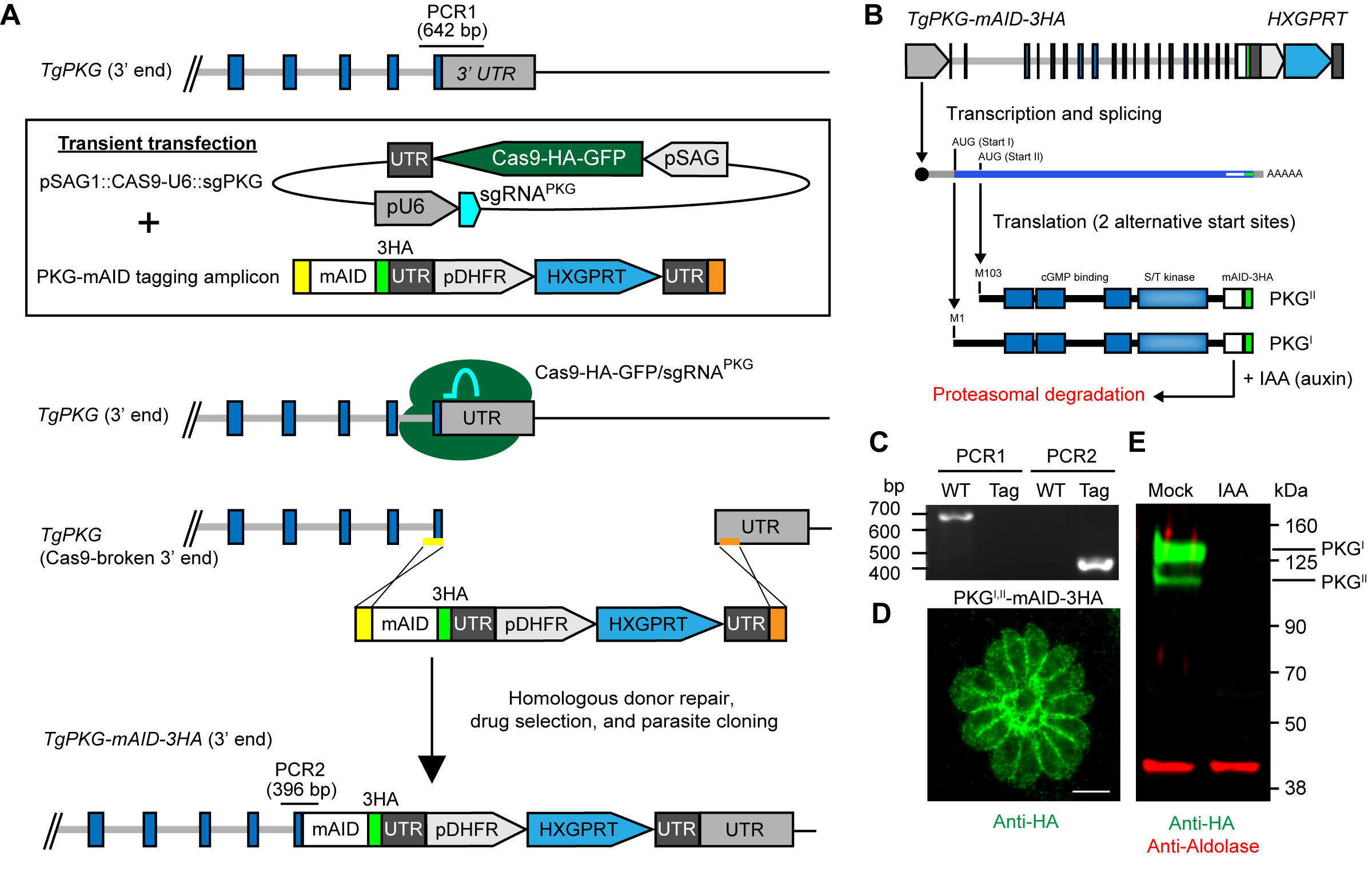
Figure 3. Generation and regulation of (m)AID protein fusions. A. CRISPR-mediated C-terminal mAID tagging in RH TIR1-3FLAG line. The example shown here is the gene encoding protein kinase G (TgPKG, reproduced with permission from [Brown et al., 2017]). Note the locations of diagnostic PCRs 1 and 2. B. Schematic of TgPKG-mAID-3HA expression showing the two PKG-mAID isoforms that are generated from alternative translation initiation sites on the transcript. Addition of IAA promotes the simultaneous degradation of both PKG-mAID isoforms. C-D. Data reproduced with modification for space limits from (Brown et al., 2017). C. Diagnostic PCRs 1 and 2 from genomic DNA samples showing successful integration of the mAID tagging construct into TgPKG (see A for PCR positions). WT (wild type), TIR1-3FLAG parent; Tag, PKGI,II-mAID-3HA parasites. D. Immunofluorescence of PKG-mAID-3HA isoforms I and II stained with anti-HA mAb and detected with Alexa Fluor 488-conjugated secondary antibody. Scale bar = 5 µm. E. Western blot assay of lysed PKGI,II-mAID-3HA parasites probed with antibodies recognizing HA (green) and aldolase (red). Parasites were treated with 500 µM IAA or the vehicle (EtOH) for 4 h prior to lysis.
- Identifying clones. After plaque development, visually check each well of the 96-well plates under an inverted-phase contrast microscope (such as a Nikon TS100) and look for wells that contain only one plaque (Refer to Ufermann et al., 2017 for high resolution examples of plaque morphology). Transfer the parasites of positive wells into 24-well plates containing confluent host cell monolayers and grow them until they naturally egress.
- Parasite transfection
- Depletion of AID-tagged proteins
- Parasite culture. Infect HFF cultures with RH TIR1-3FLAG and RH POI-(m)AID-3HA parasites. The type (e.g., dish, well plate, flask) and number of HFF monolayers to infect will depend on the experiment but at least two cultures are needed per parasite line for auxin and vehicle treatment.
- Auxin treatment. Use auxin at 1:1,000 to deplete (m)AID-tagged proteins at a final concentration of 500 µM. Mock treatment consists of an equivalent volume of 100% EtOH at a final concentration of 0.0789% (w/v). Incubate at 37 °C. Incubation times needed to achieve complete protein knockdown will vary based on protein localization, stability, and abundance. As little as 15 min has been used to achieve complete knockdown of soluble cytosolic (m)AID-fusions such as YFP (Brown et al., 2017; Long et al., 2017b). Following knockdown, parasites can be examined for phenotypes directly, harvested for follow-up assays, or lysed for analysis of (m)AID-tagged protein levels.
- Detection of AID-tagged protein levels. The 3HA portion of the (m)AID construct can be used for protein detection by immunofluorescence microscopy (Figure 3D) or Western blotting (Figure 3E) using anti-HA antibodies (Figure 2).
- Parasite culture. Infect HFF cultures with RH TIR1-3FLAG and RH POI-(m)AID-3HA parasites. The type (e.g., dish, well plate, flask) and number of HFF monolayers to infect will depend on the experiment but at least two cultures are needed per parasite line for auxin and vehicle treatment.
Data analysis
Quantification of Western blot data of (m)AID fusion knockdowns performed in triplicate can be performed using ImageJ densitometry software. We recommend viewing an online resource created by Dr. Luke Miller of San Jose State University (http://lukemiller.org/index.php/2010/11/analyzing-gels-and-western-blots-with-image-j/) for detailed guidelines for ImageJ densitometry.
- In ImageJ, open the grayscale TIF image of the (m)AID-3HA anti-HA Western blot. File -> open ->.tif
- With the rectangle tool, draw a vertical rectangle around the protein band in the first lane. The rectangle must be taller than it is wide.
- Press Ctrl + 1 to select the first lane. A ‘1’ will appear in the rectangle.
- Left click the rectangle and drag it to the next lane.
- Press Ctrl + 2 to select the second lane. A ‘2’ will appear in the second rectangle.
- Left click the newest rectangle and move to the third lane. Repeat Steps 5 and 6 until every lane has a rectangle.
- Press Ctrl + 3 to plot the data in a histogram. A new window will pop up.
- With a line drawing tool, draw a line under each peak that connects the background line on the left to the background line on the right.
- After all background lines are drawn, use the ‘magic wand’ tool to click inside of each peak, starting at the top (lane 1), and working downward.
- The density values for each peak (band) will be found in a new pop-up table.
- Repeat Steps 1-11 for the loading control blot.
- Normalize for total protein (AID-3HA density/loading control density) for each sample then transform this ratio as a percentage of the untreated control (normalized sample value/normalized control ratio x 100).
- The data from triplicate experiments can be averaged and analyzed for statistical significance using GraphPad Prism software according to manufacturer’s instructions.
Notes
The T. gondii line RH TIR1-3FLAG (genotype: RHΔhxgprtΔku80; TUB1:TIR1-3FLAG, SAG1:CAT) (Brown et al., 2017; Long et al., 2017b) has been submitted to the ATCC for distribution. To apply the auxin degron system to other T. gondii strains, the plasmid pTUB1:OsTIR1-3FLAG, SAG1:CAT (Addgene #87258) is available for transfection. Please visit http://www.addgene.org/David_Sibley/ for other AID-related plasmids.
Recipes
- 10x Tris-Acetate-EDTA (TAE) buffer
48.4 g Tris base
11.42 ml glacial acetic acid
20 ml 0.5 M EDTA pH 8.0
Q.S. to 1 L with deionized H2O
Store at room temperature - 10x Tris-Borate-EDTA (TBE) buffer
108 g Tris base
55 g boric acid
40 ml 0.5 M EDTA pH 8.0
Q.S. to 1 L with deionized H2O
Store at room temperature - 10x SDS-PAGE running buffer
30 g Tris base
144 g glycine
10 g SDS
Q.S. to 1 L with deionized H2O
Store at room temperature - 10x protein transfer buffer
30.3 g Tris base
144 g glycine
Q.S. to 1 L with deionized H2O - 1x protein transfer buffer
700 ml deionized H2O
200 ml methanol
100 ml 10x protein transfer buffer - 10x phosphate buffered saline (PBS)
2 g KCl
80 g NaCl
14.4 g Na2HPO4
2 g KH2PO4
Q.S. to 1 L with deionized H2O
Store at room temperature - Phosphate buffered saline + Tween-20 (PBST)
899 ml deionized H2O
100 ml 10x PBS
1 ml Tween-20
Store at room temperature - PCR lysis buffer
0.2 mg/ml Proteinase K in PBS - D10 medium
1 packet of DMEM powder
3.7 g sodium bicarbonate
2.38 g HEPES
10 ml of 200 mM L-glutamine
1 ml of 10 mg/ml gentamicin
100 ml Characterized fetal bovine serum
Q.S. to 1 L with deionized H2O
Filter sterilize with 1 L Stericup Filter and store at 4 °C - Hank’s balanced salt solution with HEPES and EGTA (HHE)
Hanks’ balanced salt solution containing 10 mM HEPES and 1 mM EGTA - 0.1 M KPO4 buffer pH 7.6 (for Cytomix buffer)
17.4 g K2HPO4
13.6 g KH2PO4
Q.S. to 1 L with deionized H2O - Cytomix electroporation buffer pH 7.6 (Soldati and Boothroyd, 1993)
100 ml 0.1 M KPO4 pH 7.6
8.95 g KCl
22.1 mg CaCl2
1.02 g MgCl2
5.96 g HEPES
744 mg EDTA
Q.S. to 1 L with deionized H2O
Sterile filter and store at 4 °C - 500 mM 3-indoleacetic acid (IAA/auxin) (1,000x stock)
87.59 mg 3-indoleacetic acid into 1 ml 100% EtOH
Store at -20 °C, protect from light
Acknowledgments
We would like to thank Dr. Nisha Philip and Dr. Andy Waters for helpful discussions and sharing plasmids for the development of the AID protocol in T. gondii. We have no conflicts of interest to declare.
References
- Brown, K. M., Long, S. and Sibley, L. D. (2017). Plasma membrane association by N-acylation governs PKG function in Toxoplasma gondii. MBio 8(3).
- Doench, J. G., Hartenian, E., Graham, D. B., Tothova, Z., Hegde, M., Smith, I., Sullender, M., Ebert, B. L., Xavier, R. J. and Root D. E. (2014). Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol 32(12):1263-7.
- Kreidenweiss, A., Hopkins, A.V., and Mordmuller, B. (2013). 2A and the auxin-based degron system facilitate control of protein levels in Plasmodium falciparum. PLoS One 8(11): e78661.
- Long, S., Brown, K. M., Drewry, L. L., Anthony, B., Phan, I. Q. H. and Sibley, L. D. (2017a). Calmodulin-like proteins localized to the conoid regulate motility and cell invasion by Toxoplasma gondii. PLoS Pathog 13(5): e1006379.
- Long, S., Anthony, B., Drewry, L., and Sibley, L. D. (2017b). A conserved ankyrin repeat-containing protein regulates conoid stability, motility and cell invasion in Toxoplasma gondii. Nat Commun 8(1): 2236.
- Nishimura, K., Fukagawa, T., Takisawa, H., Kakimoto, T. and Kanemaki, M. (2009). An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6(12): 917-922.
- Philip, N. and Waters, A. P. (2015). Conditional degradation of Plasmodium calcineurin reveals functions in parasite colonization of both host and vector. Cell Host Microbe 18(1): 122-31.
- Shen, B., Brown, K., Long, S. and Sibley, L. D. (2017). Development of CRISPR/Cas9 for efficient genome editing in Toxoplasma gondii. Methods Mol Biol 1498: 79-103.
- Shen, B., Brown, K. M., Lee, T. D. and Sibley, L. D. (2014). Efficient gene disruption in diverse strains of Toxoplasma gondii using CRISPR/Cas9. MBio 5(3): e01114-01114.
- Sidik, S. M., Hackett, C. G., Tran, F., Westwood, N. J. and Lourido, S. (2014). Efficient genome engineering of Toxoplasma gondii using CRISPR/Cas9. PLoS One 9(6): e100450.
- Soldati, D. and Boothroyd, J. C. (1993). Transient transfection and expression in the obligate intracellular parasite Toxoplasma gondii. Science 260(5106): 349-352.
- Starnes, G. L., Coincon, M., Sygusch, J. and Sibley, L. D. (2009). Aldolase is essential for energy production and bridging adhesin-actin cytoskeletal interactions during parasite invasion of host cells. Cell Host Microbe 5:353-364.
- Teale, W. D., Paponov, I. A. and Palme, K. (2006). Auxin in action: signalling, transport and the control of plant growth and development. Nat Rev Mol Cell Biol 7(11): 847-859.
- Ufermann, C-M., Muller, F., Frohnecke, N., Laue, M. and Seeber, F. (2017). Toxoplasma gondii plaque assays revisited: Improvements for ultrastructural and quantitative evaluation of lytic parasite growth. Exp Parasitol 180:19-26.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Brown, K. M., Long, S. and Sibley, L. D. (2018). Conditional Knockdown of Proteins Using Auxin-inducible Degron (AID) Fusions in Toxoplasma gondii. Bio-protocol 8(4): e2728. DOI: 10.21769/BioProtoc.2728.
Category
Microbiology > Microbial genetics > Mutagenesis
Molecular Biology > Protein > Targeted degradation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.