- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Generation of Chemically Induced Liver Progenitors (CLiPs) from Rat Adult Hepatocytes
Published: Vol 8, Iss 2, Jan 20, 2018 DOI: 10.21769/BioProtoc.2689 Views: 11631
Reviewed by: Nicoletta CordaniSara JohnsonXiaoyi Zheng
Original research article
The authors used this protocol in:

Jan 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Primary mature hepatocytes (MHs) or their progenitor cells are candidate cell sources for cell transplantation therapy in severe liver diseases. However, stable culture of these cells or generation of equivalent cells from pluripotent stem cells has been limited. Using a cocktail of small molecules that we previously found useful in stable culture of multiple types of stem/progenitor cells, we recently established a novel method to generate bipotent liver progenitor cells, named chemically induced liver progenitors (CLiPs), from adult rat MHs. Here, we describe a detailed protocol for the induction of rat CLiPs. We first describe the method to isolate primary rat MHs and then describe how to induce CLiPs from these MHs. In addition, we describe a method to evaluate the bipotentiality of generated CLiPs to differentiate into hepatocytes and biliary epithelial cells. We also describe how to establish stable CLiPs through long-term culture with detailed example data. Primary CLiPs can be generated within 2 weeks, and stable CLiPs, which undergo 10 passages, can be established within 2.5-4 months with batch-to-batch variability.
Keywords: HepatocyteBackground
There is a strong demand for a novel cell source to realize regenerative medicine for liver diseases. The only current treatment for end-stage liver diseases is liver transplantation, but its application is limited due to donor shortages. Recently, our group proposed an approach to generate a novel type of LPC that can stably expand in vitro and can repopulate injured liver of chronic hepatitis animal models with an extensive efficiency (Katsuda et al., 2017). Using a cocktail of three small molecules Y-27632, A-83-01 and CHIR99021 (YAC), this approach allows reprogramming of terminally committed MHs into bipotent LPCs without any genetic modification. Thus, we name these cells chemically induced liver progenitors (CLiPs). Here, we describe a step-by-step protocol to generate CLiPs from primary rat MHs and evaluate the bipotentiality of the generated CLiPs (Figure 1).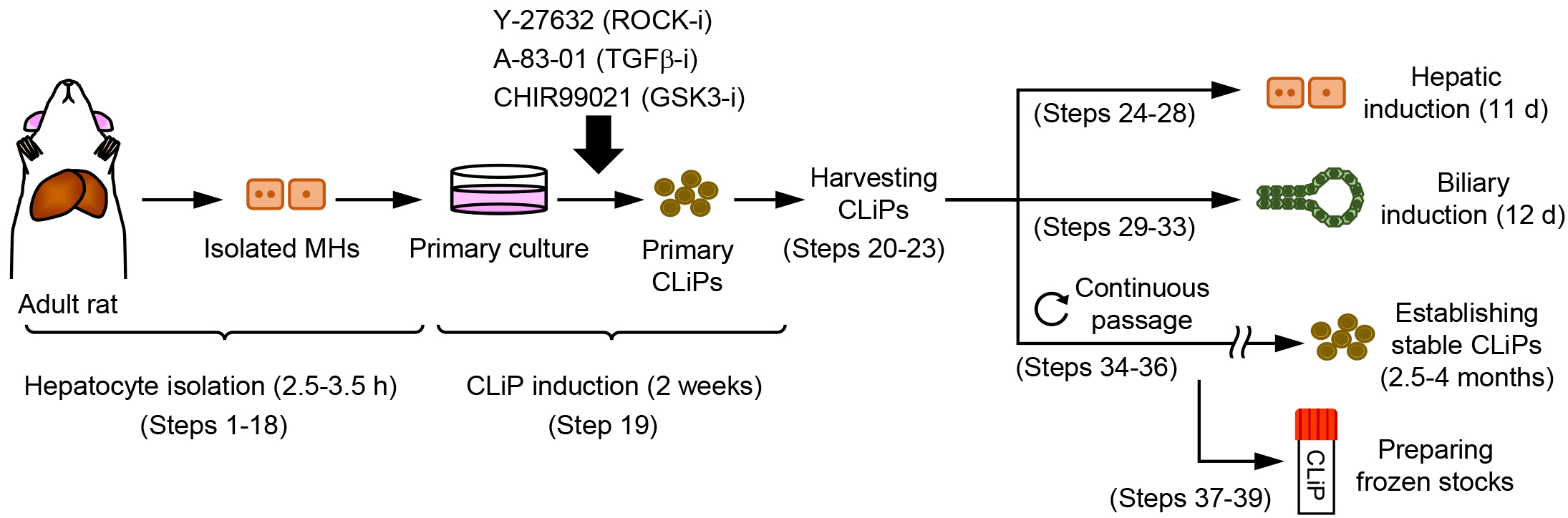
Figure 1. Overview of the experimental workflow. Protocols described in this article are composed of hepatocyte isolation (Steps 1-18), CLiP induction from the primary cultured hepatocytes (Step 19), harvesting the generated primary CLiPs (Steps 20-23), hepatic induction of CLiPs (Steps 24-28), biliary induction of CLiPs (Steps 29-33), establishment of stable CLiPs through continuous passage (Steps 34-36), and freeze stocking of CLiPs (Steps 37-39).
Experimental design and expected results:
Isolation of rat primary MHs (Steps 1-18): Rat primary MHs are isolated using the 2-step collagenase perfusion method (Seglen, 1976). In the first step, blood is eliminated from the liver by perfusing the liver with Hanks’ balanced salt solution (HBSS) supplemented with EDTA and EGTA. In the second step, the liver is digested by perfusing HBSS supplemented with collagenase. Then, the liver is extracted, minced, and further digested ex vivo with the remaining collagenase solution. MHs are collected by low-speed centrifugation at 50-60 x g (our centrifugation machine [Kubota 2800] uses 57 x g with the rotation speed at 600 rpm; researchers should adjust their rotation speed such that they can obtain 50-60 x g centrifugal force). This sequential low-speed centrifugation allows exclusion of nonparenchymal cells, such as biliary epithelial cells, sinusoidal endothelial cells, Kupffer cells and stellate cells, which weigh less than MHs, thereby enabling MH enrichment.
Induction of primary CLiPs using the small molecule cocktail YAC (Step 19): Rat CLiPs can be induced from primary MHs upon stimulation with a combination of three small molecules, YAC. An evident morphological change compared with YAC-free (YAC(-)) culture occurs approximately 4-5 days after plating (Figure 2). As previously reported, rodent MHs can divide once in vitro at approximately 2-4 days even without YAC; their proliferation completely stops thereafter (Loyer et al., 1996; Frémin et al., 2009). In contrast, MHs can continuously proliferate to produce CLiPs during 2-week culture (Figure 2). Although the emergence of CLiPs is highly reproducible (100% in our lab, n > 50), variability in the expandability of CLiPs is noted (Figures 3A and 3B). This variability is not dependent on the viability of primary MHs to be used for reprogramming (Figure 3C). In addition, when we use animals between 9 and 20 weeks of age, we do not observe a correlation between age and the expandability of CLiPs (Figure 3D). In our primary study, to avoid possible contamination of LPCs, which might reside in young animal livers, we used relatively old animals at the age of 10 to 20 weeks (Katsuda et al., 2017). However, when younger animals are used, such as animals at 4 to 8 weeks of age, increased expandability of CLiPs might be obtained.
Figure 2. Morphological change of rat MHs during reprogramming to CLiPs. Phase contrast images of rat MHs during 2-week culture in the absence or presence of YAC. On D3, smaller cells (indicated by arrowheads) appear, and the proliferation of these small cells, namely, CLiPs, becomes evident on D4-D5. On approximately D7, CLiP colonies become noticeable, and these colonies become larger thereafter. CLiPs are distinguishable from MHs because their nucleus/cytoplasm ratio is apparently larger than that of MHs. In addition, the size of CLiPs is much smaller than that of MHs.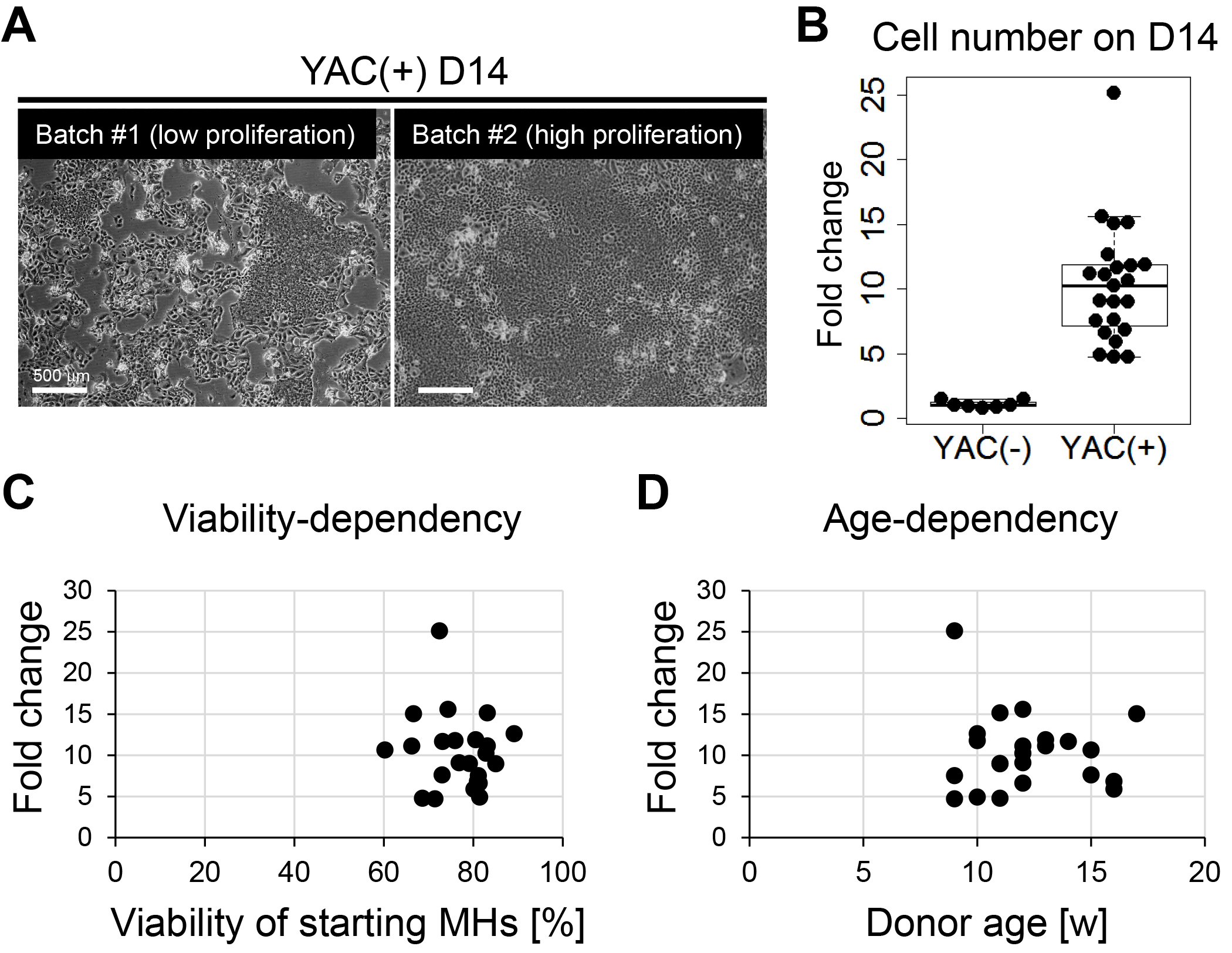
Figure 3. Inter-batch variation in proliferative capacity of primary rat CLiPs. A. Representative image of primary CLiPs on D14 with low expansion capacity (batch #1, left) and those with high expansion capacity (batch #2, right). Although reprogrammed CLiPs can be obtained in both experimental batches, the cell density on D14 is clearly different between these two batches. B. A box-and-whisker plot of cell number on D14 indicates inter-batch variation in proliferative capacity of primary CLiPs (right). The cell number was normalized with the inoculated cell number and is presented as fold change. The fold change of YAC(-) cells is presented as the control (left). n = 7 and 23 for YAC(-) and YAC(+) cells, respectively. C. No correlation was noted between the expansion capacity of primary CLiPs and the viability of inoculated MHs. D. No correlation was noted between the expansion capacity of primary CLiPs and the age (9 weeks to 17 weeks) of donor animals.
Evaluation of bipotentiality of CLiPs (Steps 25-34): Primary CLiPs can be differentiated into both hepatocytes and BECs. Hepatic differentiation is performed based on a previously reported protocol that induced hepatic maturation of foetal liver cells (Kamiya et al., 2002). This protocol consists of two steps. In the first step, CLiPs are cultured for 6 days in SHM + YAC supplemented with 20 ng/ml of oncostatin M and high concentration of dexamethasone (10-6 M). In the second step, CLiPs are further cultured for another 2 days in the presence of Matrigel. Morphological changes in Step 1 are relatively mild. The nucleus/cytoplasm ratio of hepatic-induced cells (Hep-i(+) cells) is reduced, and the cytoplasm becomes rich in granules (Figure 4A). However, morphological changes in Hep-i(+) cells become more evident in Step 2. Cell-cell boundaries become clear due to bile canaliculi formation (Figure 4A). Hepatic differentiation can be confirmed by the expression of hepatic marker genes (Figure 4B) and proteins (Figure 4C) and hepatic functionality, such as glycogen storage (Figure 4D). CLiPs can also differentiate into BECs using a two-step protocol (Figure 5A). In Step 1, CLiPs are co-cultured on cell cycle-arrested mouse embryonic fibroblasts (MEFs) in mTeSR1 + YAC for 6 days. In Step 2, CLiPs are cultured for another 6 days in 2% Matrigel-containing mTeSR1 + YAC. CLiP-derived BEC-like cells form ductal and cystic structures typically observed in Step 2 (Figure 5B, Batch #1). However, batch-to-batch variability is noted. In some experiments, CLiPs start to form ductal/cystic structures in Step 1 (Figure 5B, Batch #2). CLiP-derived ductal/cystic structures express BEC marker proteins (Figure 5C) and exhibit the capacity to transport fluorescein to their luminal spaces following incubation with fluorescein diacetate (Figure 5D).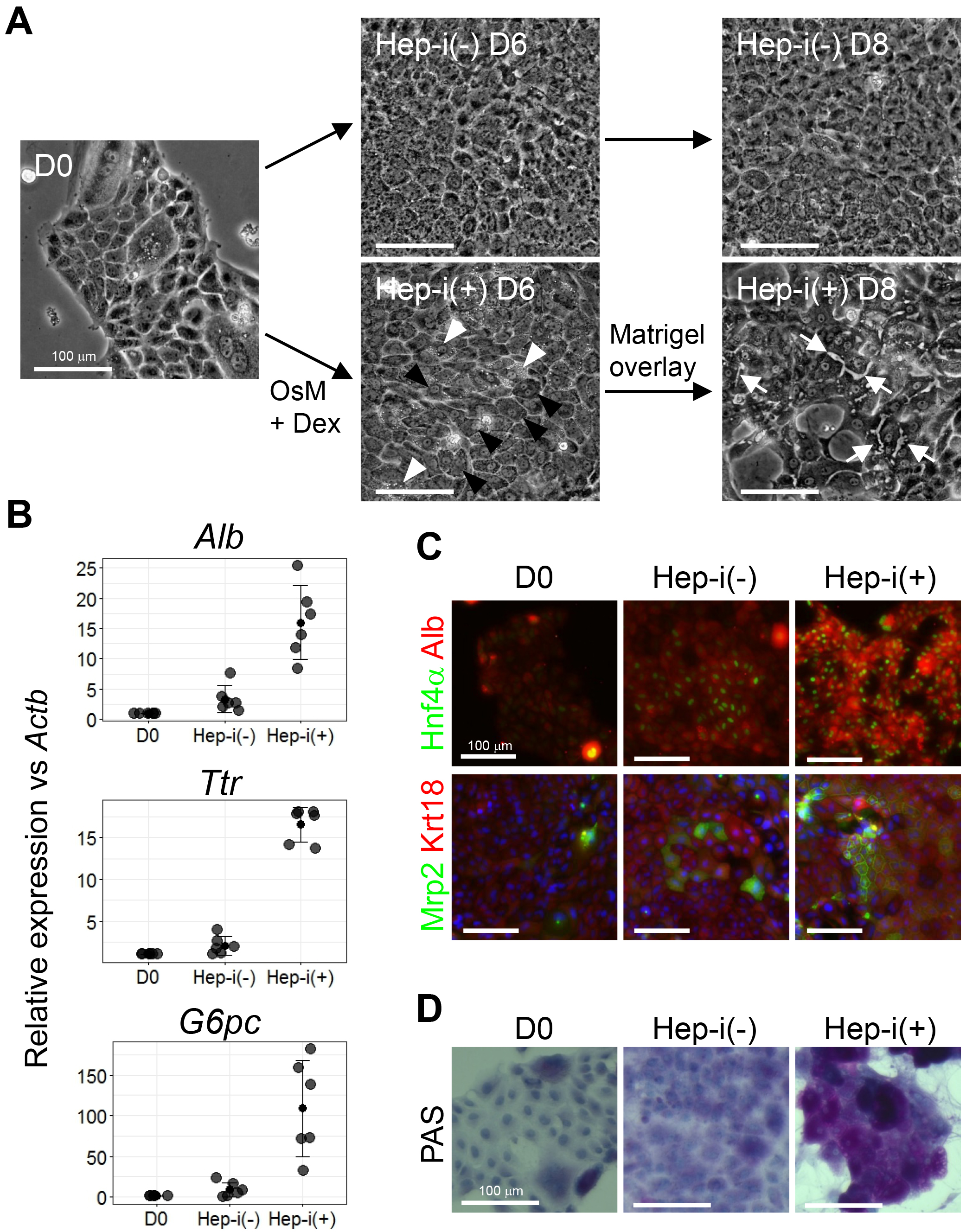
Figure 4. Characterization of CLiPs that underwent hepatic induction. A. Morphological change of CLiPs during hepatic induction. Black arrowheads indicate cells with decreased nucleus/cytoplasm ratio. White arrowheads indicate cells that are rich in granules in their cytoplasm. White arrows indicate bile canaliculi formed at the boundaries between cells that achieve hepatic maturation. B. qRT-PCR of hepatic marker genes for CLiPs before hepatic induction, Hep-i(-) and Hep-i(+) cells. The data represent the mean ± SD of 6 experiments performed with D14-16 CLiPs generated from 4 donor rats. C. ICC was performed for CLiPs before hepatic induction, Hep-i(-) and Hep-i(+) cells. Nuclei were counterstained with Hoechst 33342. The antibodies used for ICC have been previously described (Katsuda et al., 2017). D. The glycogen storage capacity was assessed by periodic acid-Schiff (PAS) staining using a commercially available staining kit (PAS kit). Nuclei were counterstained with hematoxylin.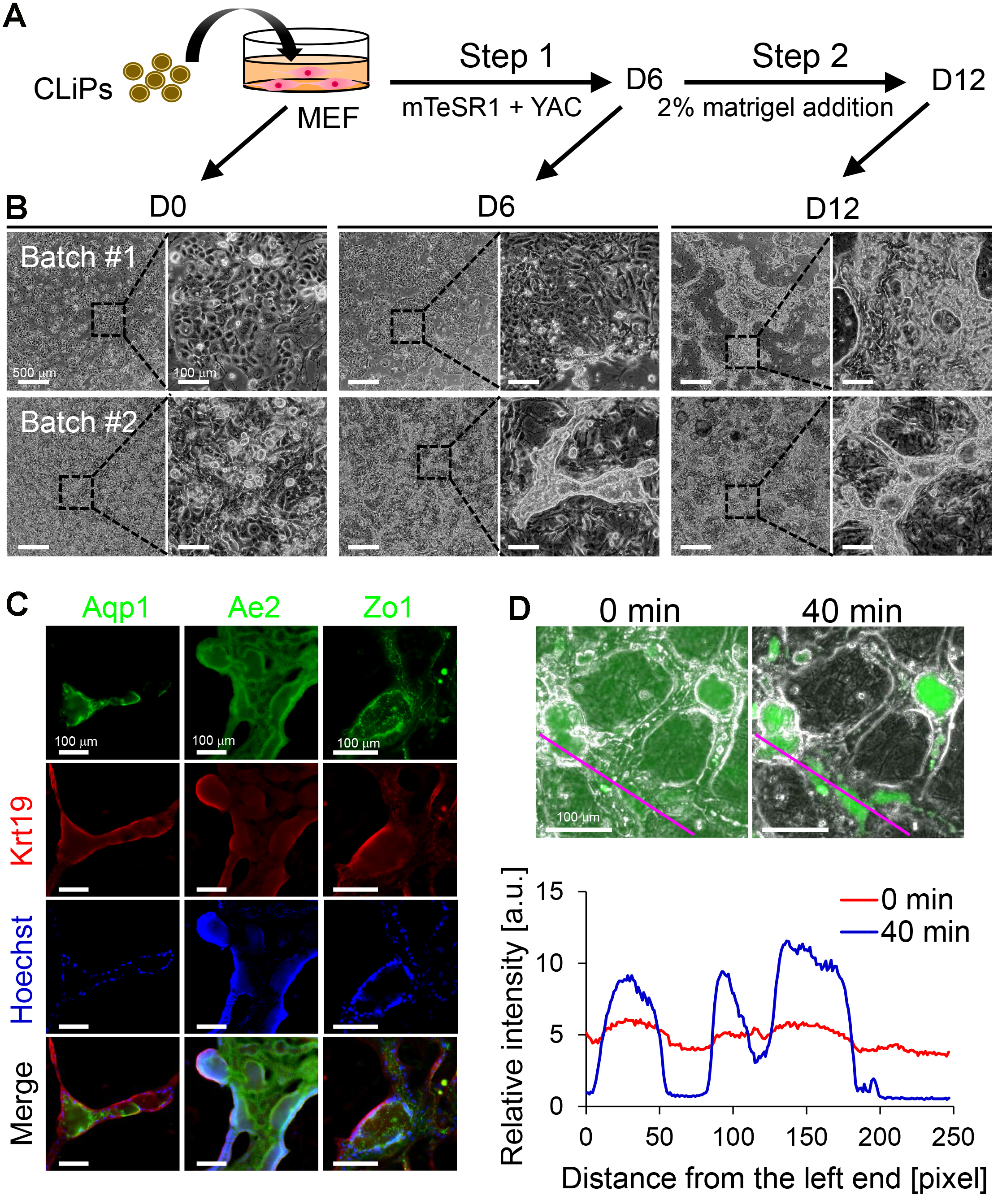
Figure 5. Characterization of CLiPs that underwent BEC induction. A. Schematic of the protocol for BEC induction from CLiPs. B. Morphological changes of CLiPs during BEC induction for 2 batches of experiments. CLiPs typically form ductal/cystic structures in Step 2 (Batch #1). In some experiments, CLiPs form ductal structures as early as Step 1 (Batch #2). C. CLiP-derived ductal/cystic structures express BEC markers, including Krt19, Aqp1 and Ae2, and the tight junction marker Zo1. D. CLiP-derived BEC-like cells exhibit the capacity to export fluorescein into their luminal space following incubation with fluorescein diacetate (FD). Phase-contrast and fluorescence images were immediately obtained after a 15-min incubation in the presence of FD (upper left panel) and an additional 40-min incubation in the absence of FD (upper right panel). The lower panel presents quantitative data of the fluorescence intensity profile along the magenta lines placed on the upper panels. The relative intensity is represented as the fluorescence intensity normalized with the total intensity along each line.
Establishment of stable CLiPs through continuous subculturing (Steps 35-36): Rat CLiPs undergo a temporal decrease in proliferative capacity between approximately passage 2 and passage 7 (Figure 6A). However, after this period, they proliferate stably. After 10 passages, a CLiP population seems to be relatively more homogeneous compared with that in earlier passages; thus, we regard stable CLiPs as the cells that undergo 10 passages. Different culture periods are required for CLiPs to reach 10 passages among experimental batches (74 days to 102 days, n = 6 biological replicates) and technical replicates (Figure 6A) (82 days to 124 days, 6 technical replicates).
We confirmed that CLiPs can proliferate on both Matrigel- and collagen-coated plates (Figure 6B), but the plating efficiency is increased when cultured on Matrigel-coated plates, thereby allowing efficient subculturing of CLiPs (Figure 6C). Thus, we recommend the usage of Matrigel-coated plates for subculturing of CLiPs.
We also confirmed that stable CLiPs, which have grown for more than 10 passages, can also undergo hepatic maturation (Figures 7B and 7C). However, we have not tried BEC induction of stable CLiPs. Stable CLiPs established from 2 of 3 donor rats exhibited biliary differentiation in vivo after transplantation into immunodeficient mice with chronic liver injury (Katsuda et al., 2017). Thus, these results suggest that CLiPs maintain bipotentiality at least partly even after serial passages.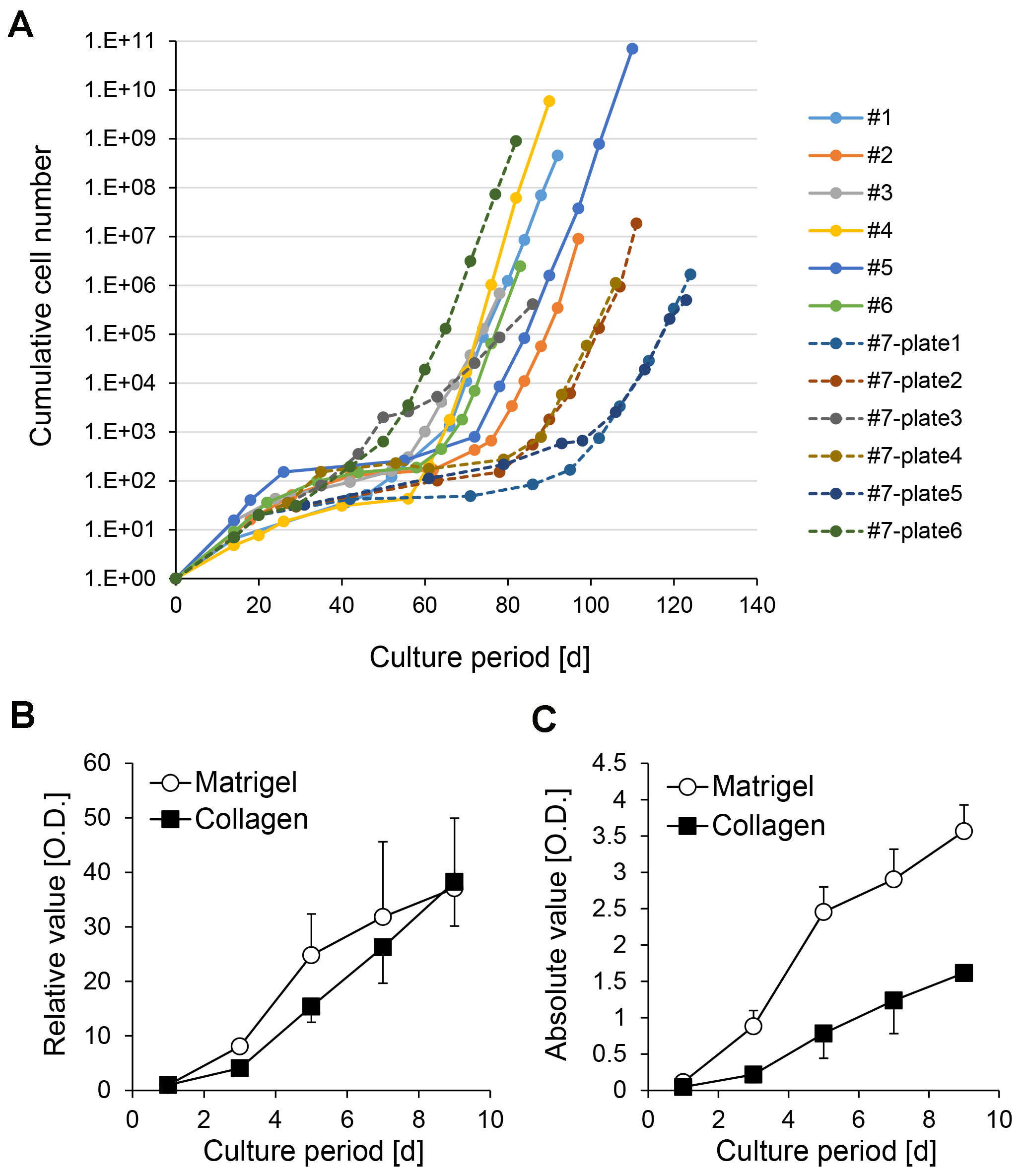
Figure 6. Inter-batch and intra-batch variation of growth rate of CLiPs during serial passages. A. Inter-batch variation of CLiP growth rates derived from 6 donor animals is presented as solid lines. The data from P0 to P9 or P10 of CLiPs are presented. Intra-batch variation of CLiP growth rate is presented for an additional donor animal (donor #7) as broken lines. Six plates (#7-plate1 to #7-plate6) were separately cultured for 10 passages. For #7 CLiPs, representative cellular numbers for P0 and P1 were determined from one plate. The cumulative cellular number was normalized to inoculated cell number in the primary culture. B. A stable CLiP cell line (#1) was seeded on collagen- or Matrigel-coated plates, and its proliferative capacity was assessed at the designated time points using the MTS assay. Signal intensity was normalized to the value of D1. The data represent the mean ± SE of 3 independent experiments. C. Absolute signal intensity for the data presented in (B). The data represent the mean ± SE of 3 independent experiments.
Limitations: As described above, the major limitation of this method is the difficulty experienced in its application to human hepatocyte reprogramming in its present form. We cultured cryopreserved human MHs in the presence of YAC and observed proliferation of epithelial cells (personal observation). These cells could be expanded. However, unlike rat and mouse MHs, they cease proliferation after approximately 4 passages. In addition, even early passage replicating cells did not exhibit hepatic differentiation capacity, suggesting that they are different from rodent CLiPs.
Another limitation is the imperfect reproducibility in the establishment of stable rat CLiPs with hepatic differentiation ability. Although the reproducibility of the induction of CLiPs from primary rat MHs is very rigid (100%, n > 50), obtaining stable CLiPs with the capacity of hepatic differentiation (namely, after 10 passages) appears to be stochastic. We have obtained stable CLiPs in all the experiments we have performed (n = 8 in Wistar rat; n = 1 in Lewis rat; n = 2 in Long Evans rat; n = 5 in Long Evans Agouti rat; n = 4 in Long Evans Cinnamon rat). However, in some of these experiments, we lost CLiPs with hepatic differentiation capacity after long-term culture. This is likely due to the heterogeneity of CLiPs. Early passage CLiPs are morphologically heterogeneous, but they become relatively homogeneous after long-term culture through the selection of limited numbers of cells in the population presumably during the period in which the proliferation rate is decreased (P2-P7). Due to this presumable selection process, CLiPs without hepatic phenotype become dominant in the culture in some experiment.
To enrich for high-quality CLiPs, namely, hepatic-competent CLiPs, the exploration of prospective surface marker(s) will be important, but we have not yet identified these markers. At present, to guarantee the securement of stable CLiPs with hepatic phenotype, we recommend preparing multiple plates or wells of culture in one experiment that are independently cultured. For example, we recommend splitting primary culture of MHs in SHM + YAC to multiple 60- or 100-mm dishes or 6- or 12-well plates and independently continue subculturing of each well/dish until CLiPs overcome the transient growth decrease (Figure 7A). In such an experiment, to reduce the labor involved in subculturing, we do not count cell numbers in every subculturing. Instead, we dilute the harvested cells at a ratio of 1:4 (during P1-P2) or 1:3-1:2 (during P3-P7, depending on the confluency of cultured wells) so that the plated cells are 20-50% confluent. When the cultured cells reach a stable growth phase, we only keep the cells that maintain LPC-like morphology (e.g., plate 1 in Figure 7A) and discard the cells without hepatic morphology (e.g., plate 3 in Figure 7A). Morphological differences can predict the hepatic differentiation capacity of stable CLiPs (Figures 7B and 7C). Figure 7 presents an example in which 3 of 6 plates (plates 1, 4 and 6) from one donor animal maintained a hepatic phenotype after 10 passages, whereas the other 3 plates (plates2, 3 and 5) exhibited a reduction in the hepatic phenotype.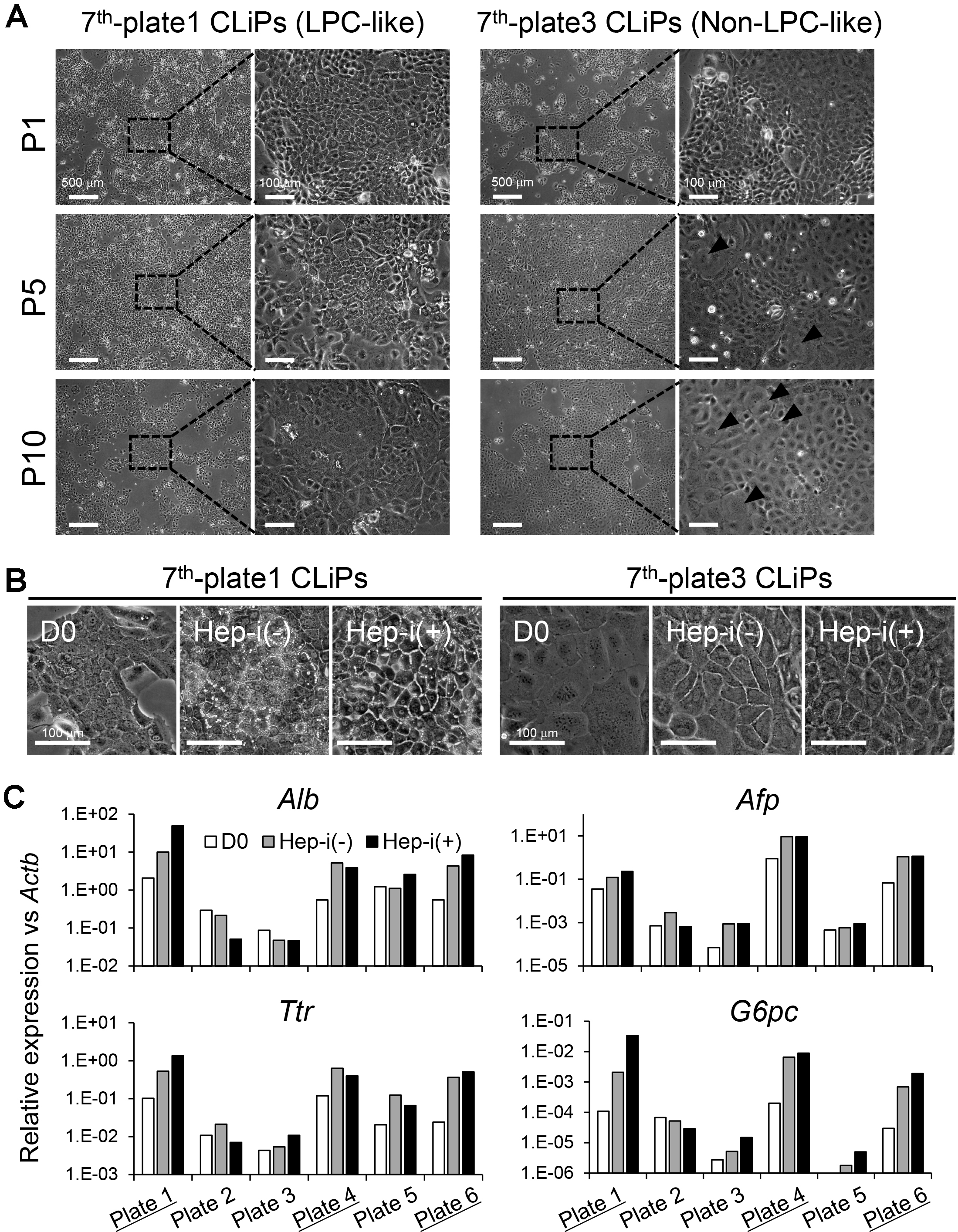
Figure 7. Morphological change of CLiPs during serial passages compared with non-LPC-like and LPC-like CLiPs. A. CLiPs cultured in two separate plates from one donor animal (donor #7) are photographed at P1, P4, P7 and P10. The morphology of non-LPC-like and LPC-like CLiPs is presented in the left and right columns, respectively. LPC-like CLiPs are small and exhibit a high nucleus/cytoplasm ratio. In contrast, non-LPC-like CLiPs are large and exhibit a relatively low nucleus/cytoplasm ratio. B. These non-LPC-like cells (plate 3) exhibit reduced responsiveness to hepatic induction. In contrast, LPC-like cells become MH-like cells with bile canaliculi-like structures following hepatic induction. C. qRT-PCR for hepatic marker genes for donor #7-derived CLiPs that were cultured individually in separate plates. CLiPs cultured in 3 of 6 plates exhibit increased hepatic maturation capacity (highlighted with underlines).
Materials and Reagents
- Paper towels
- Foam or cork board
- Silicon tube (Ø4.76 x 7.94 mm2) (TOKYO RIKAKIKAI, EYELA, catalog number: 125540 )
- Collagen type I-coated plate
96-well plate (IWAKI, catalog number: 4860-010 )
24-well plate (IWAKI, catalog number: 4820-010 )
12-well plate (IWAKI, catalog number: 4815-010 )
6-well plate (IWAKI, catalog number: 4810-010 )
100 mm dish (IWAKI, catalog number: 4020-010 ) - Non-coated tissue culture plate
96-well plate (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 167008 )
24-well plate (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 142475 )
6-well plate (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 140675 )
100 mm dish (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 172931 ) - Sterilized cotton gauze (Osaki Medical Corporation, catalog number: 15095 )
- 100 ml centrifugation tube (IWAKI, catalog number: 2355-100 )
- 50 ml conical centrifuge tube (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 339652 )
- Stainless 60 μm cell strainer (Ikemoto Scientific Technology, catalog number: 802-570-01 )
- Wide bored P200 tip (Thermo Fisher Scientific, catalog number: 118-96RNS )
- Cannula Ø1.2 mm (plastic outer needle equipped with intravenous cannula) (Top corporation, catalog number: 01151 )
- 4-0 Sterilized braid silk suture (Akiyama Medical MFG, catalog number: DEWB0404 )
- 0.22 μm filter (Stericup-GP), 250 ml (Merck, catalog number: SCGPU02RE )
- 25-ml pipette (Corning, Costar®, catalog number: 4489 )
- Wistar rats aged at 5 weeks to 20 weeks (both male and female animals can be used)
CAUTION: All animal experiments must comply with national and institutional regulations. - PAS kit (Sigma-Aldrich, catalog number: 395B-1KT )
- ISOFLURANE Inhalation Solution (Pfizer, catalog number: 871119 )
- Ethanol (Wako Pure Chemical Industries, catalog number: 054-07225 )
- E-MEM (Sigma-Aldrich, catalog number: M4655 )
- Percoll (GE Healthcare, catalog number: 17089102 )
- PBS(-) (Nissui, catalog number: 05913 )
- TrypLE Express (Thermo Fisher Scientific, GibcoTM, catalog number: 12604013 )
- Fetal bovine serum (FBS) (Gibco, catalog number not available)
Note: FBS should be heat inactivated at 56 °C for 30 min before use. - Cryopreserved MEFs (EmbryoMax® Primary Mouse Embryo Fibroblasts) (Merck, catalog number: PMEF-CF )
- CELLBANKER®1 Cell Freezing Media (Takara Bio, catalog number: CB011 )
- Liquefied nitrogen
- Sodium bicarbonate (NaHCO3) (Wako Pure Chemical Industries, catalog number: 191-01305 )
- Sodium chloride (NaCl) (Wako Pure Chemical Industries, catalog number: 192-13925 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P9541 )
- Sodium phosphate monobasic dehydrate (NaH2PO4·2H2O) (Wako Pure Chemical Industries, catalog number: 192-02815 )
- Sodium phosphate dibasic dodecahydrate (Na2HPO4·12H2O) (Wako Pure Chemical Industries, catalog number: 196-02835 )
- Glucose (Sigma-Aldrich, catalog number: G6152 )
- EGTA (DOJINDO, catalog number: 346-01312 )
- EDTA (DOJINDO, catalog number: 345-01865 )
- Phenol red (Sigma-Aldrich, catalog number: P3532 )
- HEPES (Sigma-Aldrich, catalog number: H3375 )
- Calcium chloride dihydrate (CaCl2·2H2O) (Sigma-Aldrich, catalog number: C7902 )
- Trypsin inhibitor (Sigma-Aldrich, catalog number: T9128 )
- Collagenase (Wako Pure Chemical Industries, catalog number: 032-22364 )
- L-proline (Sigma-Aldrich, catalog number: P0380 )
- Sodium hydroxide (NaOH) (Wako Pure Chemical Industries, catalog number: 197-02125 )
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A9647 )
- Human epidermal growth factor (EGF) (Sigma-Aldrich, catalog number: E9644 )
- Dexamethasone (100 mg) (Wako Pure Chemical Industries, catalog number: 047-18863 )
- Ascorbic acid-2 phosphate (Asc2P) (Wako Pure Chemical Industries, catalog number: 013-12061 )
- Y-27632 (25 mg) (RHO/ROCK pathway inhibitor; Inhibits ROCK1 and ROCK2) (Wako Pure Chemical Industries, catalog number: 251-00514 )
- A-83-01 (10 mg) (Activin/NODAL/TGF-β pathway inhibitor; Inhibits ALK5, ALK4, and ALK7) (Wako Pure Chemical Industries, catalog number: 035-24113 )
- Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, catalog number: 276855 )
- CHIR99021 (25 mg) (WNT pathway activator; Inhibits GSK3) (Axon Medchem, catalog number: 1386 )
- DMEM/F12 containing 2.4 g/L NaHCO3 and L-glutamine (Thermo Fisher Scientific, GibcoTM, catalog number: 11320082 )
- DMEM, high glucose, pyruvate (Thermo Fisher Scientific, GibcoTM, catalog number: 11995065 )
- Antibiotic/antimycotic solution (Thermo Fisher Scientific, GibcoTM, catalog number: 15240062 )
- Insulin-transferrin-serine (ITS)-X (Thermo Fisher Scientific, GibcoTM, catalog number: 51500-056 )
- Nicotinamide (Sigma-Aldrich, catalog number: N0636 )
- Recombinant Mouse Oncostatin M (OsM) (25 μg) (R&D Systems, catalog number: 495-MO-025 )
- Matrigel (Corning, catalog number: 354230 )
- 0.5% (10x) trypsin-EDTA (Thermo Fisher Scientific, GibcoTM, catalog number: 15400054 )
- mTeSRTM 1 (STEMCELL Technologies, catalog number: 85850 )
- Hydrochloric acid (HCl) (35-37%) (Wako Pure Chemical Industries, catalog number: 081-03475 )
- Leibovitz’s L-15 medium (with additives: sodium pyruvate; L-glutamine; phenol red) (Thermo Fisher Scientific, GibcoTM, catalog number: 11415064 )
- HBSS (10x), no calcium, no magnesium, no phenol red (Thermo Fisher Scientific, GibcoTM, catalog number: 14185052 )
- 7.5% NaHCO3 solution (see Recipes)
- Pre-perfusion buffer (see Recipes)
- 0.05% phenol red solution (see Recipes)
- 0.05% collagenase solution (see Recipes)
- 2 M HEPES solution (see Recipes)
- L-proline stock solution (see Recipes)
- 5 N NaOH solution (see Recipes)
- 5% BSA stock solution (see Recipes)
- 10 μg/ml EGF stock solution (see Recipes)
- 10-4 M dexamethasone (see Recipes)
- 1 M nicotinamide (see Recipes)
- 100 mM Asc2P stock solution (see Recipes)
- Y-27632 (5 mM) (see Recipes)
- A-83-01 (2.5 mM) (see Recipes)
- CHIR99021 (15 mM) (see Recipes)
- Small hepatocyte medium (SHM) (see Recipes)
- 10 μg/ml mouse OsM stock solution (see Recipes)
- Matrigel stock (see Recipes)
- Matrigel-coated plate (see Recipes)
- 0.05% (1x) trypsin-EDTA (see Recipes)
- Hepatic induction medium for Step 1 (HIM-1) (see Recipes)
- Hepatic induction medium for Step 2 (HIM-2) (see Recipes)
- BEC induction medium for Step 1 (BIM-1) (see Recipes)
- BEC induction medium for Step 2 (BIM-2) (see Recipes)
- L-15-based medium (for complete Percoll solution) (see Recipes)
- Complete Perocll solution (see Recipes)
Equipment
- Surgical scissors (any company’s product should be fine)
- Surgical tweezer (any company’s product should be fine)
- Peristaltic pump (TOKYO RIKAKIKAI, EYELA, catalog number: RP-1000 )
- Small animal anesthetizer (Muromachi Kikai, catalog number: MK-A110 )
- Air tight chamber (Muromachi Kikai, catalog number: MK-TB1 )
- Bubble trap (prepared in-house)
- CO2 incubator (Panasonic Healthcare, catalog number: MCO-170AICUVH-PJ )
- Compact Tabletop Refrigerated Centrifuge (KUBOTA, model: Model 2800 )
- Hemocytometers (Improved Neubauer) (Sunlead Glass, catalog number: A126 )
- Micropipette (Gilson, catalog numbers: F123600 for P20; F123601 for P200; F123602 for P1000)
- Unit water bath Thermominder SDminiN (TEITEC, model: SDminiN, catalog number: 0068750-000 )
- Magnetic stirrer (any company’s product should be fine)
- 150 ml plastic storage bottle (Corning, catalog number: 431175 )
- Refrigerator (any company’s product should be fine)
Procedure
Timing
Steps 1-18, preparing rat primary MHs: 2-3 h
Step 19, induction of CLiPs from primary MHs: 2 weeks
Steps 20-24, harvesting CLiPs for various applications: 0.5-1 h
Steps 25-29, hepatic induction of CLiPs: 11 d
Steps 30-34, biliary induction of CLiPs: 12 d
Steps 35-36, long-term culture of CLiPs through continuous subculturing (until reaching P10): 2.5-4 months
Steps 37-39, freeze CLiP stocks: 0.5-1 h
- Isolation of primary mature hepatocytes (MHs), TIMING 2.5-3.5 h
Notes:- Before starting, place 20 ml E-MEM at room temperature, which will be used for digesting the extracted liver.
- Before starting, warm pre-perfusion buffer and collagenase solution to 38 °C.
- Before starting, set the perfusion rate approximately at 25-30 ml/min.
- Set up the perfusion apparatus as shown in Figure 8.

Figure 8. Equipment setup for liver perfusion. The upper panel presents the complete view of the apparatus for liver perfusion. The bottom panels present detailed images from the upper panel. Pre-perfusion buffer and collagenase solution should be warmed to 37 °C. Paper cushion can be altered with any materials, such as aluminum foil. - Anaesthetize a rat by inhalation of isoflurane vapor. Place a paper towel cushion on a foam board or cork board, and lay the rat on the cushion. Sterilize the animal by wiping its fur with 70% EtOH. Open the abdomen using surgical scissors to expose the liver.
- Place a knot with sterilized suture surrounding the portal vein at the location approximately 0.5 cm from the bifurcation (Figure 9). Start drip of pre-perfusion buffer (see Recipes) by turning on the pump at 25-30 ml/min. Half-cut the portal vein at the location approximately 1.5 cm from the bifurcation of the portal vein, and insert the cannula. Tie the suture to fix the cannula.
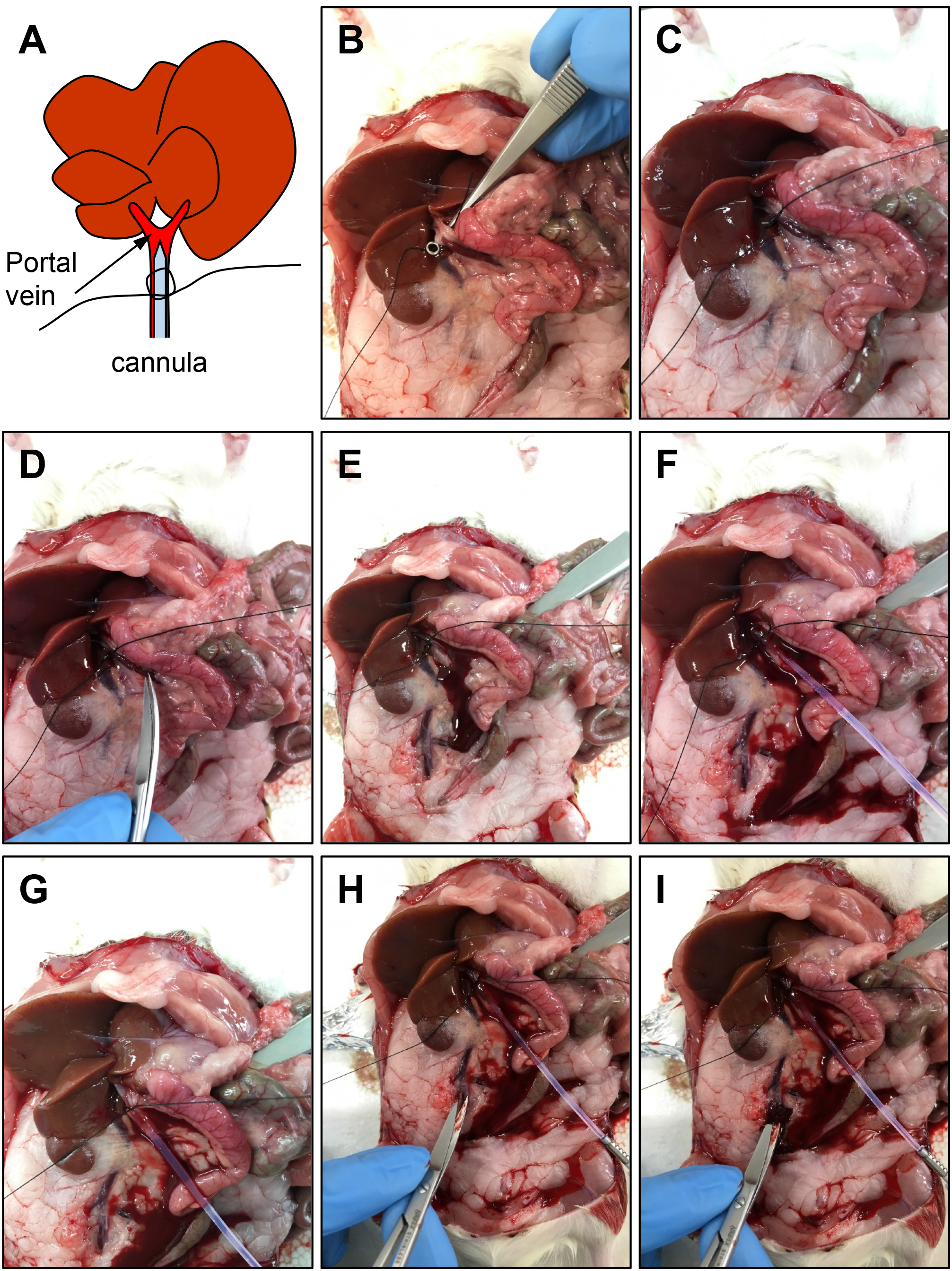
Figure 9. Cannulation of the portal vein. A. Schematic representation of the location of the cannula. B-C. Penetrate the tissue beneath the portal vein with a surgical tweezer, and make a knot with a suture surrounding the portal vein at the location approximately 0.5 cm from the bifurcation. D-E. Half-cut the portal vein with surgical scissors at the location approximately 1.5 cm from the bifurcation. F. Insert the cannula into the incision of the portal vein. G. Tie the suture to fix the cannula. H-I. Expose the vena cava, and cut it with surgical scissors to let the blood go out. - Immediately after confirming that the color of the liver changes from reddish brown to yellowish, cut the inferior vena cava (Figures 9 and 10). Perfuse approximately 450 ml pre-perfusion buffer (approximately 15 min).
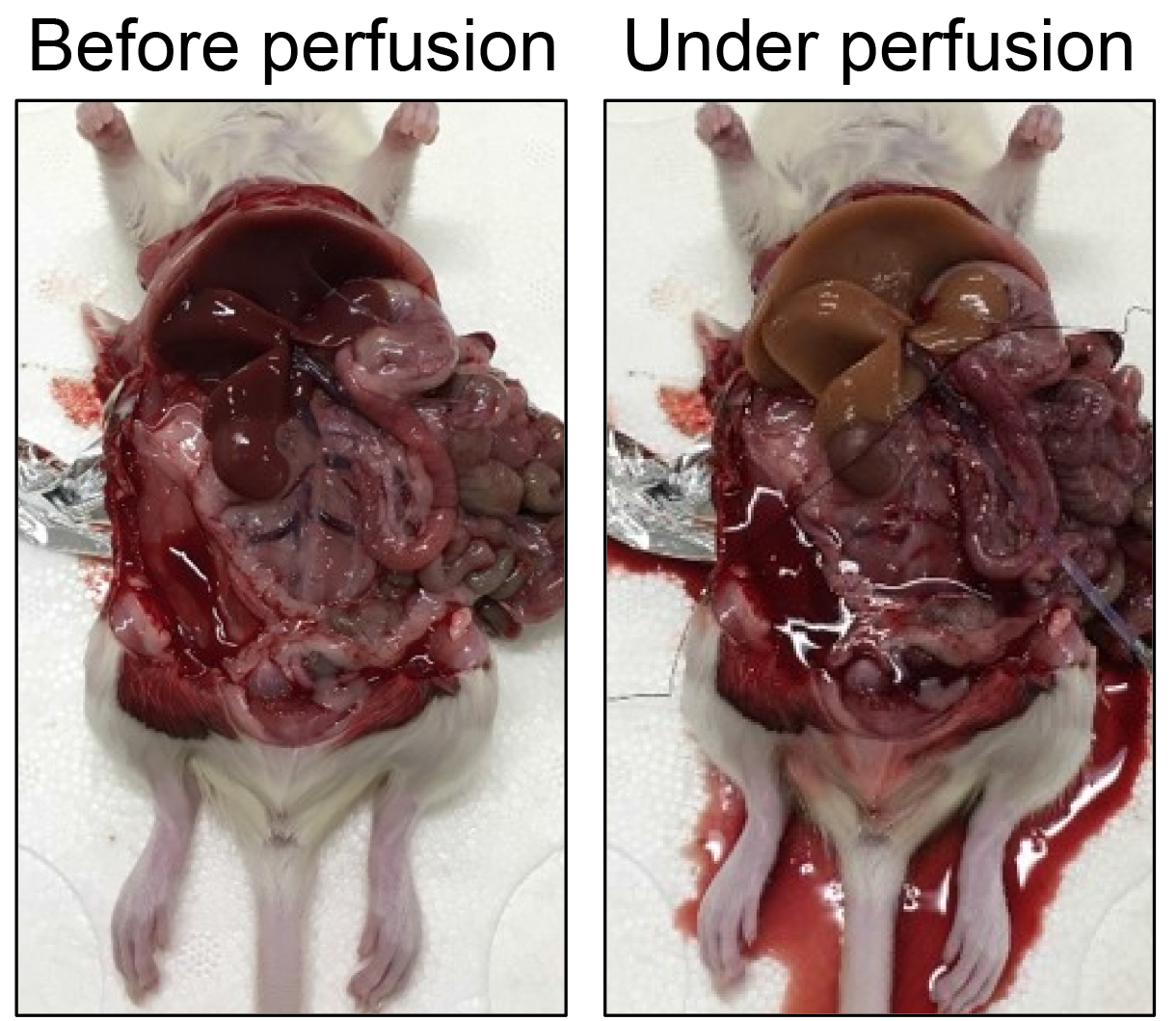
Figure 10. Colour change of the liver before and under perfusion - Pause the pump, transfer the inlet tube to the collagenase bottle, and resume perfusion. Perfuse approximately 300-350 ml collagenase (approximately 10 min).
- Stop perfusion, and remove the cannula from the portal vein. Cut the liver from the abdominal cavity, and transfer it to a 100-mm sterile plastic dish. Remove the attaching surrounding tissue, such as the diaphragm and pancreas.
Note: From this step forward, perform all procedures on a clean bench. - Add 20 ml E-MEM kept at room temperature and 20 ml of the remaining collagenase solution (see Recipes). Mince the liver with surgical scissors into pieces that are less than 3 mm in size.
- Incubate the minced tissue at 37 °C in a CO2 incubator for 15 min. After taking out the dish from the incubator, further dissociate the remaining tissue by gently pipetting the digested solution with a wide-bore pipette, such as a 25-ml pipette, several times.
Note: If large undigested fragments remain, remove them with sterilized tweezers. - Filter the undigested tissue fragments by transferring the digested solution into a 100-ml tube equipped with single-layered gauze on the top. Discard the single-layered gauze from the tube top, and transfer the filtered solution into a 100-ml tube with the double-layered gauze.
Notes:- When the gauze is clogged with undigested tissue fragments, remove the clogging materials from the filter using a pipette.
- From this step forward, all the procedures should be performed on ice, and all the reagents should be kept at 4 °C or on ice.
- When the gauze is clogged with undigested tissue fragments, remove the clogging materials from the filter using a pipette.
- Aliquot the filtered solution into two 50-ml tubes, add E-MEM into each tube up to 50 ml by decantation, and mix by gentle inversion. Centrifuge the tubes at 57 x g for 1 min at 4 °C.
- Aspirate the supernatant, and loosen the cell pellet by gently tapping the tubes. Add 10 ml E-MEM to each tube, and resuspend the cell pellet by gently pipetting several times. Decant E-MEM into each tube to 50 ml, and mix by gentle inversion. Filter the cell suspension through a double-meshed stainless cell strainer into new tubes. Centrifuge the tubes at 57 x g for 1 min at 4 °C.
Note: When pipetting the MH suspension, a wide-bore pipette should be used to avoid damaging MHs by extensive shear stress. - Aspirate the supernatant, and loosen the cell pellet by gently tapping the tubes. Add 10 ml complete Percoll solution to each tube, and resuspend the cell pellet by gently pipetting several times. Aliquot the remaining complete Percoll solution to each tube (approximately 14 ml/tube), and mix by gentle inversion. Centrifuge the tubes at 57 x g for 10 min at 4 °C.
- Aspirate the supernatant, and loosen the cell pellet by gently tapping the tubes. Add 10 ml E-MEM to each tube, and resuspend the cell pellet by gently pipetting several times. Decant E-MEM into each tube to 50 ml, and mix by gentle inversion. Centrifuge the tubes at 57 x g for 2 min at 4 °C.
- Repeat the above wash process (Step 13) once more.
- Resuspend the cells in 15 ml SHM(-), combine the 2 tubes, and measure the cell concentration.
- (Optional) If extensive aggregation disturbs cell counting, filter the cell suspension with a 40-μm cell strainer before cell counting. There is no difference in reprogramming efficiency under microscopic observation between cells isolated with and without this filtration step.
- Mix 30 μl of cell suspension with 120 μl SHM(-) to prepare a 5 fold-diluted cell suspension. Remove 30 μl from the diluted cell suspension, and mix it with 30 μl of trypan blue. Count the viable and dead cell numbers using a hemocytometer. Determine the suspension volume to be used for culture.
- Seed the isolated MHs at approximately 1 x 104 cells/cm2 in SHM(+) (if desired, SHM(-) can be also used as control) (for example, we seed cells at 2.5 x 104 cells/well, 5 x 104 cells/well and 1 x 105 cells/well in a 24-well plate, 12-well plate and 6-well plate, respectively). To obtain homogeneous seeding of MHs, shake the plates vigorously back and forth 10 times and then right and left 10 times. Let the plates stand still for 15 min so that MHs fall down to the bottom of the plate, and then, transfer the plates into a CO2 incubator.
Note: Due to the relatively heavy weight of MHs, be careful to keep the cell suspension homogeneous. Loading a large amount of the cell suspension in a pipette and releasing it to multiple plates/wells may result in inconsistent seeding density among plates/wells. We typically apply well-mixed undiluted cell suspension (generally approximately 3-8 x 106 cells/ml) using a micropipette directly to plates that are already filled with SHM(+). During the cell seeding step, ensure that the cell suspension is kept homogeneous by periodically inverting the tube to mix it.
- Before starting, place 20 ml E-MEM at room temperature, which will be used for digesting the extracted liver.
- Induction of CLiPs from primary MHs using the small molecule cocktail YAC, TIMING 2 weeks
- One day after plating primary MHs, replace the culture medium with fresh SHM(+) (if desired, SHM(-) also as control). Thereafter, renew the medium every 2-3 days. Given that proliferative YAC(+) cells are bipotential (see Steps 24-33), we designate these cells as chemically induced liver progenitors (CLiPs).
- One day after plating primary MHs, replace the culture medium with fresh SHM(+) (if desired, SHM(-) also as control). Thereafter, renew the medium every 2-3 days. Given that proliferative YAC(+) cells are bipotential (see Steps 24-33), we designate these cells as chemically induced liver progenitors (CLiPs).
- Harvesting CLiPs for various application. Steps 20-24. TIMING 0.5-1 h.
When CLiPs reach 70-100% confluency, harvest CLiPs to perform hepatic induction (Steps 25-29) or biliary induction (Steps 30-34), establish stable CLiPs (Steps 35-37), or prepare frozen stocks for future use (Steps 38-40).- Wash plates with PBS(-) twice.
- Add 0.05% trypsin-EDTA (see Recipes) or TrypLE Express (these two reagents can be used equivalently for harvesting CLiPs), and incubate at 37 °C in a CO2 incubator until the cells are detached from the plate.
Notes:- Make sure that the dissociating reagent completely covers the culture areas. We typically apply 2 ml of dissociation reagent to a 100-mm dish and use a similar ratio for other culture scales.
- CLiPs are relatively resistant to trypsinization, and it typically takes 10-15 min to detach them from plates.
- Make sure that the dissociating reagent completely covers the culture areas. We typically apply 2 ml of dissociation reagent to a 100-mm dish and use a similar ratio for other culture scales.
- Further dissociate the cells physically by pipetting with a P1000 tip to harvest as completely as possible, and transfer them to a 15-ml or 50-ml collection tube.
Note: Despite its relatively damaging effect on the cells, this procedure is indispensable. Unlike general cell lines, even if the cells exhibit round-shaped morphology following trypsinization, many of them still attach to the plate tightly and can rarely be harvested by directly applying medium onto the plates. - Collect the cells remaining in a plate with 5 ml SHM + YAC supplemented with 5%-FBS (heat inactivated) or SHM + YAC for trypsin-EDTA or TrypLE Express (TrypLE Express does not require neutralization of the enzymatic activity).
- Count the cell number using a hemocytometer, determine the suspension volume to be used for passage, transfer the required volume to a new tube, and centrifuge the tube at 200 x g at 4 °C for 5 min. Aspirate the supernatant, and loosen the cell pellet by tapping the tubes. Resuspend the cells with medium to be used for each application.
- Wash plates with PBS(-) twice.
- Hepatic induction of CLiPs (at P1). Steps 25-29. TIMING 0.5-1 h for passage, 2 d for culturing in SHM(+), and 8 d for hepatic induction
For primary CLiPs, we typically use cells cultured for 2 weeks (14-16 days).- Harvest the cells following Steps 20-24, and obtain the required volume of the cell suspension for hepatic induction. For early passage CLiPs, we seed the cells at approximately 5 x 104 cells/cm2 (1 x 105 cells/well and 2 x 105 cells/well for 24- and 12-well plates, respectively). Centrifuge the tube at 200 x g at 4 °C for 5 min.
Note: The cell density can be reduced to 1-2.5 x 104 cells/cm2 when stable CLiPs are used for hepatic induction. - Aspirate the supernatant, resuspend the cells in 0.5 or 1 ml SHM + YAC supplemented with 5% FBS, and seed to a well of collagen-coated 24- or 12-well plates, respectively.
Note: To increase the attachability of primary CLiPs on culture plates, we recommended using 5% FBS-supplemented SHM + YAC. - One day after seeding, replace the medium with fresh SHM + YAC, and culture the cells for 2 days.
- Replace the medium with HIM-1 (see Recipes), and continue culturing for another 6 days. Replace the medium with fresh HIM-1 every other day.
- Replace the medium with HIM-2 (see Recipes), and continue culturing for another 2 days.
Note: Prepare HIM-2 immediately prior to use. Use P1000 or wide-bore 200 μl tips chilled at 4 °C or on ice.
- Harvest the cells following Steps 20-24, and obtain the required volume of the cell suspension for hepatic induction. For early passage CLiPs, we seed the cells at approximately 5 x 104 cells/cm2 (1 x 105 cells/well and 2 x 105 cells/well for 24- and 12-well plates, respectively). Centrifuge the tube at 200 x g at 4 °C for 5 min.
- Biliary induction of CLiPs. Steps 30-34. TIMING 0.5 h for thawing MEFs, 1 d for culturing MEFs, 0.5-1 h for passage of CLiPs, 13 d for culturing CLiPs for BEC induction
Passage primary CLiPs for differentiation into BECs.- One day before harvesting CLiPs, seed cell cycle-arrested MEFs on 12-well collagen plates at 5 x 104 cells/well. Briefly, thaw cryopreserved MEF cells in a 37 °C water bath, and dilute the suspension in 10 ml 10%FBS-DMEM. Following centrifugation at 200 x g at 4 °C for 5 min, resuspend in 10%FBS-DMEM at 5 x 104 cells/ml, and seed 1 ml/well to a 12-well collagen plate.
- Harvest the primary CLiPs following Steps 20-24, and obtain the desired volume of the cell suspension for hepatic induction. We seed the cells at 5 x 105 cells/well in 12-well plates. Centrifuge the tube at 200 x g at 4 °C for 5 min.
- Aspirate the supernatant, resuspend the cells in 1 ml SHM + YAC supplemented with 5% FBS, and seed onto the pre-inoculated MEFs.
Note: To increase the attachability of primary CLiPs on culture plates, we recommend using 5% FBS-supplemented SHM + YAC. - (Step 1) On the following day, start biliary induction by replacing the medium with BIM-1 (see Recipes). Culture the cells for 6 days while replacing the medium with fresh BIM-1 every other day.
- (Step 2) Replace the medium with BIM-2 (see Recipes). Culture the cells for another 6 days while replacing the medium with fresh BIM-2 every other day.
Note: Prepare BIM-2 immediately prior to use. Use P1000 or wide-bore 200 ml tips chilled at 4 °C or on ice.
- One day before harvesting CLiPs, seed cell cycle-arrested MEFs on 12-well collagen plates at 5 x 104 cells/well. Briefly, thaw cryopreserved MEF cells in a 37 °C water bath, and dilute the suspension in 10 ml 10%FBS-DMEM. Following centrifugation at 200 x g at 4 °C for 5 min, resuspend in 10%FBS-DMEM at 5 x 104 cells/ml, and seed 1 ml/well to a 12-well collagen plate.
- Long-term culture of CLiPs through continuous subculturing. Steps 35-36. TIMING 2.5-4 months (until reaching P10)
CLiPs can be cultured stably through continuous passages; however, the proliferation rates transiently decrease from approximately P2 to P7. During long-term culture, the cells can be frozen for backup or future use.
Notes:- CLiPs undergo a transient decrease in their proliferative capacity between P2 and P7 (if not they enter complete senescence). Thus, during this period, it takes 15-40 days until the cells obtain 30-60% confluence, depending on experimental batches (Figure 5).
- Prepare a Matrigel-coated plate before starting passage (we typically use 35-mm, 60-mm, and 10-cm dishes for cell maintenance).
- Harvest the cells following Steps 20-24, and obtain the desired volume of the cell suspension for passage. Centrifuge the tube at 200 x g at 4 °C for 5 min.
- Resuspend the cells in an appropriate volume of SHM + YAC, seed on a desired plate coated with Matrigel, and culture the cells in a CO2 incubator. Medium should be replaced every 1-3 days.
- CLiPs undergo a transient decrease in their proliferative capacity between P2 and P7 (if not they enter complete senescence). Thus, during this period, it takes 15-40 days until the cells obtain 30-60% confluence, depending on experimental batches (Figure 5).
- Freeze stocking of CLiPs. Steps 37-39. TIMING 0.5-1 h
- Harvest the cells following Steps 20-24, and obtain a desired volume of the cell suspension for preparation of frozen stocks (typically 0.5-10 x 106 cells/vial). Centrifuge the tube at 200 x g at 4 °C for 5 min.
- Aspirate the supernatant, and resuspend the cells in a desired volume of CELLBANKER®1. The volume of CELLBANKER®1 used is 0.5 ml/vial.
- Aliquot to serum tubes equipped with screw caps. Seal the caps tightly, and store at -80 °C for at least one day. Then, transfer samples to liquefied nitrogen.
- Harvest the cells following Steps 20-24, and obtain a desired volume of the cell suspension for preparation of frozen stocks (typically 0.5-10 x 106 cells/vial). Centrifuge the tube at 200 x g at 4 °C for 5 min.
Notes
Trouble shooting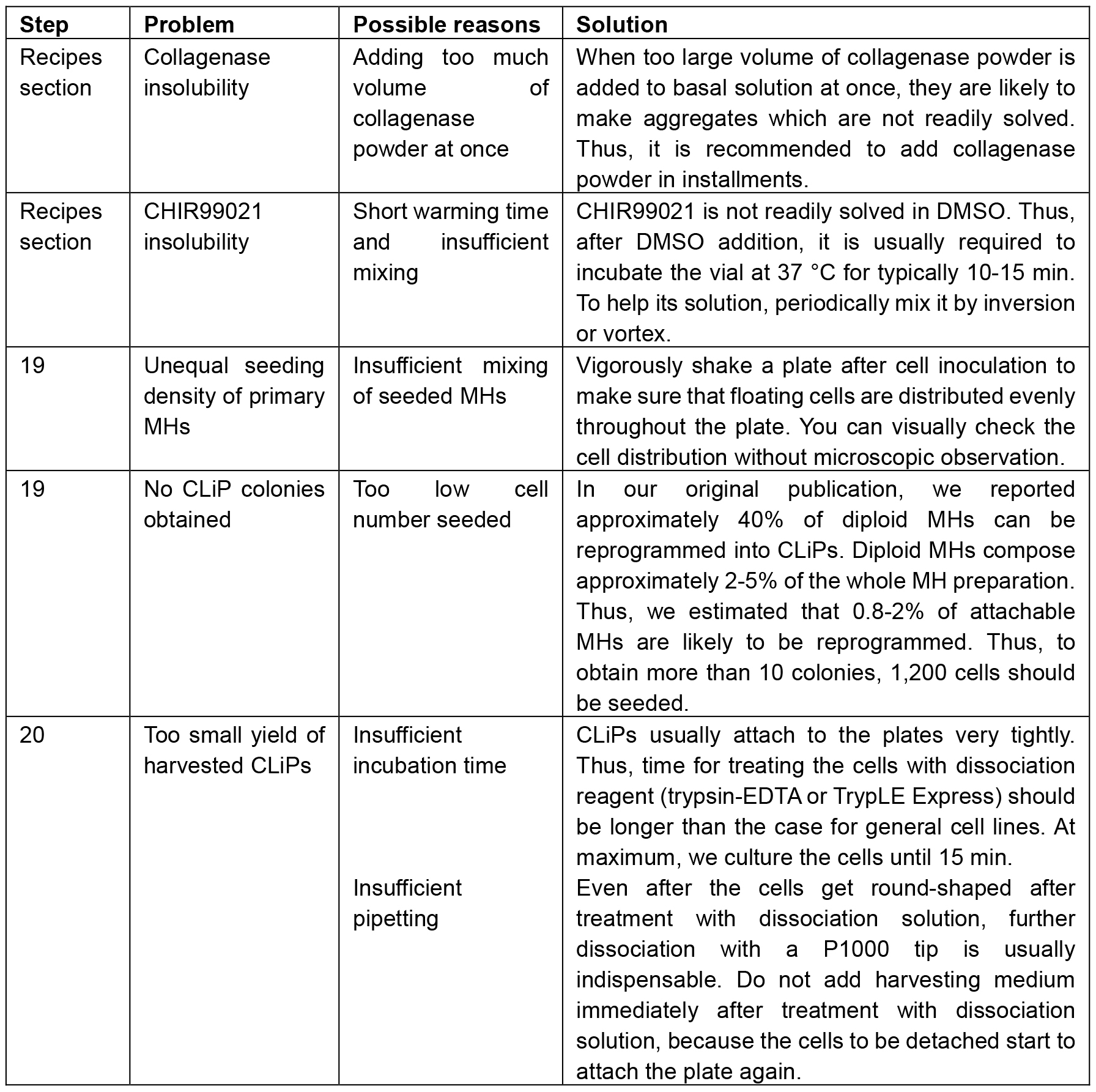
Recipes
- 7.5% NaHCO3 solution
Dissolve 7.5 g of NaHCO3 in 100 ml dH2O
Filter with a 0.22-μm filter, and store at 4 °C until use. Use within 1 month - Pre-perfusion buffer
- Prepare 10x pre-perfusion buffer stock by dissolving the following reagents in 500 ml distilled water: 40 g NaCl, 2.0 g KCl, 0.39 g NaH2PO4·2H2O, 0.76 g Na2HPO4·12H2O, 4.5 g glucose, 1.9 g EGTA, 3.7 g EDTA, 0.0030 g phenol red, and 11.9 g HEPES
- This stock solution can be stored at room temperature. Before use, make a 1x solution by diluting 50 ml of 10x stock solution with 450 ml dH2O
- After autoclaving, add 2.35 ml of 7.5% NaHCO3 solution and 2 ml of 1 N NaOH solution
- Prepare 10x pre-perfusion buffer stock by dissolving the following reagents in 500 ml distilled water: 40 g NaCl, 2.0 g KCl, 0.39 g NaH2PO4·2H2O, 0.76 g Na2HPO4·12H2O, 4.5 g glucose, 1.9 g EGTA, 3.7 g EDTA, 0.0030 g phenol red, and 11.9 g HEPES
- 0.05% phenol red solution
Dissolve 0.05 g of phenol red in 100 ml dH2O
Store at room temperature
Note: If the solution contains precipitates, mix the solution with a magnetic stirrer prior to each use. - 0.05% collagenase solution
Prepare 1.2 L of collagenase basal solution as below:- Dissolve the following reagents in 1.2 L of dH2O supplemented with 14.4 ml of 0.05% phenol red solution: 9.6 g NaCl, 0.48 g KCl, 0.094 g NaH2PO4·2H2O, 0.18 g Na2HPO4·12H2O, 2.86 g HEPES, 0.888 g CaCl2·2H2O and 0.42 g NaHCO3
- Store this basal solution at 4 °C until use
- For perfusion, pour 400 ml into a beaker and adjust pH to 7-7.4 with 5 N NaOH and/or 6 N HCl
- Then, dissolve 0.02 g trypsin inhibitor and 0.2 g collagenase
- After adjusting the pH to 7.6 with 5 N NaOH and/or 6 N HCl, filter with a 0.22-μm filter, and store at 4 °C until use
- Dissolve the following reagents in 1.2 L of dH2O supplemented with 14.4 ml of 0.05% phenol red solution: 9.6 g NaCl, 0.48 g KCl, 0.094 g NaH2PO4·2H2O, 0.18 g Na2HPO4·12H2O, 2.86 g HEPES, 0.888 g CaCl2·2H2O and 0.42 g NaHCO3
- 2 M HEPES solution
Dissolve 47.7 g HEPES in 100 ml dH2O
Filter with a 0.22-μm filter, and store at 4 °C until use - L-proline stock solution
Dissolve 0.3 g of L-proline in 10 ml of PBS(-)
Filter with a 0.22-μm filter, and store at 4 °C until use - 5 N NaOH solution
- Weigh approximately 20 g of NaOH and immediately record the precise weight
- Calculate the required volume of sterilized dH2O (for 20 g of NaOH, 100 ml is required)
- Add NaOH to approximately 80 ml of sterilized dH2O while stirring the solution
- After completely dissolving NaOH, increase the volume to the required amount with sterilized dH2O, transfer to a plastic storage bottle, and store at room temperature
- Because NaOH readily becomes moist, measure the weight as soon as possible.
- Given that dense NaOH solution dissolves glass, use a plastic bottle for storage
- Weigh approximately 20 g of NaOH and immediately record the precise weight
- 5% BSA stock solution
Dissolve 5 g BSA in 100 ml of PBS(-)
Filter with a 0.22-μm filter. Protect from light and store at 4 °C until use - 10 μg/ml EGF stock solution
- Prepare 20 ml of 0.1% BSA solution by diluting 0.4 ml of 5% BSA with 19.6 ml PBS(-), and dissolve 0.2 mg EGF
- Store 500-μl aliquots at -20 °C until use
- Prepare 20 ml of 0.1% BSA solution by diluting 0.4 ml of 5% BSA with 19.6 ml PBS(-), and dissolve 0.2 mg EGF
- 10-4 M dexamethasone
- Prepare 10-2 M primary stock solution by dissolving 100 mg dexamethasone in 25.4 ml ethanol, and store 500-μl aliquots at -20 °C until use
- To prepare 50 ml of working stock solution (10-4 M), dilute 500 μl of primary stock with 49.5 ml dH2O
- Store the working stock solution at 4 °C in the dark
- Prepare 10-2 M primary stock solution by dissolving 100 mg dexamethasone in 25.4 ml ethanol, and store 500-μl aliquots at -20 °C until use
- 1 M nicotinamide
Dissolve 12.21 g NA in 100 ml PBS(-)
Filter with a 0.22-μm filter, and store at 4 °C until use - 100 mM Asc2P stock solution
Dissolve 2.9 g of Asc2P in 100 ml PBS(-)
Filter with a 0.22-μm filter
Protect from light and store at 4 °C until use - Y-27632 (5 mM)
Dissolve 25 mg of Y-27632 in 14.8 ml sterilized dH2O
Store 1-ml aliquots at -20 °C until use - A-83-01 (2.5 mM)
- Prepare a primary stock solution (25 mM) by dissolving 10 mg of A-83-01 in 949 μl DMSO, and store 100-μl aliquots at -20 °C
- To prepare a working stock solution (2.5 mM), add 900 μl DMSO to one vial of primary stock, and store 100-μl aliquots at -20 °C
- Prepare a primary stock solution (25 mM) by dissolving 10 mg of A-83-01 in 949 μl DMSO, and store 100-μl aliquots at -20 °C
- CHIR99021 (15 mM)
Add 3.58 ml DMSO into the vial containing 25 mg CHIR99021, and store 100-μl aliquots at -20 °C - Small hepatocyte medium (SHM)
- Remove 27.3 ml from a 500-ml bottle of DMEM/F12
- The removed DMEM/F12 can be used to prepare Matrigel-coated plates (see below)
- To the remaining DMEM/F12, add 1.25 ml of 2 M HEPES, 500 μl L-proline, and 5 ml antibiotic/antimycotic
- Then, add 250 μl of 5 N NaOH to adjust pH to approximately 7.5. Then, add 5 ml of 5% BSA, 500 μl of 10 μg/ml of EGF, 5 ml of ITS-X, 500 μl of 10-4 M dexamethasone, 5 ml of 1 M nicotinamide and 5 ml of 100 mM Asc2P
- Store at 4 °C
- Remove 27.3 ml from a 500-ml bottle of DMEM/F12
- 10 μg/ml mouse OsM stock solution
- Prepare 2.5 ml of 0.1% BSA/PBS(-) by diluting 50 µl of 5% BSA with 2.45 ml PBS(-)
- Dissolve 25 μg mouse OsM in 2.5 ml of 0.1% BSA solution
- Store 100-μl aliquots at -20 °C until use
- Prepare 2.5 ml of 0.1% BSA/PBS(-) by diluting 50 µl of 5% BSA with 2.45 ml PBS(-)
- Matrigel stock
- Thaw a bottle of Matrigel in a refrigerator overnight. Prepare 0.5-1 ml aliquots on ice
- Store aliquots at 4 °C for short-term use (within 2 weeks) or at -20 °C for later use
- Thaw a bottle of Matrigel in a refrigerator overnight. Prepare 0.5-1 ml aliquots on ice
- Matrigel-coated plate
- Dilute Matrigel at a ratio of 16 µl per ml of DMEM/F12
- Add approximately 0.1-0.2 ml/cm2 of the diluted Matrigel solution (we typically add 0.5 ml, 1 ml, 2.5 ml and 5 ml to a well of 12- and 24-well plates, a 35-mm dish, a 60-mm dish and a 100-mm dish, respectively)
- Incubate at 37 °C for 20 min
- Wash once with an equal volume of SHM(-) before seeding cells (we typically add 0.5 ml, 1 ml, 2.5 ml and 5 ml to a well of 12- and 24-well plates, a 35-mm dish, a 60-mm dish and a 100-mm dish, respectively)
- Dilute Matrigel at a ratio of 16 µl per ml of DMEM/F12
- 0.05% (1x) trypsin-EDTA
Dilute 5 ml of 0.5% trypsin-EDTA with 45 ml PBS(-), and store at 4 °C until use - Hepatic induction medium for Step 1 (HIM-1)
Add 100 μl of 10 μg/ml OsM and 500 μl of 10-4 M dexamethasone into 50 ml of SHM + YAC. Final concentrations of OsM and dexamethasone are 20 ng/ml and 10-6 M, respectively - Hepatic induction medium for Step 2 (HIM-2)
Notes:- Prepare this medium on ice immediately before use.
- Use P1000 tips or wide-bore P200 tips chilled at 4 °C or on ice when working with Matrigel.
Mix HIM-1 and Matrigel at 7:1 volume ratio. For example, mix 62.5 μl Matrigel and 437.5 μl HIM-1 (500 μl in total) and 125 μl Matrigel and 875 μl HIM-1 (1 ml in total) for one well of a 24- or 12-well plate, respectively - Prepare this medium on ice immediately before use.
- BEC induction medium for Step 1 (BIM-1)
BIM-1 is mTeSRTM 1 medium supplemented with YAC. Prepare complete mTeSRTM 1 medium by adding the 100 ml of supplement into 400 ml basal medium, and add each vial of YAC stock to the solution - BEC induction medium for Step 2 (BIM-2)
BIM-2 is BIM-1 supplemented with 2% Matrigel. Prepare this medium immediately before use. Add 1 volume of Matrigel to 49 volumes of BIM-1 - L-15-based medium (for complete Percoll solution)
Dissolve the following reagents in 99 ml of Leibovitz’s L-15 medium (Gibco, with additives: sodium pyruvate; L-glutamine; phenol red): 0.2 g bovine serum albumin (BSA) (Sigma-Aldrich) and 0.0429 g HEPES. Filter with a 0.22-μm filter, add 1 ml of antibiotic/antimycotic (Gibco) and store at 4 °C until use - Complete Perocll solution
Prepare complete Perocoll solution in a 50 ml conical tube just before use by mixing 25 ml of L-15-based medium, 2.4 ml of HBSS (10x) and 21.6 ml of Percoll solution
Acknowledgments
We thank Ayako Inoue for her technical assistance, and Yusuke Sakai and Takanobu Hara for helping us prepare photos used in this manuscript. This research was partly supported by Grant-in-Aid for Young Scientists B (16K16643) and the Research Program on Hepatitis (16fk0310505h0005) from the Japan Agency for Medical Research and Development (AMED). This protocol has been used in Cell Stem Cell (Katsuda et al., 2017). The authors have declared no conflict of interest.
References
- Frémin, C., Bessard, A., Ezan, F., Gailhouste, L., Regeard, M., Le Seyec, J., Gilot, D., Pages, G., Pouyssegur, J., Langouet, S. and Baffet, G. (2009). Multiple division cycles and long-term survival of hepatocytes are distinctly regulated by extracellular signal-regulated kinases ERK1 and ERK2. Hepatology 49(3): 930-939.
- Kamiya, A., Kojima, N., Kinoshita, T., Sakai, Y. and Miyaijma, A. (2002). Maturation of fetal hepatocytes in vitro by extracellular matrices and oncostatin M: induction of tryptophan oxygenase. Hepatology 35(6): 1351-1359.
- Katsuda, T., Kawamata, M., Hagiwara, K., Takahashi, R. U., Yamamoto, Y., Camargo, F. D. and Ochiya, T. (2017). Conversion of terminally committed hepatocytes to culturable bipotent progenitor cells with regenerative capacity. Cell Stem Cell 20(1): 41-55.
- Loyer, P., Cariou, S., Glaise, D., Bilodeau, M., Baffet, G. and Guguen-Guillouzo, C. (1996). Growth factor dependence of progression through G1 and S phases of adult rat hepatocytes in vitro. Evidence of a mitogen restriction point in mid-late G1. J Biol Chem 271(19): 11484-11492.
- Seglen, P. O. (1976). Preparation of isolated rat liver cells. Methods Cell Biol 13: 29-83.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Katsuda, T., Hosaka, K. and Ochiya, T. (2018). Generation of Chemically Induced Liver Progenitors (CLiPs) from Rat Adult Hepatocytes. Bio-protocol 8(2): e2689. DOI: 10.21769/BioProtoc.2689.
Category
Cell Biology > Cell engineering > Partial reprogramming
Cell Biology > Cell isolation and culture > Cell isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.