- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Assessing Rates of Long-distance Carbon Transport in Arabidopsis by Collecting Phloem Exudations into EDTA Solutions after Photosynthetic Labeling with [14C]CO2
Published: Vol 7, Iss 24, Dec 20, 2017 DOI: 10.21769/BioProtoc.2656 Views: 7161
Reviewed by: Rebecca Van AckerAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2016
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Phloem loading and transport of photoassimilate from photoautotrophic source leaves to heterotrophic sink organs are essential physiological processes that help the disparate organs of a plant function as a single, unified organism. We present three protocols we routinely use in combination with each other to assess (1) the relative rates of sucrose (Suc) loading into the phloem vascular system of mature leaves (Yadav et al., 2017a), (2) the relative rates of carbon loading and transport through the phloem (this protocol), and (3) the relative rates of carbon unloading into heterotrophic sink organs, specifically roots, after long-distance transport (Yadav et al., 2017b), We propose that conducting all three protocols on experimental and control plants provides a reliable comparison of whole-plant carbon partitioning, and minimizes ambiguities associated with a single protocol conducted in isolation (Dasgupta et al., 2014; Khadilkar et al., 2016). In this protocol, [14C]CO2 is photoassimilated in source leaves and phloem loading and transport of photoassimilate is quantified by collecting phloem exudates into an EDTA solution followed by scintillation counting.
Keywords: ArabidopsisBackground
The allocation of reduced carbon and other compounds from photoautotrophic source tissues to heterotrophic sink tissues through the phloem is a crucial physiological process influencing growth and yield of plants. Because of this central role, there is interest in analyzing and quantifying phloem content from many areas of plant biology. However, collecting authentic phloem sap is difficult because the translocation stream is generally under high hydrostatic pressure and sieve elements have a rapid self-sealing mechanisms to prevent loss when damaged. Several collection techniques have emerged, but there is not currently a single or combination of methods that provide a complete and artifact-free measure of translocating phloem sap. Here, we briefly describe alternative techniques before detailing our approach to collecting phloem exudates into solutions containing low concentrations of ethylenediaminetetraacetic acid (EDTA) after photosynthetically labeling shoots with [14C]CO2. Turgeon and Wolf provide a comprehensive review of alternative techniques and their limitations (Turgeon and Wolf, 2009).
Phloem feeding insects, including aphids, scale insects, and planthoppers, evolved mechanisms to feed by directly drawing phloem sap from a plant’s vascular system and evade the self-sealing mechanism. Severing the feeding insect from a stylet penetrated into the phloem–referred to as stylectomy–can provide nanoliter to microliter quantities of sap that may most accurately reflect phloem content. Limitations of the technique are that it is technically challenging, works with only specific insect/plant combinations, the insects are selective for phloem with desired content, and insect saliva injected into the plant influences phloem content (Will et al., 2007; Hewer et al., 2011).
Another technique is to use plants that exude solution from cut stems without apparent sealing. Cucurbits, legumes, Ricinus communis, and some trees are well known for this and have become model systems for studying metabolites and signaling compounds in the phloem. However, the sap collected by this technique generally has low sugar concentrations, suggesting significant dilution and contamination from non-phloem sources and, particularly in the case of cucurbits, may be derived from specialized extrafascicular phloem elements rather than canonical fascicular phloem within vascular bundles (Zhang et al., 2010 and 2012).
The most common technique to sample phloem contents, and the one described in this protocol, is to collect phloem exudates from cut stems or petioles into solutions containing low concentrations of chelating agents, such as EDTA. Cations, particularly calcium (Ca2+), are involved in the rapid self-sealing mechanism of sieve tubes. Therefore, application of chelating agents to the cut ends of phloem-containing tissues limits sealing and permits phloem exudations for long periods from most, if not all, plants (King and Zeevaart, 1974; van Bel and Hess, 2008; Liu et al., 2012; Tetyuk et al., 2013). Although this method is most commonly used, it has important limitations and is not without controversy. Exudates are not collected directly, but are diluted into the EDTA solution during the collection period. This method therefore does not provide a measure of concentration, but rather a rate of exudation (i.e., unit of quantity per unit of time). As with the use of plants that naturally continue to exude, it is not clear how much the exudate is diluted by other sources in the plant or how much of the exudate is derived from the fascicular phloem involved in long distance translocation of photoassimilate in phloem sap. Enzymes, such as invertase from damaged cells, can enter the exudate solution and alter the profile of molecules exuded, as discussed at length in several articles (van Bel and Hess, 2008; Liu et al., 2012). A particularly important pitfall of this approach is that EDTA is toxic to cells and promotes membrane leakage which exacerbates contamination of the exudates with the contents of damaged cells and alteration of metabolite composition by leaked enzymes (Turgeon and Wolf, 2009). To minimize these impacts, we use the lowest EDTA concentration that still prevents sieve element sealing and as little tissue as possible is submersed in the EDTA solution (van Bel and Hess, 2008). In addition, we limit EDTA uptake via the xylem by conducting exudations in darkness and high humidity to promote stomatal closure and limit transpiration. Importantly, this procedure uses photosynthetic labeling of rosette leaves with [14C]CO2, and exudates are collected from cut petioles that have negligible photosynthetic activity and are well shaded by the mature rosette above. Therefore, counting 14C in the exudate solution is a quantitative representation of photoassimilate translocated from the leaves. When combined with other protocols (Yadav et al., 2017a and 2017b), this contributes to a reliable comparison of whole-plant carbon partitioning, and minimizes ambiguities associated with a single protocol conducted in isolation (Dasgupta et al., 2014; Khadilkar et al., 2016).
Materials and Reagents
- Labeling chambers derived from clear plastic deli containers (e.g., 16.5 cm long, 13 cm wide, and 5 cm deep ‘B16-Double Hot Dog Clamshell Case’ from Douglas Stephens Plastics, Paterson 640 NJ)
- Potting mixture (Sun Gro Horticulture, catalog number: Fafard 3B Mix ; or similar)
- 10 ml syringe (without a needle attached) (Luer-Lok Tip, BD, catalog number: 309604 )
- Dow Corning high vacuum grease
- Modeling clay (American Art Clay)
- Scotch Removable Mounting Putty
- 5 ml plastic syringe barrel and plunger (Luer-Lok Tip, BD, catalog number: 309646 )
- Syringe needle, 1.5-2.0 inches, 18 gauge (Monoject Needle, Covidien, catalog number: 1188818112 )
- High-humidity chambers derived from the clear plastic deli containers described above, lined with wet paper towel
- Petri dishes (100 x 25 mm) (Fisher Scientific, catalog number: FB0875711 )
- 24-well culture plates (Greiner Bio One International, catalog number: 662160 )
- Microcentrifuge tubes (Fisher Scientific, catalog number: 05-408-138 )
- Microcentrifuge tubes, screw cap with O-rings (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 3464 )
- 20 ml scintillation vials (Fisher Scientific, catalog number: 12383317 )
- Double edge razor blades (PERSONNA brand) (Electron Microscopy Sciences, catalog number: 72000 )
- Plant material (e.g., Arabidopsis thaliana Col-0, control and experimental material); > 12 healthy plants for each treatment with 3 WT and 3 of each experimental plant in each labeling chamber
- Sodium bicarbonate [14C]NaHCO3 (MP Biomedicals; catalog number: 0117441H ; 40-60 mCi/mmol; 2 mCi/ml; 5 mCi; 185 MBq)
- Lactic acid (85%) (Fisher Scientific, catalog number: A162-500 )
- Soda lime (LI-COR, catalog number: 9964-090 ) loosely packed to fill a column (e.g., Bio-Rad Econo-Column, 1.5 x 20 cm; Bio-Rad Laboratories, catalog number: 7371522 )
- Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, catalog number: E9884 )
- Potassium hydroxide (KOH) for pH adjustment (Fisher Scientific)
- Ethanol absolute (Pharmco-AAPER, catalog number: 111000200 )
- Commercial bleach (Sodium hypochlorite, NaClO)
- Ecolume scintillation fluid (MP Biomedicals, catalog number: 0188247004 )
- 5 mM EDTA solution (see Recipes)
Equipment
- Environmental growth chambers for control and experimental plants
- Personal safety equipment: lab coat, nitrile gloves (or similar), and eye protection
- Lamp suitable for photosynthetic labeling, such as a 400 W metal halide lamp (SYLVANIA 64490 - 400 Watt - BT37 - Metal Halide)
- Fume hood with appropriate support for metal halide lamp (Figure 1)
- Light meter (LI-COR 1400)
- Cork borer (The Science Company, catalog number: NC-11269 )
- Slim line micro blowers for air circulation (optional; Exton PA, Pelonis Technologies, catalog number: RFB3004 ; powered by four 1.5 volt D-cell batteries in sequence to provide 6 volts)
- Small air pump with inlet and outlet (e.g., Airpo, Barcodable, catalog number: UPC 045635496699 )
- Clear plastic tubing (4 and 6 mm internal diameter)
- Scissors (Fisher Scientific, catalog number: 08-951-20 )
- Forceps (Fisher Scientific, catalog number: 22-327379 )
- Balance with readability to at least 1 mg (METTLER TOLEDO, model: AE100 )
- Pipettes (Bioexpress, GeneMate, P20-P1000)
- Microcentrifuge
- Rotary platform shaker (Orbital Shaker Variable, BioExpress, GeneMate, catalog number: S-3200-LS )
- Geiger counter (Ludlum Measurements, model: Model 3 )
- Scintillation counter (Beckman Counter, model: LS 6000IC )
Procedure
- Preparing a work area suitable for [14C]CO2 photoassimilation
- Working with any radionuclide requires special consideration and approval from the appropriate institutional office. Clearance can take a long time (months or more) and the application process should be started early, or collaborations should be established with groups that already have approvals in place.
- [14C]NaHCO3 stocks will release gaseous [14C]CO2. To minimize this, commercial stocks are buffered to pH 9.5. Stocks should be stored at 4 °C and not -20 °C: We recommend aliquoting stock into 1.5 ml screw cap microcentrifuge tubes with O-rings. Receipt and use of stocks should be recorded as required by the institute where the experiments are conducted.
- In this procedure, [14C]CO2 gas is created by acidification of [14C]NaHCO3. Establishing a work area in a dedicated fume hood is recommended (with appropriate traps for [14C]CO2; see below).
- Within the fume hood, a 400 W metal halide lamp is suspended 60 to 90 cm above the work area. We constructed a simple frame from 2 x 4 lumber inside our fume hood to suspend the lamp (Figure 1).
- The distance between the lamp and the working surface will impact the photon flux density to which the plants are exposed. Our lamp is 90 cm above the fume hood bench top, and we work on a 15 cm platform so the plants are 75 cm from the lamp. At this distance, we can expose six of the indicated clamshell containers with equal illumination of 130 µmol photons m-2 sec-1, as determined with a PAR (photosynthetically active radiation) sensor.
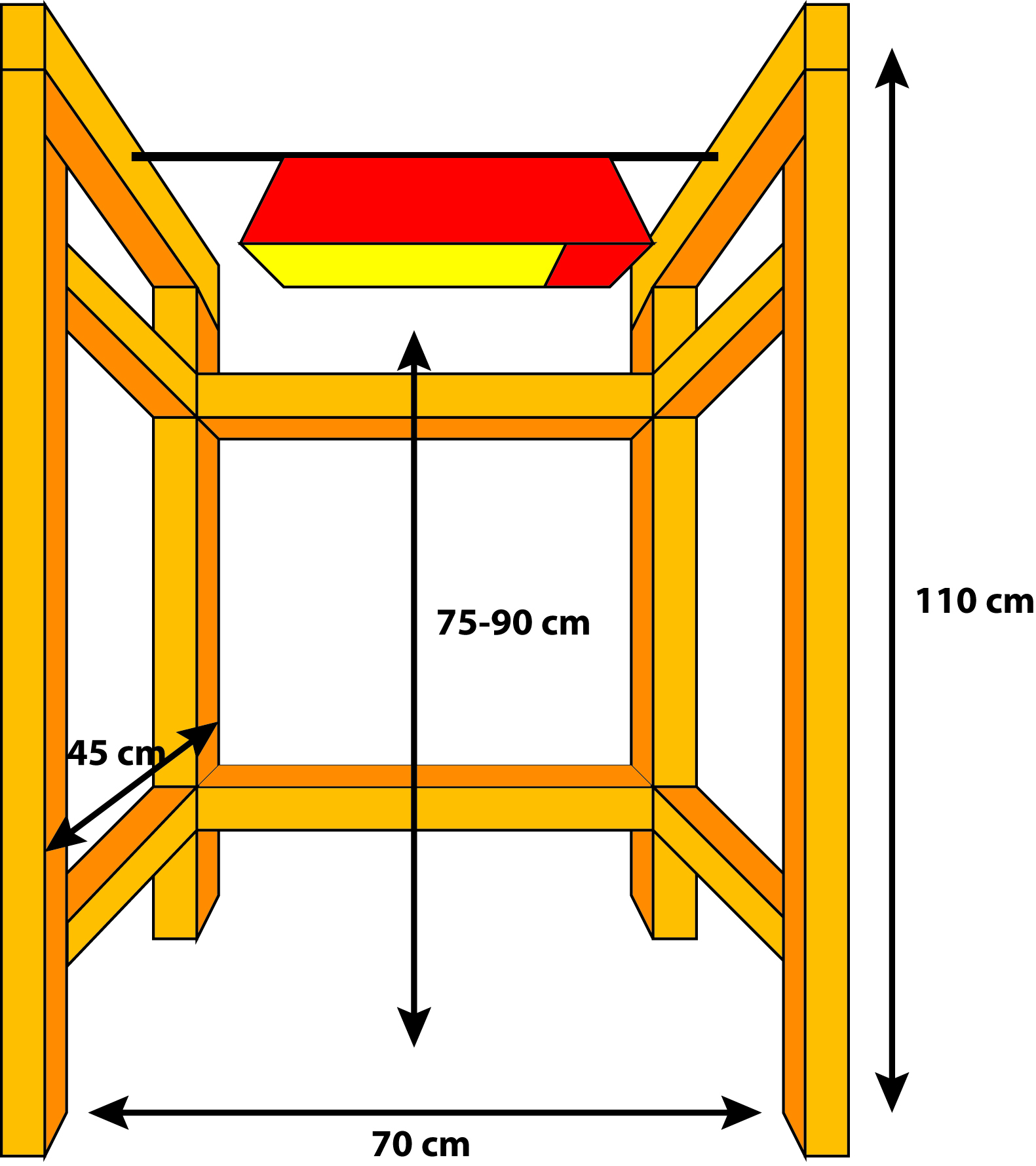
Figure 1. A representation of a frame that fits inside a fume hood and allows a metal halide lamp to be supported above the labeling work surface. The frame is made of standard 2 x 4 lumber (orange), and the 400 W metal halide light fixture (red/yellow) is supported by a metal rod (black).
- Working with any radionuclide requires special consideration and approval from the appropriate institutional office. Clearance can take a long time (months or more) and the application process should be started early, or collaborations should be established with groups that already have approvals in place.
- Strategy to germinate and grow plants
- Germinate seeds in potting mixture in transparent, clamshell deli containers. Use a 6 mm cork borer to make drainage holes in the bottom of clamshell deli containers for aeration and drainage. Fill the bottom half of the clamshell container with potting mix, 3.5 cm deep and water the potting mix. Add seeds and germinate by standard procedures for growth on potting mixture (Dasgupta et al., 2014; Khadilkar et al., 2016).
Notes:- Each clamshell deli container will be an independent [14C]CO2 labeling chamber. Therefore, controls and experimental plants should be grown together in the same chamber. In the clamshell containers we use, nine plants can be grown without crowding for 21-28 days. We typically grow WT controls and two experimental lineages in a randomized design in a single container, and have six replicate containers.
- The hypocotyl will be cut and submerged in EDTA solution to collect exudates. Germinating seeds in darkness, such as in a light-tight cabinet, drawer or box, for 4 to 5 days will promote hypocotyl elongation and make it easier to work with.
- Each clamshell deli container will be an independent [14C]CO2 labeling chamber. Therefore, controls and experimental plants should be grown together in the same chamber. In the clamshell containers we use, nine plants can be grown without crowding for 21-28 days. We typically grow WT controls and two experimental lineages in a randomized design in a single container, and have six replicate containers.
- Grow plants under a 12 h light (22 °C)/12 h dark (20 °C) diurnal cycle at 130 μmol photons m-2 sec-1, or other conditions suitable to the specific experiment. Keep the lids of the chamber open and maintain watering and fertilizer levels as appropriate for plants grown on potting mix. Grow the plants for 21-28 days to produce rosettes 3 to 4 cm in diameter.
Note: The transition from vegetative to reproductive growth impacts source/sink relationships. We plan our experiments so all plants are only vegetative during labeling and exudation.
- Germinate seeds in potting mixture in transparent, clamshell deli containers. Use a 6 mm cork borer to make drainage holes in the bottom of clamshell deli containers for aeration and drainage. Fill the bottom half of the clamshell container with potting mix, 3.5 cm deep and water the potting mix. Add seeds and germinate by standard procedures for growth on potting mixture (Dasgupta et al., 2014; Khadilkar et al., 2016).
- Preparation for [14C]CO2 photosynthetic labeling
- Photograph the plants on the day they are to be labeled. Turn on the 400 W metal halide light in the fume hood about an hour before labeling, so it reaches a stable intensity. Acclimate the plants under the metal halide lamp for about an hour before labeling, with the lid of the labeling chamber open.
Notes:- Photosynthesis, carbon partitioning into different metabolic pools, and long-distance transport to sink organs fluctuate through the diurnal cycle. For consistency, we generally label 6 h into the illuminated period.
- Although six clamshell containers will fit under the light and receive equal light intensity, it is impractical to process this number of plants in a single day (see time frame Table 1). With two people working together, we process three clamshell containers on one day, and the other three the following day. It is better to spread the experiment over two days and conduct labeling in the same phase of the diurnal cycle than it is to have the first clamshell container and the last clamshell container processed at significantly different times of day.

- Photosynthesis, carbon partitioning into different metabolic pools, and long-distance transport to sink organs fluctuate through the diurnal cycle. For consistency, we generally label 6 h into the illuminated period.
- Prepare the chambers for labeling. Make two holes in the lid of each clamshell container: a small one in the front side to inject [14C]CO2, and one slightly bigger in the opposite side to remove remaining [14C]CO2 after labeling (see flow chart, Figure 2A). The end of a paper clip, heated with a Bunsen burner, works well. To improve the seal of the labeling chamber, fill a 10 ml syringe (without a needle attached) with vacuum grease and apply a bead around the internal margin of the cover, but do not close and seal the cover until just before labeling. Use a small ball of modeling clay to cover the inject and exhaust holes.

Figure 2. A pictorial flow chart for photosynthetic labeling Arabidopsis plants with [14C]CO2 and collecting and quantifying 14C exudates into EDTA solutions. A. Grow Arabidopsis plants for 3-4 weeks in clear plastic deli containers containing potting mixture and with holes in the bottom for drainage and aeration, and two holes in the top to inject and exhaust [14C]CO2 (circles with arrows–blue for injection and red for exhaust). Acclimate the plants in the fume hood under the metal halide lamp for about an hour. Keep the lids open until just before labeling, and then seal by applying a bead of vacuum grease around the lid; place a small patch of modeling clay over the inject and exhaust holes. B. Prepare a syringe barrel for injecting [14C]CO2. A barrel cut to ~3 cm is easier to work with than an intact barrel. Place 2.5 µl of [14C]NaHCO3 in the barrel near the needle (indicated with yellow arrow) and a 15 µl of lactic acid ~1 cm away (indicated with gray arrow). C. Gently insert the plunger inside the barrel without disturbing the separate drops of [14C]NaHCO3 and lactic acid. D. Insert the needle through the inject hole and create [14C]CO2 by mixing [14C]NaHCO3 with lactic acid; pump the plunger 2-3 times to mix thoroughly and push the labeled gas into the chamber. E. Allow photosynthesis to proceed under the metal halide lamp for a 20 min ‘pulse’. F. Remove remaining unassimilated [14C]CO2 through the exhaust hole for 5 min using Tygon tubing connected to a vacuum pump and a column containing soda lime. G. Open the cover of labeling chamber and let photosynthesis continue for another 35 min ‘chase’. H. Gently cut the stem below the soil surface. I. Clean soil particles from the root surface, record the fresh weight. J. Recut through the hypocotyl under EDTA solution to prevent sieve element sealing. K. Transfer the shoot to a 24-well culture plate containing EDTA solution, such that only ~2 mm of hypocotyl is in the solution. Place the plate in a humidity chamber and then place in the dark for 10 min. Similarly collect subsequent exudations in 24-well culture plates with EDTA solution for the 1st and 2nd h. L. After collecting exudates, store the plates on ice to minimize enzyme activity until they can be conveniently processed further (image shows the plate on a bed of ice). Transfer phloem exudates into scintillation vials and add scintillation fluid (M) for scintillation counting. The labeled rosette should also be counted. - Create a schedule (Table 1) to organize processing of three replicate containers.
- With three replicate containers arranged and acclimated under the light source, close the lid of the first chamber to be labeled and ensure a tight seal. To create [14C]CO2 for labeling, pipette 2.5 μl of [14C]NaHCO3 (2 μCi/μl) in a droplet near the syringe needle junction of a syringe barrel cut to ~3 cm (Figure 2B). Place a 15 µl droplet of 85% lactic acid in the barrel, being careful to keep this droplet separate from the droplet of [14C]NaHCO3. Gently insert the plunger just inside the barrel (Figure 2C). Insert the needle through the injection hole of the clamshell container, and arrange the modeling clay around it (Figure 2D and 2E) to seal the hole; make sure the exhaust hole is also covered. Keep the droplets of [14C]NaHCO3 and lactic acid separate during these steps. Push the plunger gently to mix the lactic acid with [14C]NaHCO3 and release the [14C]CO2 into the air space of the chamber. Move the plunger back and forth to pump the [14C]CO2 gas into the clamshell container; avoid injecting fluids into the chamber since the lactic acid can damage the plants. Remove the needle and cover the injection hole with modeling clay.
Note: (Optional) For improved [14C]CO2 circulation, a small blower fan, such as one typically used for cooling small electronic equipment (e.g., Pelonis Technologies Cat. No. RFB3004), can be oriented inside the labeling chamber to blow air across the plants. We typically use Scotch Removable Mounting Putty to hold the blower to the top or side of the chamber, with the wires emerging through the vacuum grease used to seal top and bottom halves of the chamber. Power is provided by D-cell batteries. - Allow plants to do photosynthesis in the presence of [14C]CO2 for 20 min. This is the ‘pulse’ phase (Figure 2E).
- While plants in the first chamber are being labeled, prepare and label the second chamber as indicated in Table 1, and then the third chamber, etc.
- 20 min after injecting [14C]CO2, use the larger hole to vent the chamber through soda lime and to capture unassimilated [14C]CO2 (Figure 2F). A column filled with soda lime connected with tubing to an air pump works well (Figure 3). After ~5 min of venting through soda lime, open the chamber lid (Figure 2G) to allow photosynthesis to continue for another 35 min. This provides 40 min of total ‘chase’ time.
Note: To ensure effective capture of unassimilated CO2 by the soda lime, it should be fresh and well-sealed between uses. Old soda lime should be discarded as 14C labeled dry waste.
Figure 3. Exhaust system to remove and capture unassimilated [14C]CO2. A column, such as a Bio-Rad chromatography column, is filled with soda lime to capture residual [14C]CO2. One end of the column has tubing (4 mm internal diameter) to insert into the labeling chamber (red arrow) to vent out the remaining [14C]CO2. The other end is connected to the air intake of a small air pump, and the outlet is vented into the exhaust stream of the fume hood. Both ends of the soda lime column are equipped with stopcocks to minimize soda lime exposure to ambient CO2 when the column is not in use.
- Photograph the plants on the day they are to be labeled. Turn on the 400 W metal halide light in the fume hood about an hour before labeling, so it reaches a stable intensity. Acclimate the plants under the metal halide lamp for about an hour before labeling, with the lid of the labeling chamber open.
- Collection of phloem exudates
- Prior to initiating labeling, prepare three 24-well culture plates for each clamshell labeling chamber. These will be used for the initial wash step, the 1st h of exudation, and the 2nd h of exudation, respectively. Fill nine, well-spaced wells with 500 µl 5 mM EDTA solution, and set aside until needed. Prepare high-humidity chambers: prepare clamshell containers (one for each labeling chamber) with a bead of vacuum grease as described above; line the bottom half of the containers with wet paper towels. During exudation the 24-well culture plates will be placed in these high-humidity chambers and the chambers will be placed in the dark to stimulate stomatal closure and limit transpiration.
- At T = 60 min, use small, sharp scissors to cut below the hypocotyls in the uppermost part of the root. Remove any soil clinging to the plant and record the rosette fresh weight (Figures 2H and 2I).
Note: This weight will include a small portion of roots that are removed in the next step (D3), but we considered this small amount to be negligible and it is important to conduct Step D3 quickly without reweighing the tissue. - Submerge the cut end of the stem in a Petri dish containing 5 mM EDTA. Keeping the stem submerged, use one half of a double-edged razor blade, snapped in half lengthways, to cleanly slice the stem in the hypocotyl ~5 mm up from the first cut (Figures 2I and 2J).
Note: Double-edged razor blades are sharper than single edged blades. To work with them safely, fold the blade lengthways down the center while it is still in the protective wrapper and it will snap in half to provide a safe single edged blade. - Transfer each rosette immediately to the first prepared 24-well culture plate containing 500 µl 5 mM EDTA solution so that ~2 mm of cut stem is submerged. Minimize EDTA exposure to the rest of the plant tissues (Figure 2K).
- Once all nine plants are processed, place the 24-well culture plate into the clamshell humidity chamber and close the chamber to maintain near 100% humidity (Figure 2K). Place the humidity chamber in complete darkness, such as a drawer or box, to promote stomatal closure.
Notes:- Steps D1 to D5 should be carried out carefully but efficiently. Table 1 allows for ~10 min to process the nine plants in each labeling chamber, and ~10 min for the wash step in EDTA solution. This initial wash step is intended to remove enzymes released from the cut and damaged surface.
- Leave the plants in high humidity and darkness until further processing. Throughout this wash step and subsequent collection steps, the plants should be in high humidity and darkness unless they are being actively worked on.
- Steps D1 to D5 should be carried out carefully but efficiently. Table 1 allows for ~10 min to process the nine plants in each labeling chamber, and ~10 min for the wash step in EDTA solution. This initial wash step is intended to remove enzymes released from the cut and damaged surface.
- Transfer each rosette to a second 24-well culture plate, submerging only ~2 mm of cut stem in the EDTA solution. Place this culture plate into a humidity chamber, seal the lid, and store in darkness for 1 h. If exudates from the first plate (i.e., the EDTA wash solution) are to be counted, store the plate on ice to minimize potential enzyme activity until it can be conveniently processed for scintillation counting (Figure 2L). Transfer the solutions to microcentrifuge tubes and store in the freezer for longer term storage.
- After 60 min exudation, transfer the rosettes to the third 24-well culture plate, submerging only ~2 mm of cut stem in the EDTA solution. Place this third plate in the humidity chamber and store in the dark for another 60 min. Place the second plate (i.e., exudations from the first hour) on ice to minimize potential enzyme activity until it can be conveniently processed for scintillation counting (Figure 2L). Transfer the solutions to microcentrifuge tubes and store in the freezer for longer term storage.
- After 60 min, remove the rosettes and chill this third plate (i.e., exudations from the second hour) on ice to minimize potential enzyme activity until it can be conveniently processed for scintillation counting (Figure 2L), or transfer the solutions to microcentrifuge tubes and store in the freezer for longer term storage.
- Prior to initiating labeling, prepare three 24-well culture plates for each clamshell labeling chamber. These will be used for the initial wash step, the 1st h of exudation, and the 2nd h of exudation, respectively. Fill nine, well-spaced wells with 500 µl 5 mM EDTA solution, and set aside until needed. Prepare high-humidity chambers: prepare clamshell containers (one for each labeling chamber) with a bead of vacuum grease as described above; line the bottom half of the containers with wet paper towels. During exudation the 24-well culture plates will be placed in these high-humidity chambers and the chambers will be placed in the dark to stimulate stomatal closure and limit transpiration.
- Analyzing phloem exudates by scintillation counting
- Transfer 400 µl of each EDTA solution to scintillation vials (Figure 2M) and add 5 ml scintillation fluid for aqueous samples. Mix thoroughly by shaking the vial to create a monophasic solution. Document counts per min (cpm) or disintegrations per min (dpm) with a scintillation counter and a program suitable for 14C. Calculate 14C in 500 µl exudations as a rate per rosette mass: cpm or dpm (fresh weight)-1 h-1. The first and second hour of exudations should be analyzed by scintillation counting and should give reasonably consistent counts. Remaining EDTA solution should be collected and disposed as 14C liquid waste.
- To assess the remaining quantity of labeled photoassimilate in shoots, submerge the rosette in 1 ml 80% ethanol, and gently agitate on a shaker for ~1 h to extract pigments and metabolites (a greater volume may be required for larger rosettes). Add 500 µl of commercial bleach and agitate for ~1 h to destroy the pigments. Add sufficient scintillation fluid and mix thoroughly to get a clear, single-phase solution; 5-10 volumes of scintillation fluid to 1 volume of ethanol/bleach solution should suffice.
- (Optional) To measure individual metabolites by HPLC or enzymatic assay, add 500 µl chloroform to exudates, mix and centrifuge to separate the phases. Recover the aqueous phase to fresh microcentrifuge tubes for polar metabolites. Retain the chloroform phase if required to analyze non-polar metabolites.
- The exudates can be stored at -20 °C for further analysis if desired, bearing in mind that exudates are isotopically labeled with 14C.
- Transfer 400 µl of each EDTA solution to scintillation vials (Figure 2M) and add 5 ml scintillation fluid for aqueous samples. Mix thoroughly by shaking the vial to create a monophasic solution. Document counts per min (cpm) or disintegrations per min (dpm) with a scintillation counter and a program suitable for 14C. Calculate 14C in 500 µl exudations as a rate per rosette mass: cpm or dpm (fresh weight)-1 h-1. The first and second hour of exudations should be analyzed by scintillation counting and should give reasonably consistent counts. Remaining EDTA solution should be collected and disposed as 14C liquid waste.
Data analysis
Experimental design for data analysis
Each chamber constitutes an independent labeling experiment. Values obtained from experimental plants in each plate should be standardized to a percent value of WT controls in the same chamber. Standardized values from separate chambers are then combined as independent replicates. This removes chamber to chamber variation in labeling efficiency to provide a more accurate representation of the differences in photoassimilation, distribution and transport between controls and experimental plants (Khadilkar et al., 2016).
Recipes
- EDTA solution (0.5 M stock 500 ml) (FW 292.24)
Dissolve 73.06 g EDTA into 300 ml of ddH2O
Adjust pH to 8.0 with 5 N KOH. Make the final volume 500 ml
Autoclave in liquid cycle for 15 min
Store at room temperature
For working concentration, dilute 100x to prepare 5 mM of the required volume and adjust to pH 6.0
Acknowledgments
This protocol is based on methods published in (Dasgupta et al., 2014; Khadilkar et al., 2016). Work on phloem loading and long distance transport in B.G. Ayre’s laboratory is/was supported by the National Science Foundation 0344088, 0922546, 1121819, and 1558012. The authors report no conflicts of interest or competing interests.
References
- Dasgupta, K., Khadilkar, A. S., Sulpice, R., Pant, B., Scheible, W. R., Fisahn, J., Stitt, M. and Ayre, B. G. (2014). Expression of sucrose transporter cDNAs specifically in companion cells enhances phloem loading and long-distance transport of sucrose but leads to an inhibition of growth and the perception of a phosphate limitation. Plant Physiol 165(2): 715-731.
- Hewer, A., Becker, A. and van Bel, A. J. (2011). An aphid's Odyssey--the cortical quest for the vascular bundle. J Exp Biol 214(Pt 22): 3868-3879.
- Khadilkar, A. S., Yadav, U. P., Salazar, C., Shulaev, V., Paez-Valencia, J., Pizzio, G. A., Gaxiola, R. A. and Ayre, B. G. (2016). Constitutive and companion cell-specific overexpression of AVP1, encoding a proton-pumping pyrophosphatase, enhances biomass accumulation, phloem loading, and long-distance transport. Plant Physiol 170(1): 401-414.
- King, R. W. and Zeevaart, J. A. (1974). Enhancement of phloem exudation from cut petioles by chelating agents. Plant Physiol 53(1): 96-103.
- Liu, D. D., Chao, W. M. and Turgeon, R. (2012). Transport of sucrose, not hexose, in the phloem. J Exp Bot 63(11): 4315-4320.
- Slewinski, T. L., Zhang, C. and Turgeon, R. (2013). Structural and functional heterogeneity in phloem loading and transport. Front Plant Sci 4: 244.
- Tetyuk, O., Benning, U. F. and Hoffmann-Benning, S. (2013). Collection and analysis of Arabidopsis phloem exudates using the EDTA-facilitated method. J Vis Exp (80): e51111.
- Turgeon, R. and Wolf, S. (2009). Phloem transport: cellular pathways and molecular trafficking. Annu Rev Plant Biol 60: 207-221.
- van Bel, A. J. E. and Hess, P. H. (2008). Hexoses as phloem transport sugars: the end of a dogma? J Exp Bot 59: 261-272.
- Will, T., Tjallingii, W. F., Thonnessen, A. and van Bel, A. J. (2007). Molecular sabotage of plant defense by aphid saliva. Proc Natl Acad Sci U S A 104(25): 10536-10541.
- Yadav, U. P., Khadilkar, A. S., Shaikh, M. A., Turgeon, R. and Ayre, B. G. (2017a). Quantifying the capacity of phloem loading in leaf disks with [14C]sucrose. Bio Protoc 7(24): e2658.
- Yadav, U. P., Khadilkar, A. S., Shaikh, M. A., Turgeon, R. and Ayre, B. G. (2017b). Assessing long-distance transport from photosynthetic source leaves to heterotrophic sink organs with [14C]CO2. Bio Protoc 7(24): e2657 .
- Zhang, B., Tolstikov, V., Turnbull, C., Hicks, L. M. and Fiehn, O. (2010). Divergent metabolome and proteome suggest functional independence of dual phloem transport systems in cucurbits. Proc Natl Acad Sci U S A 107(30): 13532-13537.
- Zhang, C., Yu, X., Ayre, B. G. and Turgeon, R. (2012). The origin and composition of cucurbit "phloem" exudate. Plant Physiol 158(4): 1873-1882.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Yadav, U. P., Khadilkar, A. S., Shaikh, M. A., Turgeon, R. and Ayre, B. G. (2017). Assessing Rates of Long-distance Carbon Transport in Arabidopsis by Collecting Phloem Exudations into EDTA Solutions after Photosynthetic Labeling with [14C]CO2. Bio-protocol 7(24): e2656. DOI: 10.21769/BioProtoc.2656.
- Khadilkar, A. S., Yadav, U. P., Salazar, C., Shulaev, V., Paez-Valencia, J., Pizzio, G. A., Gaxiola, R. A. and Ayre, B. G. (2016). Constitutive and companion cell-specific overexpression of AVP1, encoding a proton-pumping pyrophosphatase, enhances biomass accumulation, phloem loading, and long-distance transport. Plant Physiol 170(1): 401-414.
Category
Plant Science > Plant physiology > Nutrition
Plant Science > Plant physiology > Photosynthesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.